

Abstract
Impaired mitochondrial maintenance in disparate cell types is a shared hallmark of many human pathologies and ageing1,2,3,4,5,6,7,8. How mitochondrial biogenesis coordinates with the removal of damaged or superfluous mitochondria to maintain cellular homeostasis is not well understood. Here we show that mitophagy, a selective type of autophagy targeting mitochondria for degradation, interfaces with mitochondrial biogenesis to regulate mitochondrial content and longevity in Caenorhabditis elegans. We find that DCT-1 is a key mediator of mitophagy and longevity assurance under conditions of stress in C. elegans. Impairment of mitophagy compromises stress resistance and triggers mitochondrial retrograde signalling through the SKN-1 transcription factor that regulates both mitochondrial biogenesis genes and mitophagy by enhancing DCT-1 expression. Our findings reveal a homeostatic feedback loop that integrates metabolic signals to coordinate the biogenesis and turnover of mitochondria. Uncoupling of these two processes during ageing contributes to overproliferation of damaged mitochondria and decline of cellular function.
Similar content being viewed by others


Main
A common denominator of numerous pathological conditions and ageing is a deregulation of cellular mitochondrial content1,2,3,4,5,6,7,8. To uncover the cellular and molecular underpinnings of mitochondrial mass homeostasis, we examined the involvement of mitochondria-selective autophagy (mitophagy) in regulating mitochondrial content of C. elegans during ageing. Mitochondria gradually accrue with age in wild-type nematodes1 (Fig. 1a and Extended Data Fig. 1a–d). Depletion of the Atg6/Vps30/Beclin1 homologue BEC-1, the main autophagy regulator in worms9, recapitulates the effect of ageing on mitochondrial mass in young adult animals (Fig. 1b). Thus, failure of autophagy impairs the removal of mitochondria and may contribute to progressive accumulation of mitochondria during ageing.

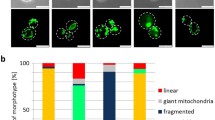
Transgenic animals expressing mitochondria-targeted GFP in the intestine were monitored (a) throughout adulthood for mitochondrial content. Indicative images are shown on the right for days 1, 3, 5 and 10 (n = 90; ***P < 0.001; one-way analysis of variance (ANOVA)). b, BEC-1 deficiency increases mitochondrial mass (n = 100; ***P < 0.001; unpaired t-test). c, Knockdown of either DCT-1, PINK-1 or PDR-1 increases mitochondrial mass (n = 100; ***P < 0.001; one-way ANOVA). Scale bar, 100 μm. d, Depletion of either DCT-1, PINK-1 or PDR-1 alters mitochondrial network morphology in body wall muscle cells (n = 120; ***P < 0.001; one-way ANOVA). Scale bar, 20 μm. Anterior, left; posterior, right. e, Induction of mitophagy is indicated by co-localization of GFP and DsRed (for each group of images, mitochondria are shown in green on top, autophagosomes in red below, with a merged image at the bottom). Scale bars, 20 μm. Images were acquired using ×5 and ×40 objective lenses. Error bars, s.e.m.
To target mitophagy specifically, we knocked down dct-1 (DAF-16/FOXO Controlled, germline Tumour affecting-1)10,11, a putative orthologue to the mammalian NIX/BNIP3L and BNIP3 (Nip3-like protein X/Bcl-2 and adenovirus E1B interacting protein; Extended Data Fig. 2a), which act as mitophagy receptors in mammals12,13,14,15. DCT-1 is widely expressed throughout development as an integral membrane protein and localized on the outer mitochondrial membrane (Extended Data Fig. 2b–g). Expression of dct-1 is partly controlled by the FOXO transcription factor DAF-16 and is elevated upon low insulin/IGF-1 signalling10,11 (Extended Data Fig. 1e, f). Similarly to BEC-1, DCT-1 deficiency increases intestinal and muscle cell mitochondrial content in young adult animals and distorts mitochondrial network morphology (Fig. 1c, d and Extended Data Figs 1g–k and 3a–c). DCT-1 encompasses a WXXL motif and co-localizes with the nematode Atg8/LC3 autophagosome protein LGG-1 on the outer mitochondrial membrane12,13,14,15 (Extended Data Figs 2a and 4a). These findings indicate that DCT-1 is the nematode orthologue of NIX/BNIP3L and functions as a key regulator of mitophagy.
To confirm that impairment of mitophagy underlies mitochondrial accumulation in worms, we developed two composite systems for monitoring mitophagy in vivo. First, we generated transgenic animals expressing mitochondria-targeted Rosella (mtRosella) biosensor that combines a fast-maturing pH-insensitive DsRed fused to a pH-sensitive green fluorescent protein (GFP) variant. To validate this tool, we examined animals carrying mtRosella, under normal and mitophagy-inducing conditions (heat stress, carbonyl cyanide m-chlorophenylhydrazone (CCCP) and paraquat exposure). All treatments reduce the GFP/DsRed ratio of Rosella fluorescence, indicating stimulation of mitophagy. Knockdown of lgg-1 gene, a core component of the general autophagic machinery, blocks mitophagy (Extended Data Fig. 4f–h). Additionally, we generated transgenic animals expressing a mitochondria-targeted GFP, together with the autophagosomal marker LGG-1 fused with DsRed in body wall muscle cells, and looked for co-localization under normal and mitophagy-inducing conditions. Mitophagy-inducing stimuli induce the formation of autophagosomes that extensively co-localize with mitochondria (Extended Data Fig. 5a, b). Pronounced induction of mitophagy was also observed in long-lived daf-2 mutants (Fig. 1e and Extended Data Figs 4f, h and 5b). In all cases, induction of mitophagy is entirely dependent on DCT-1 (Fig. 1e and Extended Data Figs 4f, g and 5a). Additionally, we tested the requirement of the mitochondrial phosphatase and tensin (PTEN)-induced kinase 1 (PINK-1) and the cytosolic E3 ubiquitin ligase Parkin, two critical conserved components of mitophagy16. Consistent with their previously described role, PINK-1 and PDR-1 (the nematode Parkin homologue) deficiency blocks mitophagy under stress or low insulin/IGF-1 signalling, and destabilizes mitochondrial network in wild-type C. elegans cells (Fig. 1c–e and Extended Data Figs 1i, j, 4f, g and 5a). Increased mitochondrial mass in mitophagy-depleted animals is not a compensatory protective mechanism against extensive dysfunctional mitochondria or due to enhanced mitochondrial biogenesis, impaired proteasomal activity or altered tissue-specific promoter activity of the reporters (Extended Data Fig. 6a–h). Furthermore, mitochondrial morphology defects upon mitochondrial dynamics impairment are epistatic to morphological aberrations occurring upon mitophagy inhibition, suggesting the latter are secondary events (Extended Data Fig. 3a–c).
Depletion of CED-9, the nematode homologue of cell death inhibitor Bcl-2 and physical interactor of DCT-1 (refs 17, 18), causes mitochondrial mass increase without affecting DCT-1 mitochondrial localization (Extended Data Fig. 7a, b). Interestingly, CED-9 is required for mitophagy upon oxidative and mitochondrial stress (Extended Data Fig. 7c, d). Simultaneous knocking down of ced-9/dct-1, ced-9/pink-1 and ced-9/pdr-1 does not further increase mitochondrial content, indicating that ced-9, dct-1, pink-1 and pdr-1 act in the same genetic pathway to regulate mitophagy (Extended Data Fig. 7b).
To investigate the contribution of mitophagy to longevity, we knocked down DCT-1, PINK-1 and PDR-1 in wild-type and long-lived mutants. Notably, DCT-1, PINK-1 and PDR-1 impairment functions non-redundantly to abrogate elevated autophagy in long-lived daf-2 mutants and remarkably shortens their lifespan, whereas it does not alter basal autophagy or lifespan of wild-type animals (Fig. 2a–c, Extended Data Figs 4c–e and 8a–f and Supplementary Table 1). Moreover, dct-1 or pink-1 knockdown shortens lifespan extension of mutants with moderated mitochondrial dysfunction (isp-1 and clk-1), or the inherently caloric-restricted eat-2 mutants (Fig. 2d, Extended Data Fig. 8g, h and Supplementary Table 1). We conclude that mitophagy is a common longevity assurance process, underlying the pro-survival effects of diverse manipulations that extend lifespan.

a, Quantification of GFP::LGG-1-positive foci in seam cells of L4 wild-type and daf-2(e1370) animals upon knockdown of either dct-1, pink-1 or pdr-1 (n = 150 cells per assay; ***P < 0.001; one-way ANOVA). Error bars, s.e.m. b, Knockdown of either dct-1, pink-1, pdr-1 or lgg-1 compared with animals subjected to simultaneous knockdown of lgg-1 and dct-1 or lgg-1 and pink-1 or lgg-1 and pdr-1 does not affect the lifespan of otherwise wild-type animals. c, d, Knockdown of either dct-1 or pink-1 shortens the lifespan of long-lived daf-2(e1370), or isp-1(qm150) mutants. Lifespan values are given in Supplementary Table 1; assays were performed at 20 °C.
To elucidate the mechanism by which mitophagy contributes to longevity, we tested mitophagy-depleted animals for stress resistance. The dct-1, pink-1 and pdr-1 mutants are substantially more sensitive to various stressors (Fig. 3a–e and Extended Data Fig. 9a, b), whereas DTC-1 overexpression confers PINK-1- and PDR-1-dependent protection against stress. Interestingly, the WXXL motif of DTC-1 is required for DCT-1-dependent stress resistance (Extended Data Figs 8l and 9g–k). We hypothesized that diminished stress resistance may originate from the accumulation of dysfunctional mitochondria in mitophagy mutants. Indeed, in mitophagy-deficient animals, although mitochondrial DNA (mtDNA) levels remain relatively unchanged (Extended Data Fig. 9f), we noted decreased ATP levels, elevated mitochondrial reactive oxygen species (ROS) generation, mitochondrial membrane depolarization, increased oxygen consumption and cytoplasmic Ca2+ elevation that becomes exacerbated under stress (Fig. 3f–j and Extended Data Fig. 9c–e). These findings, combined with the distorted mitochondrial network morphology noted above, indicate that mitophagy impairment precipitates pronounced mitochondrial dysfunction.

Mutants dct-1(tm376), pink-1(tm1779), pdr-1(gk448) and wild-type animals incubated a, at 37 °C for 7 h (n = 150; ***P < 0.001; one-way ANOVA), b, in CCCP for 2 h (n = 150; ***P < 0.001; one-way ANOVA), or c, exposed to 500 J m−2 of ultraviolet radiation (n = 150; ***P < 0.001; one-way ANOVA), d, treated with 8 mM paraquat (n = 150; ***P < 0.001; one-way ANOVA). e, Survival after 5 days of starvation (n = 150; ***P < 0.001; one-way ANOVA). f, ATP levels are decreased in mitophagy mutants (***P < 0.001; one-way ANOVA). g, Mitophagy-deficient animals display increased mitochondrial ROS formation (n = 180; ***P < 0.001; one-way ANOVA). h, Mitophagy mutants display decreased ratio of dysfunctional mitochondrial to total mitochondrial mass (n = 170; **P < 0.01; one-way ANOVA). i, Oxygen consumption rates (***P < 0.001; one-way ANOVA). j, Transgenic animals expressing the Ca2+-reporter upon knockdown of either dct-1 or pink-1 incubated at 37 °C for 3 h, with or without the presence of CCCP (n = 120; ***P < 0.001, **P < 0.01; one-way ANOVA). Error bars, s.e.m.
To examine the molecular interaction between DCT-1, PINK-1 and PDR-1, we immunoprecipitated DCT-1 upon mitophagy stimulation, and analysed tryptic peptides by mass spectrometry. We found that DCT-1 is ubiquitinated on lysine 26, a modification that becomes enriched under mitophagy-inducing conditions and depends on PINK-1 (Extended Data Fig. 9l–o). Additionally, DCT-1 co-localizes with PDR-1, indicating that DCT-1 ubiquitination upon mitophagy is PDR-1-dependent (Extended Data Fig. 4b).
Despite the aforementioned defects, mitophagy-depleted animals show normal lifespan in the absence of stress stimuli (Fig. 2b–d and Extended Data Fig. 8a–h). To resolve this apparent paradox, we considered whether accumulation of dysfunctional mitochondria upon mitophagy impairment signals a compensatory retrograde response that induces expression of nuclear genes encoding mitochondrial proteins. Accumulation of damaged mitochondria induces oxidative stress. In mammalian cells, the nuclear factor-erythroid 2-related factor 2 (NRF2/NFE2L2) is activated under oxidative stress conditions and orchestrates the expression of several genes promoting survival19,20. SKN-1 (SKiNhead-1), the nematode homologue of NRF2, was recently found to associate with outer mitochondrial membrane proteins21. SKN-1 transcriptional activity is pivotal for mitohormesis-mediated longevity22,23, and is implicated in the maintenance of mitochondrial homeostasis24,25,26. Interestingly, SKN-1 becomes specifically activated upon mitophagy inhibition (Fig. 4a and Extended Data Fig. 10a–d). SKN-1 is required for the expression of several mitochondrial biogenesis genes as well as the mitophagy mediator dct-1 (Fig. 4b and Extended Data Fig. 10e, h). In addition to DAF-16, dct-1 expression is also controlled by SKN-1 (Fig. 4c). These findings suggest that SKN-1 may control mitophagy through regulation of dct-1 transcription. Indeed, skn-1 knockdown impairs mitochondrial network morphology, causes mitochondrial membrane depolarization and increases cytoplasmic Ca2+ concentration (Fig. 4d–f and Extended Data Fig. 10g), similarly to DCT-1 deficiency. Notably, SKN-1 depletion reduces mtDNA content, further highlighting the role of SKN-1 in the maintenance of mitochondrial integrity (Extended Data Fig. 10f). Furthermore, induction of mitophagy under low insulin/IGF-1 signalling or stress is diminished in the absence of SKN-1 (Fig. 4g and Extended Data Fig. 10i, j, l). DAF-16 activity is also required for mitophagy stimulation. Simultaneous depletion of either DAF-16 and DCT-1, or SKN-1 and DCT-1, does not cause a more pronounced mitophagy defect (Extended Data Fig. 10j), indicating that DAF-16, SKN-1 and DCT-1 are components of a common genetic pathway regulating mitophagy. Knockdown of either dct-1 or pink-1 shortens the lifespan of skn-1 mutant in contrast to wild-type animals (Extended Data Fig. 10k and Supplementary Table 1), indicating that SKN-1 activation compensates the reduced basal mitophagy to maintain normal lifespan. Treatment of mitophagy-deficient animals with the ROS scavengers N-acetyl-l-cysteine (NAC) and butylated hydroxyanisole (BHA)27,28 does not change their lifespan, ruling out the involvement of mitohormetic feedback signalling (Extended Data Fig. 8i, j and Supplementary Table 1). Surprisingly, supplementation of ethylene glycol tetraacetic acid (EGTA), a calcium-specific chelating agent, shortens the lifespan of mitophagy-deficient worms without influencing lifespan of wild-type animals (Extended Data Fig. 8k). Furthermore, SKN-1 activation is diminished in mitophagy-depleted animals upon EGTA treatment (Extended Data Fig. 10n). Elevated cytoplasmic calcium induces mitochondrial biogenesis through calcium/calmodulin-dependent protein kinase II (CaMKII), p38 mitogen activated protein kinase (MAPK), and PGC-1a29,30. Indeed, UNC-43 (the nematode homologue of CaMKII) regulates the transcriptional activity of SKN-1 upon mitophagy inhibition, where cytoplasmic calcium levels are increased (Extended Data Fig. 10m, o). Our observations suggest that cytoplasmic calcium elevation ensuing upon mitophagy impairment mediates SKN-1 activation, which overcomes the cost of basal mitophagy reduction on longevity.

a, Fluorescence intensity of transgenic animals expressing the pgst-4GFP transgene subjected to either dct-1, pink-1 and pdr-1 knockdown (n = 180; ***P < 0.001; one-way ANOVA). Scale bars, 100 μm. b, Messenger RNA (mRNA) levels of atp-5, gas-1, hmg-5, tim-17, W09C5.8 and dct-1 in wild-type and SKN-1-depleted animals (*P < 0.01; one-way ANOVA). c, mRNA levels of dct-1 in animals with the indicated genetic backgrounds (***P < 0.001, *P < 0.01; one-way ANOVA). d, Knockdown of SKN-1 alters mitochondrial network morphology in body wall muscle cells (n = 120; ***P < 0.001; unpaired t-test). Scale bar, 20 μm. e, TMRE staining of wild-type and SKN-1-deficient animals (n = 150; ***P < 0.001; one-way ANOVA). f, Transgenic animals expressing the Ca2+-reporter upon knockdown of skn-1 (n = 120; ***P < 0.001; unpaired t-test). g, Mitophagy is not activated in SKN-1-deficient animals under low insulin signalling conditions (for each group of images mitochondria are shown in green on top, autophagosomes in red below, with a merged image at the bottom). Scale bars, 20 μm. h, The contribution of the DCT-1 mitophagy receptor and SKN-1 to mitochondrial physiology and consequently to cellular homeostasis is depicted. DCT-1 is an essential mitophagy mediator that is transcriptionally regulated by both DAF-16 and SKN-1. Under normal conditions, basal-level mitophagy maintains the mitochondrial pool and regulates mitochondrial number, depending on the metabolic state of the cell. Mitophagy is triggered under stress conditions to eliminate damaged mitochondria and promote cell survival. Concomitant activation of SKN-1 enhances mitophagy and promotes mitochondrial biogenesis by stimulating expression of DCT-1 and several mitochondrial genes. Error bars, s.e.m. Images were acquired using ×5 and ×40 objective lenses.
Overall, our analysis uncovers a key regulatory mechanism that interfaces mitochondrial biogenesis with mitochondrial clearance through selective autophagy, to preserve energy homeostasis in C. elegans. A central node of the pathway is the mitophagy mediator DCT-1/NIX, which integrates insulin/IGF-1 signals via DAF-16 and SKN-1, in addition to other stress signals, relayed through SKN-1, to control mitophagy (Fig. 4h). Accumulation of damaged or superfluous mitochondria causes oxidative stress, initiating a bipartite retrograde response that is mediated by SKN-1 and involves the coordinated induction of both mitochondrial biogenesis and mitophagy genes. This closed feedback loop preserves mitochondrial quality by neutralizing damaged, and generating fresh mitochondria (Fig. 4h). Moreover, coordination of biogenesis and turnover of mitochondria allows cells to adjust their mitochondrial content in response to physiological demands, stress and other intracellular or environmental stimuli. In this context, age-related decline of mitophagy both hinders removal of damaged mitochondria and impairs mitochondrial biogenesis to instigate progressive mitochondrial accumulation. The tight evolutionary conservation and ubiquitous expression of the regulatory factors involved in this closely orchestrated response suggest that similar pathways might uphold mitochondrial homeostasis across different taxa.
Methods
No statistical methods were used to predetermine sample size.
Strains and genetics
We followed standard procedures for C. elegans strain maintenance31. Nematode rearing temperature was kept at 20 °C, unless noted otherwise. The following strains were used in this study: N2: wild-type Bristol isolate, CB1370: daf-2(e1370)III, CB4876: clk-1(e2519)III, MQ887: isp-1(qm150)IV, DA465: eat-2(ad465)II, CF1038: daf-16(mu86)I, pink-1(tm1779)II, dct-1(tm376)X, VC1024: pdr-1(gk448)III, N2;Ex[pmyo-3INVOM::RFP], SJ4103: N2;Is[pmyo-3mtGFP], SJ4143: N2;Is[pges-1mtGFP], CL2166: N2;Is[pgst-4GFP], CF1553: N2;Is[psod-3GFP], SJ4005: N2;Is[phsp-4GFP]V, SJ4058: N2;Is[phsp-60GFP]V, EU1: skn-1(zu67) IV/nT1(IV;V), EU31: skn-1(zu135)IV/nT1(IV;V), EU40: skn-1(zu129)IV/nT1(IV;V). To investigate the localization of DCT-1, we examined IR1431: N2;Ex001[pdct-1DCT-1::GFP] and IR1607: N2;Is[pmyo-3INVOM::RFP];Ex001[pdct-1DCT-1::GFP] transgenic animals. The following strains were used to monitor mitophagy process: IR1284: N2;Is[pmyo-3mtGFP];Ex011[plgg-1DsRed::LGG-1], IR1511: N2;Ex001[pmyo-3DsRed::LGG-1;pdct-1DCT-1::GFP] and IR1631: N2;Ex003[pmyo-3TOMM-20::Rosella]. To examine mitochondrial morphology and mitochondrial mass under mitophagy-deficient conditions, we used the SJ4103: N2;Is[pmyo-3mtGFP], IR1298: dct-1(tm376);Is[pmyo-3mtGFP] and SJ4143: N2;Is[pges-1mtGFP], IR1505: N2;Ex001[punc-119CTS-1::mCherry] strains. To monitor autophagy induction after inhibition of mitophagy, we used the DA2123: N2;Is[plgg-1GFP::LGG-1], MAH14: daf-2(e1370);Is[plgg-1GFP::LGG-1] and IR621: N2;Ex002[plgg-1DsRed::LGG-1] strain. To monitor DCT-1 and PDR-1 interaction in vivo, we used the following transgenic animal: IR1739: N2;Ex001[pmyo-3PDR-1::DsRed;pdct-1DCT-1::GFP]. To investigate the protective role of global expression of dct-1 under mitophagy inducing conditions, we examined the following transgenic animals: IR1431: N2;Ex001[pdct-1DCT-1::GFP], IR1487: pink-1(tm1779);Ex001[pdct-1DCT-1::GFP], IR1733: pdr-1(gk448);Ex001[pdct-1DCT-1::GFP], IR1666: dct-1(tm376);Ex002[plet-858DCT-1], IR1668: dct-1(tm376);Ex001[plet-858DCT-1(ΔMER)], IR1667: dct-1(tm376);Ex003[plet-858DCT-1(ΔBH3)], IR1669: dct-1(tm376);Ex003[plet-858DCT-1(ΔWXXL)]. The following strain was used for monitoring cytoplasmic calcium levels: IR1155: N2;Ex001[plet-858GCaMP2.0]. To monitor the levels of ROS, such as H2O2, in mitophagy-deficient animals, we used the JRIS1: N2;Is[prpl-17HyPer] strain32.
Molecular cloning
To generate the pdct-1DCT-1::GFP reporter construct, we fused a KpnI fragment, containing the coding sequence of dct-1 and 2 kb upstream of the coding sequence, amplified from C. elegans genomic DNA using the primers 5′-GGATCCCATCTTCTGTACCACGCA-3′ and 5′-GGTACCGCTTGACGATGATGTTTGAT-3′, at the amino (N) terminus of GFP, in the pPD95.77 plasmid vector. The translational pdct-1DCT-1::GFP fusion construct was co-injected with pRF4 (contains the rol-6(su1006) dominant transformation marker) into the gonads of wild-type animals. To generate the plet-858GCaMP2.0 reporter construct, we removed the mec-17 promoter form pmec-17GCaMP2.0 described previously33, and inserted a HindIII/SmaI fragment (derived from the L3786 plasmid vector), containing the sequence of the let-858 promoter upstream of GCaMP2.0. The resulting plasmid construct was co-injected with pRF4 into the gonad of wild-type animals. To generate pmyo-3DsRed::LGG-1, we inserted an AgeI/EcoRI fragment (derived from plgg-1DsRed::LGG-134), containing the coding sequence of DsRed downstream of the myo-3 promoter of the pPD96.52 plasmid vector. We then fused an EcoRI fragment (derived from plgg-1DsRed::LGG-134) containing the coding sequence of lgg-1 at the carboxy (C) terminus of DsRed of the pmyo-3DsRed. The translational pmyo-3DsRed::LGG-1fusion construct was co-injected with pmyo-2GFP transformation marker. To generate pmyo-3TOMM-20::Rosella, we inserted an EcoRI fragment (derived from the mtRosella yeast vector35,36) containing the coding sequence of Rosella biosensor in pPD96.52 under myo-3 promoter. We then inserted a BamHI fragment containing the two first exons of tomm-20 gene downstream of the myo-3 promoter and upstream of the Rosella coding sequence. The tomm-20 fragment was amplified from C. elegans genomic DNA using the primers 5′-GGATCCATGTCGGACACAATTCTTGG-3′ and 5′-GGATCCTTCGCGAGTCGACGGC-3′. The translational pmyo-3TOMM-20::Rosella fusion construct was co-injected with pRF4 into the gonads of wild-type animals. To generate pmyo-3PDR-1::DsRed, we inserted an AgeI/BamHI fragment containing the full length pdr-1 gene without the stop codon, amplified from C. elegans genomic DNA using the primers 5′-GTCGACACTGCAAGCAGGCCCGC-3′ and 5′-ACCGGTGGATTAAACCAATGGTCCCATTG-3′, at the N terminus of DsRed, in the pPD96.52 plasmid vector. The translational pmyo-3PDR-1::DsRed fusion construct was co-injected with pRF4 into the gonads of wild-type animals. To generate punc-119CTS-1::mCherry, we inserted a XbaI/XmnI fragment containing the first two exons of cts-1 gene, amplified from C. elegans genomic DNA using the primers 5′-ATGTCGCTCTCTGGAATGGC-3′ and 5′-GGTGCTACCGTGCTCTGTGC-3′, at the N terminus of GFP, in the pPD95.77 plasmid vector. We then inserted a HindIII/XbaI fragment, amplified from C. elegans genomic DNA using the primers 5′-CTCTCAAAATTCAGTTTTTTAAACCAC-3′ and 5′-ATATGCTGTTGTAGCTGAAAATTTTG-3′ containing the sequence of the unc-119 promoter upstream of CTS-1::GFP. punc-119CTS-1 was inserted as a KpnI fragment upstream of mCherry in the pPD95.77 plasmid vector. The translational punc-119CTS-1::mCherry fusion construct was co-injected with pRF4 into the gonads of wild-type animals. To generate plet-858DCT-1 rescue plasmid, we inserted an XbaI/XhoI fragment containing dct-1 complementary DNA (cDNA) sequence in pPD96.52. The dct-1 fragment was amplified from C. elegans cDNA using the primers 5′-TCTAGAATGTCCTCATTTCTTGAGTTTGC-3′ and 5′-CTCGAGTTGACGATGATGTTTGATTAGTTTC-3′. We then removed the myo-3 promoter from pPD96.52 and inserted a HindIII/XmaI fragment (derived from the L3786 plasmid vector), containing the let-858 promoter, upstream of dct-1. The translational plet-858DCT-1 fusion construct was co-injected with pRF4 into the gonads of dct-1(tm376) animals. To generate the plet-858DCT-1(ΔWXXL) plasmid that contains a deletion of WXXL motif, we inserted an XbaI/AgeI fragment fused with an AgeI/XhoI fragment of the dct-1 cDNA sequence in pPD96.52, spanning the sequence that encodes for WXXL motif. The dct-1(ΔWXXL) fragment was amplified from C. elegans cDNA using the primers 5′-TCTAGAATGTCCTCATTTCTTGAGTTTGC-3′ and 5′-ACCGGTCGATTCTGACATACCAGGTG-3′, 5′-ACCGGTGCACCAAGCCGTACCAGTT-3′ and 5′-CTCGAGTTGACGATGATGTTTGATTAGTTTC-3′. We the removed the myo-3 promoter from pPD96.52 and we inserted a HindIII/XmaI fragment (derived from the L3786 plasmid vector), containing the sequence of the let-858 promoter upstream of DCT-1(ΔWXXL). The translational plet-858DCT-1(ΔWXXL) fusion construct was co-injected with pRF4 into the gonads of dct-1(tm376) animals. To generate plet-858DCT-1(ΔMER) plasmid that contains a deletion of the MER domain, we inserted an XbaI/AgeI fragment fused with an AgeI/XhoI fragment of dct-1 cDNA sequence in pPD96.52 spanning the sequence that encodes for MER domain. dct-1(ΔMER) fragment was amplified from C. elegans cDNA using the primers 5′-TCTAGAATGTCCTCATTTCTTGAGTTTGC-3′ and 5′-ACCGGTGACGCTACTGCACAAACT-3′, 5′-ACCGGTGTCTCAATCGCACAATCTC-3′ and 5′-CTCGAGTTGACGATGATGTTTGATTAGTTTC-3′. We then removed the myo-3 promoter from pPD96.52 and inserted a HindIII/XmaI fragment (derived from the L3786 plasmid vector), containing the let-858 promoter upstream of dct-1(ΔMER). The translational plet-858DCT-1(ΔMER) fusion construct was co-injected with pRF4 into the gonads of dct-1(tm376) animals. To generate plet-858DCT-1(ΔBH3) plasmid that contains a deletion of the BH3 domain, we inserted an XbaI/AgeI fragment fused with an AgeI/XhoI fragment of dct-1 cDNA sequence in pPD96.52, spanning the sequence that encodes for the BH3 domain. dct-1(ΔBH3) fragment was amplified from C. elegans cDNA using the primers 5′-TCTAGAATGTCCTCATTTCTTGAGTTTGC-3′ and 5′-ACCGGTGTACTTTACTTGTTCTAAGCTCTC-3′, 5′-ACCGGTTGGAGCAGTCGGCCA-3′ and 5′-CTCGAGTTGACGATGATGTTTGATTAGTTTC-3′. We then removed the myo-3 promoter from pPD96.52 and inserted a HindIII/XmaI fragment (derived from the L3786 plasmid vector), containing the let-858 promoter upstream of dct-1(ΔBH3). The translational plet-858DCT-1(ΔBH3) fusion construct was co-injected with pRF4 into the gonads of dct-1(tm376) animals. For engineering the skn-1, dct-1, pdr-1, ced-9, pbs-5, rpn-6, fzo-1, drp-1, isp-1, clk-1 and unc-43 RNAi constructs, gene-specific fragments of interest were obtained by PCR amplification directly from C. elegans genomic DNA using the following sets of primers 5′-TCAGTTCACCATCGTCCAACACCTC-3′ and 5′-CTGCTGCTCCAACGACAAAGCTG-3′, 5′-TTGCGTCCGGCGAGAAGACG-3′ and 5′-CTGCTGCTCCAACGACAAAGCTG-3′, 5′-GTCGACACTGCAAGCAGGCCCGC-3′ and 5′-ACCGGTGGATTAAACCAATGGTCCCATTG-3′, 5′-ATGACACGCTGCACGGCGG-3′ and 5′-CTTCAAGCTGAACATCATCCGCCC-3′, 5′-ACCGGTATGTGGGGCGAGACATTCG-3′and 5′-GGGCCCACGTCATCAACACCCAGCC-3′, 5′-GAAACGAGTTCACGAGAAGACACTA-3′ and 5′-GGAAGGTCAAGCATCACCTGAA-3′, 5′-ATTCGCGTCGTGCTGCCG-3′ and 5′-ATGGCGTTGGCGGAGAGTCC-3′, 5′-ATGGAAAATCTCATTCCTGTCGTC-3′ and 5′-CGCTCTTTCTCCGGTTGATG-3′, 5′-ATGGCTTCTCTTGCTAGATCTGGAGG-3′ and 5′-AGTGAGATCCGTGACAAGGGCAG-3′, 5′-ATGTTCCGTGTAATAACCCGTG-3′ and 5′-TCTCAGCAATCGCAATAGCTC-3′, 5′-CCAAGTTTAGTGACAATTACGATGTG-3′ and 5′-CTTCTTGAGAACTTCTGGCGACA-3′ respectively. The PCR-generated fragments were subcloned into the pL4440 plasmid vector. The resulting constructs were transformed into HT115(DE3) Escherichia coli bacteria deficient for RNase III. Bacteria carrying an empty vector were used in control experiments. For skn-1, pbs-5 and rpn-6 RNAi, the effect can be severe, leading to embryonic lethality of the animals.
mRNA quantification
To quantify dct-1, atp-5, hmg-5, gas-1, tim-17, W09C5.8, pink-1, pdr-1, ges-1, myo-3 and unc-119 mRNA levels, total RNA was extracted using the TRIzol reagent (Invitrogen). The following sets of primers were used, respectively: 5′-GGCTCCAACCTTACCACTCC-3′ and 5′-GCAAATCCTACTGCTGCTCC-3′, 5′-ACTGGTCGAAGCTCGCCGAG-3′ and 5′-ACTCGGCTGGAACCTCTCCG-3′, 5′-CGTCCAAGTGTTCCTCCAAGTG-3′ and 5′-CTTCGCTTCGTCTGTGTACTTCTTT-3′, 5′-CTGGATGTTCGAGGAACGTG-3′ and 5′-AGCTGACACAAGTCCGATGTC-3′, 5′-TGCGAGAAGTACGAATGAGAAGCAC-3′ and 5′-CGAGACGAATCCTTTCCATCTTTGG-3′, 5′-ATGCTGCCACGTTTGGCTCTCC-3′ and 5′-ACGGAAGCTGTAGCGGTACAAGAGC-3′, 5′-AGCATATCGAATCGCAAATGAGTTAG-3′ and 5′-TCGACCGTGGCGAGTTACAAG-3′, 5′-AGCCACCGAGCGATTGATTGC-3′ and 5′-GTGGCATTTTGGGCATCTTCTTG-3′, 5′-TCGCCAAGAGGTATGCTTCACAAG-3′ and 5′-TGCTGCTCCTGCACTGTATCCC-3′, 5′-AGCCAGACGGTGAGGAAGATGC-3′ and 5′-TGCGTCAAGGGTCTTGTTACATCTGG-3′, 5′-AACAACAACAATCGATCGCACC-3′ and 5′-CACGTGTTCCGTGTCGAGGTC-3′. Results were normalized to genomic DNA using the following primers specific for ama-1: 5′-CCTACCTACACTCCAAGTCCATCG-3′ and 5′-GGTGAAGCTGGCGAATACGTTG-3′ and pmp-3: 5′-ATGATAAATCAGCGTCCCGAC-3′ and 5′-TTGCAACGAGAGCAACTGAAC-3′. For cDNA synthesis, mRNA was reverse transcribed using a iScriptTM cDNA Synthesis Kit (BioRad) and PrimeScriptTM Reverse Transcriptase (Takara). Quantitative PCR was performed in triplicate using a Bio-Rad CFX96 Real-Time PCR system (Bio-Rad).
Ca2+ monitoring
For intracellular Ca2+ monitoring experiments, transgenic animals expressing the Ca2+ reporter GCaMP2.0 globally were examined under a Zeiss AxioImager Z2 epifluorescence microscope. L4 larvae were grown on RNAi plates with or without the presence of 15 μm CCCP. RNAi-treated animals against dct-1 and pink-1 were monitored on day 4 of adulthood. For heat stress assay, L4 larvae were grown on RNAi plates. On day 4 of adulthood, worms were monitored after 3 h of incubation at 37 °C. Worms were immobilized with levamisole before mounting on 2% agarose pads for microscopic examination with a Zeiss AxioImager Z2 epifluorescence microscope. Images were acquired under the same exposure. Average pixel intensity values were calculated by sampling images of different animals. We calculated the mean and maximum pixel intensity for each animal in these images using the ImageJ software (http://rsb.info.nih.gov/ij/). Mean values were compared using unpaired t-tests. For multiple comparisons, we used the one-factor (ANOVA) variance analysis corrected by the post hoc Bonferroni test. For each experiment, at least 100 animals were examined for each strain/condition. Each assay was repeated at least three times. We used the Prism software package (GraphPad Software) for statistical analyses.
Mitochondrial imaging
For MitoTracker staining, 500 μl heat-inactivated OP50 (65 °C, 30 min) were mixed with 100 μl MitoTracker Red FM (catalogue number M-22425; Molecular Probes, Invitrogen) stock solution (100 μM) and seeded on agar plates. Animals were incubated for 2 days with the compound. Stained and washed worms were immobilized with levamisole before mounting on 2% agarose pads for microscopic examination with a Zeiss AxioImager Z2 epifluorescence microscope. For the quantification of mitochondrial ROS, 500 μl heat-inactivated OP50 (65 °C, 30 min) were mixed with 100 μl MitoTracker Red CM-H2X ROS (catalogue number M-7513; Molecular Probes, Invitrogen) stock solution (100 μM) and seeded on agar plates. Animals were incubated for 2 days with corresponding compound. Stained and washed worms were immobilized with levamisole before mounting on 2% agarose pads for microscopic examination with a Zeiss AxioImager Z2 epifluorescence microscope. For TMRE staining (tetramethylrhodamine, ethyl ester, perchlorate; a dye that accumulates in intact, respiring mitochondria), 1-day-adult animals were grown at 20 °C or 25 °C in the presence of 150 nM TMRE for 24 h. Stained and washed worms were immobilized with levamisole before mounting on 2% agarose pads for microscopic examination with a Zeiss AxioImager Z2 epifluorescence microscope. Images were acquired under the same exposure. Average pixel intensity values were calculated by sampling images of different animals. We calculated the mean and maximum pixel intensity for each animal in these images using the ImageJ software (http://rsb.info.nih.gov/ij/). For each experiment, at least 150 animals were examined for each strain/condition. Each assay was repeated at least three times. We used the Prism software package (GraphPad Software) for statistical analyses.
Stress assays
To evaluate thermotolerance (heat stress assay), 4-day-old adult hermaphrodites were placed on pre-warmed (37 °C) NGM plates and incubated at 37 °C. After incubation for 7 h, plates were removed; worms were recovered at 20 °C for 1 h. Then, worms were scored for motility, provoked movement and pharyngeal pumping. Worms failing to display any of these traits were scored as dead. Three distinct populations of 150 adults (for each strain) were scored repeatedly over the assay period. For CCCP stress assays, 4-day-old adult hermaphrodites were incubated for 2 h at 20 °C in M9 buffer containing CCCP at a final concentration of 15 μm. Animals were scored for survival after a 1-h recovery period at 20 °C. Three distinct populations of 150 adults (for each strain) were scored repeatedly over the assay period. For ultraviolet stress assays, 4-day-old adult hermaphrodites were exposed to 500 J m−2 ultraviolet irradiation using a ultraviolet crosslinker (BIO-LINK-BLX-E365, Vilber Lourmat). Three days after exposure, animals were scored for survival. Three distinct populations of 150 adults (for each strain) were scored repeatedly over the assay period. For oxidative stress assays, 7-day-old adult hermaphrodites were exposed to paraquat at a final concentration of 8 mM. Animals were scored for survival after 2 days at 20 °C. Three distinct populations of 150 adults (for each strain) were scored repeatedly over the assay period. For the starvation assay, worm eggs were collected upon hypochlorite treatment of gravid adults, resuspended in M9 buffer and incubated at 20 °C. Newly hatched worms were arrested in L1 larval stage owing to lack of food. Survival under starvation was assessed every 2 days by taking aliquots and scoring live animals. Three distinct populations (for each strain) were scored repeatedly over the assay period. We used the Prism software package (GraphPad Software) for statistical analyses.
ATP measurements
To determine ATP content, 50 4-day-old adult hermaphrodites were collected in 50 ml of M9 buffer and frozen at −80 °C. Frozen worms were immersed in boiling water for 15 min, cooled and centrifuged to pellet insoluble debris. The supernatant was moved to a fresh tube and diluted tenfold before measurement. ATP content was determined by using the Roche ATP bioluminescent assay kit HSII (Roche Applied Science) and a TD-20/20 luminometer (Turner Designs). ATP levels were normalized to total protein content. We used the Prism software package (GraphPad Software) for statistical analyses.
Mitochondrial DNA quantification
mtDNA was quantified using quantitative real time PCR as described previously38. We used the primers 5′-GTTTATGCTGCTGTAGCGTG-3′ and 5′-CTGTTAAAGCAAGTGGACGAG-3′ (Mito1 set) for mtDNA. The results were normalized to genomic DNA using the following primers specific for ama-1: 5′-TGGAACTCTGGAGTCACACC-3′ and 5′-CATCCTCCTTCATTGAACGG-3′. Quantitative PCR was performed using a Bio-Rad CFX96 Real-Time PCR system, and was repeated three times.
Oxygen consumption rate measurements
Oxygen consumption rates were measured as previously described37,38 using a Clark-type electrode with some minor modifications (Hansatech Instruments). Young adult worms were washed and collected in M9 buffer. Approximately 100 μl of slurry pellet of worms were delivered into the chamber in 1 ml of M9 buffer. The chamber was kept at 20 °C, and measurements were done for 10–15 min, depending on the oxygen consumption rate. The slope of the straight portion of the plot was used to derive the oxygen consumption rate. Worms were recovered after respiration measurements and collected for protein quantification. Rates were normalized to protein content. We performed three independent measurements per strain. We used the Prism software package (GraphPad Software) for statistical analysis.
Western blotting
Protein samples were analysed by 8% Tricine-SDS–polyacrylamide gel electrophoresis (SDS–PAGE), transferred on nitrocellulose membrane and blotted against various antibodies. The antibodies used in the study were anti-GFP (rabbit polyclonal, Minotech Biotechnology), anti-HSP-60 (mouse monoclonal-Developmental Studies Hybridoma Bank) and anti-β-tubulin (mouse monoclonal-Abcam). For total worm protein extraction, protein samples were produced by directly boiling worms in 2× Laemmli sample buffer with β-mercaptoethanol supplemented with 5 mM PMSF and complete mini proteinase inhibitor cocktail (Roche).
Mitochondria isolation
Mixed populations of worms were incubated in 10 mM DTT in M9 for 30 min at 4 °C with rotation. DTT was washed off with three successive centrifugations and dilutions in M9. Worm bodies were homogenized by hand with a 3 ml Potter-Elvehjem homogenizer with PTFE pestle and glass tube (Sigma-Aldrich) in isolation buffer (50 mM Tris-HCl pH 7.4, 210 mM mannitol, 70 mM sucrose, 0.1 mM EDTA, 2 mM PMSF, and complete mini protease inhibitor cocktail (Roche), the last two being added just before use). The progress of homogenization was monitored by visual inspection under a stereoscope. The suspension was centrifuged at 200g for 1 min and the supernatant fraction kept separately while the pellet was resuspended in isolation buffer and homogenized as previously. The homogenate was centrifuged again at 200g for 1 min. Supernatants were combined and centrifuged once more at low speed (200g) for 1 min and then at high speed (12,000g). The pellet was resuspended in isolation buffer and centrifuged at 200g for 1 min. An aliquot of the supernatant was kept separately for protein concentration determination. High-speed centrifugation followed, and mitochondrial pellets were used directly for proteinase K treatment and carbonate extraction and/or SDS–PAGE.
Proteinase K treatment
Fifty micrograms of crude mitochondria from mixed worm population were digested with 100 μg ml−1 proteinase K (Sigma-Aldrich) in isolation buffer minus protease inhibitors for 30 min. The reaction was inhibited with 5 mM PMSF for 10 min on ice. Proteinase K-shaved mitochondria were analysed by SDS–PAGE and western blotting.
Sodium carbonate extraction
Fifty micrograms of crude mitochondria from mixed worm population were diluted in 0.1 M sodium carbonate (pH 11.5), and incubated on ice for 30 min. The suspension was centrifuged for 30 min at 55,000g in a Beckman Optima TLX-120 ultracentrifuge. Soluble and membrane fractions were analysed by western blotting.
Immunoprecipitation
For immunoprecipitation, mixed populations of worms were treated for 3 h with 10 μΜ MG132 (Sigma-Aldrich) in M9 buffer and subsequently with 10 μm MG132 and 60 mM paraquat (methyl viologen dochliride, Sigma Aldrich) in M9 buffer. Worm lysis was performed by hand with pellet pestle (Sigma-Aldrich) in lysis buffer (50 mM HEPES-KOH, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 1% Triton-X100, 1% NP-40, 0.1% SDS) supplemented with 1 mM DTT, 5 mM PMSF (Sigma-Aldrich), protease and phosphatase inhibitor cocktails (cOmplete mini protease inhibitors cocktail tablets-ROCHE, phosphatase inhibitors cocktail-Cell Signaling), 20 μm NEM (Sigma-Aldrich) and 10 μm MG132 just before use. The lysate was incubated on ice for 30 min and then centrifuged for 15 min at 16,000g at 4 °C. Supernatant was diluted three times in dilution buffer (50 mM HEPES-KOH, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol) supplemented with 5 mM PMSF (Sigma-Aldrich), protease and phosphatase inhibitor cocktails (cOmplete mini protease inhibitors cocktail tablets (Roche), phosphatase inhibitors cocktail (Cell Signaling), 20 μm NEM (Sigma-Aldrich) and 10 μm MG132 just before use. An equal amount of protein from each sample was incubated for 4 h at 4 °C with Sepharose-Protein G beads (Amersham–GE Healthcare) pre-loaded with rabbit anti-GFP antibody (Minotech Biotechnology), cross-linked onto protein G with BS3 cross-linker (Thermo Scientific). The immunoprecipitated proteins were digested by trypsin for mass spectrometry analysis.
Sample preparation and mass spectrometry analysis
Sepharose beads carrying the immunoprecipitated material were washed twice in 50 mM ammonium bicarbonate buffer. Subsequently, 1 μg of trypsin (sequencing grade (Roche Applied Science)) was added per sample and digestion was performed on beads for 14 h at 37 °C. Tryptic peptides were dried and analysed by nanoflow liquid chromatography-electrospray ionization with tandem mass spectrometry (nLC-ESI–MS/MS) on an EASY-nLC system (Thermo Scientific, software version 2.7.6 #1) coupled with an LTQ-Orbitrap XL ETD (Thermo Scientific) through a nES ion source (Thermo Scientific). Data were acquired with Xcalibur software (LTQ Tune 2.5.5 sp1, Thermo Scientific). The nLC-MS/MS raw data were loaded in Proteome Discoverer 1.3.0.339 (Thermo Scientific) and run using both Mascot 2.3.02 (Matrix Science) and Sequest (Thermo Scientific) search algorithms against the C. elegans theoretical proteome (last modified 11 June 2014, version 108). Mass spectrometry was performed in-house in the Proteomics Facility at Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology - Hellas (http://profi.imbb.forth.gr).
Lifespan assays
Lifespan assays were performed at 20 °C unless noted otherwise. Synchronous animal populations were generated by hypochlorite treatment of gravid adults to obtain tightly synchronized embryos that were allowed to develop into adulthood under appropriate, defined conditions. The antioxidant NAC was administered at a final concentration of 10 mM. NAC was added into NGM media from a concentrated stock solution (500 mM) before plate pouring. The antioxidant butylated hydroxyanisole (BHA) was administered at a final concentration of 25 μm. BHA was added into NGM media from a concentrated stock solution (50 mM) before plate pouring. Ethidium bromide (EtBr) was administered at a final concentration of 5 μg ml−1. EtBr was added into NGM media from a concentrated stock solution (10 mg ml−1) before plate pouring. Ethylene glycol tetraacetic acid (EGTA) was administered at a final concentration of 10 mM. EGTA concentration was prepared by dilutions in 150 μl of sterilized water, from a concentrated stock solution (0.5 M), and applied to the top of the agar medium. Plates were then gently swirled to allow the drug to spread to the entire NGM surface. Identical drug-free water solutions were used for the control plates. For RNAi lifespan experiments, worms were placed on NGM plates containing 2 mM IPTG and seeded with HT115(DE3) bacteria transformed with either the pL4440 vector or the test RNAi construct. Progeny were grown at 20 °C unless noted otherwise, through the L4 larval stage and then transferred to fresh plates in groups of 10–20 worms per plate for a total of 100–150 individuals per experiment. The day of egg collection and initiation of RNAi was used as t = 0. Animals were transferred to fresh plates every 2–4 days thereafter and examined every day for touch-provoked movement and pharyngeal pumping, until death. Worms that died owing to internally hatched eggs, an extruded gonad or desiccation due to crawling on the edge of the plates were censored and incorporated as such into the data set. Each survival assay was repeated at least twice and figures represent typical assays. Survival curves were created using the product-limit method of Kaplan and Meier. The log-rank (Mantel–Cox) test was used to evaluate differences between survivals and determine P values. We used the Prism software package (GraphPad Software) for statistical analysis and to determine lifespan values1.
References
Artal-Sanz, M. & Tavernarakis, N. Prohibitin couples diapause signalling to mitochondrial metabolism during ageing in C. elegans. Nature 461, 793–797 (2009)
Campbell, G. R., Ohno, N., Turnbull, D. M. & Mahad, D. J. Mitochondrial changes within axons in multiple sclerosis: an update. Curr. Opin. Neurol. 25, 221–230 (2012)
Kaeberlein, M. Lessons on longevity from budding yeast. Nature 464, 513–519 (2010)
Malpass, K. Neurodegenerative disease: defective mitochondrial dynamics in the hot seat-a therapeutic target common to many neurological disorders? Nature Rev. Neurol. 9, 417 (2013)
Preston, C. C. et al. Aging-induced alterations in gene transcripts and functional activity of mitochondrial oxidative phosphorylation complexes in the heart. Mech. Ageing Dev. 129, 304–312 (2008)
Vafai, S. B. & Mootha, V. K. Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383 (2012)
Wallace, D. C. Diseases of the mitochondrial-DNA. Annu. Rev. Biochem. 61, 1175–1212 (1992)
Wredenberg, A. et al. Increased mitochondrial mass in mitochondrial myopathy mice. Proc. Natl Acad. Sci. USA 99, 15066–15071 (2002)
Melendez, A. et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387–1391 (2003)
Oh, S. W. et al. Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nature Genet. 38, 251–257 (2006)
Pinkston-Gosse, J. & Kenyon, C. DAF-16/FOXO targets genes that regulate tumor growth in Caenorhabditis elegans. Nature Genet. 39, 1403–1409 (2007)
Sandoval, H. et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235 (2008)
Schweers, R. L. et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl Acad. Sci. USA 104, 19500–19505 (2007)
Zhang, J. et al. A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy 8, 1325–1332 (2012)
Zhu, Y. et al. Modulation of serines 17 and 24 in the LC3-interacting region of BNIP3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 288, 1099–1113 (2013)
Pickrell, A. M. & Youle, R. J. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273 (2015)
Xue, D. & Horvitz, H. R. Caenorhabditis elegans CED-9 protein is a bifunctional cell-death inhibitor. Nature 390, 305–308 (1997)
Yasuda, M., D’Sa-Eipper, C., Gong, X. L. & Chinnadurai, G. Regulation of apoptosis by a Caenorhabditis elegans BNIP3 homolog. Oncogene 17, 2525–2530 (1998)
Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426 (2013)
Yun, J. & Finkel, T. Mitohormesis. Cell Metab. 19, 757–766 (2014)
Paek, J. et al. Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab. 16, 526–537 (2012)
Schmeisser, S. et al. Mitochondrial hormesis links low-dose arsenite exposure to lifespan extension. Aging Cell 12, 508–517 (2013)
Zarse, K. et al. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial l-proline catabolism to induce a transient ROS signal. Cell Metab. 15, 451–465 (2012)
Ghose, P., Park, E. C., Tabakin, A., Salazar-Vasquez, N. & Rongo, C. Anoxia-reoxygenation regulates mitochondrial dynamics through the hypoxia response pathway, SKN-1/Nrf, and stomatin-like protein STL-1/SLP-2. PLoS Genet. 9, e1004063 (2013)
Staab, T. A., Evgrafov, O., Knowles, J. A. & Sieburth, D. Regulation of synaptic nlg-1/neuroligin abundance by the skn-1/Nrf stress response pathway protects against oxidative stress. PLoS Genet. 10, e1004100 (2014)
Weimer, S. et al. D-glucosamine supplementation extends life span of nematodes and of ageing mice. Nature Commun. 5, 3563 (2014)
Schmeisser, K. et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nature Chem. Biol. 9, 693–700 (2013)
Schulz, T. J. et al. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 6, 280–293 (2007)
Ventura-Clapier, R., Garnier, A. & Veksler, V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1α. Cardiovasc. Res. 79, 208–217 (2008)
Wright, D. C., Geiger, P. C., Han, D. H., Jones, T. E. & Holloszy, J. O. Calcium induces increases in peroxisome proliferator-activated receptor gamma coactivator-1α and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J. Biol. Chem. 282, 18793–18799 (2007)
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974)
Back, P. et al. Exploring real-time in vivo redox biology of developing and aging Caenorhabditis elegans. Free Radic. Biol. Med. 52, 850–859 (2012)
Kourtis, N., Nikoletopoulou, V. & Tavernarakis, N. Small heat-shock proteins protect from heat-stroke-associated neurodegeneration. Nature 490, 213–218 (2012)
Samara, C., Syntichaki, P. & Tavernarakis, N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 15, 105–112 (2008)
Mijaljica, D., Prescott, M. & Devenish, R. J. A fluorescence microscopy assay for monitoring mitophagy in the yeast Saccharomyces cerevisiae. J. Vis. Exp.. http://dx.doi.org/10.3791/2779 (2011)
Rosado, C. J., Mijaljica, D., Hatzinisiriou, I., Prescott, M. & Devenish, R. J. Rosella: a fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy 4, 205–213 (2008)
Braeckman, B. P., Houthoofd, K., De Vreese, A. & Vanfleteren, J. R. Assaying metabolic activity in ageing Caenorhabditis elegans. Mech. Ageing Dev. 123, 105–119 (2002)
Cristina, D., Cary, M., Lunceford, A., Clarke, C. & Kenyon, C. A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genet. 5, e1000450 (2009)
Acknowledgements
We thank A. Pasparaki for technical support with experiments, N. Kourtis for the GCaMP2.0-expressing C. elegans strain, N. Charmpilas for the neuronal CTS-1::mCherry-expressing C. elegans strain and the unc-43 RNA interference (RNAi) plasmid, and K. Kounakis for the rpn-6 RNAi plasmid. We thank B. P. Braeckman for the Hyper-expressing C. elegans strain and D. Sieburth for the INVOM::RFP-expressing C. elegans strain. We thank R. Devenish for providing the pAS1NB-CS-Rosella plasmid. Mass spectrometry analysis was performed at the Institute of Molecular Biology and Biotechnology Proteomics Facility. We are grateful to M. Aivaliotis for the characterization and relative quantification of DCT-1 tryptic peptides by nanoflow liquid chromatography with tandem mass spectrometry. Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the National Center for Research Resources of the National Institutes of Health, and S. Mitani (National Bioresource Project) in Japan. We thank A. Fire for plasmid vectors. This work was funded by grants from the European Research Council, the European Commission 7th Framework Programme and the Greek General Secretariat for Research and Technology.
Author information
Authors and Affiliations
Contributions
K.P., E.L. and N.T. designed and performed experiments. K.P. and N.T. analysed data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper. Correspondence and requests for materials should be addressed to N.T. (tavernarakis@imbb.forth.gr).
Extended data figures and tables
Extended Data Figure 1 Mitochondrial accumulation during ageing and upon mitophagy depletion.
a, Stained wild-type animals with MitoTracker Red FM dye were monitored throughout adulthood for mitochondrial content. Indicative images are shown on the right for days 1, 3, 5 and 10 (n = 90; ***P < 0.001; one-way ANOVA). b, Total worm lysates from transgenic animals expressing a mitochondria-tagged GFP in the intestine on day 1, 3, 5 and 10 of adulthood were loaded on Tricine-SDS–PAGE and immunoblotted against the indicated proteins. c, Transgenic animals expressing mitochondria-targeted GFP in the intestine were monitored throughout adulthood for mitochondrial content. (n = 100; ***P < 0.001; one-way ANOVA). d, Transgenic animals expressing mitochondria-targeted mCherry in neurons were monitored throughout adulthood for mitochondrial content. Indicative images are shown on the right for days 1, 3, 5 and 10 (n = 70; ***P < 0.001; one-way ANOVA). e, mRNA levels of dct-1 in wild-type animals, dct-1(tm376) mutants, dct-1(RNAi) animals, daf-2(e1370) and daf-16(mu86) mutants. Expression of dct-1 is transcriptionally regulated by DAF-16 (***P < 0.001; one-way ANOVA). f, Efficacy of dct-1 silencing by RNAi. Transgenic animals expressing a full-length pdct-1DCT-1::GFP reporter fusion subjected to dct-1 RNAi. g, mRNA levels in ges-1 and myo-3 in wild-type animals, dct-1(RNAi) and pink-1(RNAi) animals. Expression of ges-1 and myo-3 is not changed upon mitophagy inhibition (NS, P > 0.05; one-way ANOVA). h, Mutant dct-1(tm376) animals expressing mitochondria-targeted GFP in body wall muscle cells display increased mitochondrial mass (n = 100; ***P < 0.0001; unpaired t-test). Scale bars, 100 μm. Images were acquired using a ×5 objective lens. i, Knockdown of either DCT-1 or PINK-1 increases mitochondrial mass in worms that express a mitochondria-targeted GFP in body wall muscles (n = 120; ***P < 0.001; one-way ANOVA). Anterior, left; posterior, right. j, Stained wild-type animals and dct-1(tm376), pink-1(tm1779) and pdr-1(gk448) mutants with MitoTracker Red FM dye were monitored for mitochondrial content (n = 120; ***P < 0.001; one-way ANOVA). Scale bars, 20 μm. Images were acquired using a ×10 objective lens. k, Mitochondrial network morphology is altered in dct-1(tm376) mutants. The mitochondrial network in wild-type animals is well-organized and runs parallel with the myofilament lattice. By contrast, dct-1(tm376) mutant animals display fragmented and disorganized mitochondrial network morphology. Scale bar, 20 μm. Images were acquired using a ×40 objective lens. Error bars, s.e.m.
Extended Data Figure 2 DCT-1 expression and sub-cellular localization.
a, DCT-1 is the homologue of the mammalian BNIP3 and NIX/BNIP3L in C. elegans. Alignment of C. elegans DCT-1, M. musculus BNIP3 and M. musculus NIX/BNIP3L proteins. The predicted transmembrane domain of DCT-1, BNIP3 and NIX/BNIP3L is depicted in green. The conserved WXXL motif that is essential for interaction with the autophagosomal protein LC3/Atg8 is shown in red. The conserved MER and BH3 domains are depicted in blue and yellow, respectively. b, Spatiotemporal expression and sub-cellular localization of DCT-1. Images of transgenic animals expressing a full-length pdct-1DCT-1::GFP reporter fusion. Expression of dct-1 occurs in all somatic tissues of adult animals, including neurons, the pharynx, the intestine, body wall muscles and vulva muscles (indicated by the arrows). Scale bars, 50 μm. c, Expression is detectable in embryos and remains high during all postembryonic developmental stages, throughout adulthood. Scale bars, 50 μm. Images were acquired using × 10 and × 40 objective lenses. d, e, DCT-1::GFP co-localizes with mitochondria in different tissues. Transgenic animal expressing a full-length pdct-1DCT-1::GFP reporter fusion localizing in mitochondria. Animals were stained with Mitotracker Red FM (Invitrogen, Molecular Probes). Scale bars, 20 μm. Images were acquired using a ×100 objective lens. f, Transgenic animal co-expressing a full-length pdct-1DCT-1::GFP reporter fusion together with pmyo-3INVOM::RFP outer mitochondrial membrane reporter. DCT-1::GFP co-localizes with INVOM::RFP, demonstrating that DCT-1 is an outer mitochondrial membrane protein. Scale bars, 10 μm and 5 μm. Images were acquired using a ×100 objective lens. g, Isolated mitochondria from transgenic animals expressing a full-length pdct-1DCT-1::GFP reporter fusion were untreated (lane 1, M), treated with proteinase K (lane 2, MPK), or treated with sodium carbonate (lanes 3 and 4) to separate soluble from membrane material. DCT-1::GFP is accessible and degraded by proteinase K while HSP-60, a mitochondrial matrix protein, is protected. Soluble (lane 3, S) and membrane (lane 4, P) fractions after carbonate extraction are separated by ultracentrifugation. DCT-1::GFP is detected in the membrane fraction (P) while soluble HSP-60 protein is found in the soluble material, indicating that DCT-1 is an integral membrane protein.
Extended Data Figure 3 Mitophagy inhibition does not alter intestinal or muscle cell mitochondrial network morphology of FZO-1- and DRP-1-depleted animals.
a, Mitophagy inhibition by RNAi against dct-1, pink-1 and pdr-1 does not affect mitochondrial network morphology in FZO-1- and DRP-1-depleted animals that express a mitochondria-targeted GFP in the intestine. The mitochondrial network in wild-type animals is well-organized and highly connected. By contrast, animals subjected to RNAi against dct-1, pink-1, pdr-1 and fzo-1 display fragmented and disorganized mitochondrial network morphology. Simultaneous knocking down of fzo-1/dct-1, pink-1/fzo-1 and fzo-1/pdr-1 does not alter the already fragmented mitochondrial network of FZO-1-deficient worms. DRP-1-depleted animals display disorganized aggregated and globular mitochondria compared with wild-type worms. Simultaneous knocking down of drp-1/dct-1, pink-1/drp-1 and pdr-1/drp-1 does not change the impaired mitochondrial network of DRP-1-depleted animals. Scale bar, 20 μm. Images were acquired using a ×40 objective lens. b, Transgenic animals expressing a mitochondria-targeted GFP in muscle cells subjected to RNAi against dct-1, pink-1, pdr-1 and fzo-1. The mitochondrial network in wild-type animals is well organized and runs parallel with the myofilament lattice. By contrast, DCT-1, PINK-1, PDR-1 and FZO-1 depleted animals display fragmented and disorganized mitochondrial network morphology. Simultaneous knocking down of fzo-1/dct-1, pink-1/fzo-1 and fzo-1/pdr-1 does not alter the already fragmented mitochondrial network of FZO-1-deficient worms (n = 120; ***P < 0.001 and NS, P > 0.05; one-way ANOVA). c, DRP-1-deficient animals present disorganized and aggregated mitochondrial morphology. Simultaneous knocking down of drp-1/dct-1, pink-1/drp-1 and pdr-1/drp-1 does not change the impaired mitochondrial network of DRP-1-depleted animals (n = 100; ***P < 0.001 and NS, P > 0.05; one-way ANOVA). Error bars, s.e.m.
Extended Data Figure 4 Μitophagy is induced under several stress conditions.
a, DCT-1 specifically localizes on the outer mitochondrial membrane (shown for muscle cells), where it co-localizes with the autophagosomal protein LGG-1/Atg8. Confocal images of body wall muscle cells of wild-type animals expressing fluorescently tagged DCT-1::GFP along with the autophagosomal marker LGG-1/Atg8 (tagged with DsRed). b, DCT-1 co-localizes with the E3 ubiquitin ligase PDR-1/Parkin. Confocal images of body wall muscle cells in wild-type animals expressing fluorescently tagged DCT-1::GFP and PDR-1::DsRed. Scale bar, 20 μm. Images were acquired using a ×40 objective lens. c, C. elegans transgenic embryos expressing a full-length plgg-1GFP::LGG-1 fusion protein indicative of autophagic activity subjected to single daf-2, dct-1, pink-1 RNAi or subjected simultaneous daf-2/dct-1 and pink-1/daf-2 RNAi (n = 70; NS, P > 0.05, ***P < 0.001; one-way ANOVA). d, C. elegans transgenic animals expressing a full-length plgg-1DsRed::LGG-1 fusion protein indicative of autophagic activity subjected to single daf-2, dct-1, pink-1, pdr-1 RNAi or subjected simultaneous daf-2/dct-1, pink-1/daf-2, daf-2/pdr-1 RNAi. Autophagosome number monitored in the pharynx of the animals (n = 100; NS, P > 0.05, ***P < 0.001; one-way ANOVA). e, The daf-2(e1370) mutant subjected to RNAi against dct-1, pink-1, pdr-1 and lgg-1 or subjected simultaneous against lgg-1/dct-1, pink-1/lgg-1, lgg-1/pdr-1 RNAi. Lifespan values are given in Supplementary Table 1; assays were performed at 20 °C. f, Transgenic animals expressing the mtRosella biosensor in body wall muscle cells, were subjected to daf-2(RNAi), treated with paraquat or CCCP and exposed to heat stress (37 °C). Mitophagy stimulation is signified by the reduction of the ratio between pH-sensitive GFP to pH-insensitive DsRed. DCT-1, PINK-1 and PDR-1 are required for mitophagy. Mitophagy is not activated under stress conditions when transgenic animals are subjected to RNAi against dct-1, pink-1 and pdr-1 (n = 120; NS, P > 0.05, ***P < 0.001; one-way ANOVA). Scale bars, 20 μm. Images were acquired using a ×10 objective lens. g, Mitophagy is not further reduced under oxidative stress conditions in transgenic animals subjected to RNAi against dct-1, pdr-1 or subjected to simultaneous pdr-1/dct-1 RNAi (NS, P > 0.05, ***P < 0.001; one-way ANOVA). h, Mitophagy inhibition under stress conditions in transgenic animals subjected to RNAi against lgg-1 (NS, P > 0.05, ***P < 0.001; one-way ANOVA). Error bars, s.e.m.
Extended Data Figure 5 Monitoring mitophagy in vivo.
Transgenic animals co-expressing a mitochondria-tagged GFP (mtGFP) in body wall muscle cells and the autophagosomal protein LGG-1 fused with DsRed, were exposed to heat stress (37 °C), treated with CCCP, paraquat or subjected to RNAi against daf-2. Mitophagy induction is signified by co-localization of GFP and DsRed signals (for each group of images mitochondria are shown in green on top, autophagosomes in red below, with a merged image at the bottom). a, DCT-1 and PINK-1 are required for mitophagy. Mitophagy inhibition under stress conditions when transgenic animals are subjected to RNAi against dct-1 and pink-1. Scale bars, 20 μm. Images were acquired using a ×40 objective lens. b, High-magnification images showing induction of mitophagy. Scale bars, 10 μm. Images were acquired using a ×40 objective lens.
Extended Data Figure 6 Mitochondrial content is not affected upon mitochondrial dysfunction.
a, Knockdown of either PHB-1, PHB-2, CLK-1 or ISP-1 does not affect the mitochondrial mass in transgenic worms that express a mitochondria-targeted GFP in the intestine (n = 100; NS, P > 0.05; one-way ANOVA). b, c, Simultaneous RNAi against phb-1/dct-1, phb-1/pink-1, phb-1/pdr-1, phb-2/dct-1, pink-1/ phb-2, phb-2/pdr-1, isp-1/dct-1, pink-1/isp-1, pdr-1/isp-1, clk-1/dct-1, pink-1/clk-1, clk-1/pdr-1 increases mitochondrial mass (n = 100; ***P < 0.001; one-way ANOVA). d, Knockdown of either DCT-1, PINK-1 or PDR-1 increases mitochondrial mass in transgenic worms that express a mitochondria-targeted GFP in the intestine. Transgenic worms expressing mitochondria-tagged GFP in the intestine were raised over one generation in the presence of EtBr to block mitochondrial biogenesis. Knockdown of either DCT-1, PINK-1 or PDR-1 increases mitochondrial content of animals exposed to EtBr (n = 100; ***P < 0.001; one-way ANOVA). e, Knockdown of either PBS-5 or RPN-6 promotes fragmentation of mitochondrial network and decreases mitochondrial content in worms that express a mitochondria-targeted GFP in the intestine (n = 100; ***P < 0.001; one-way ANOVA). Scale bar, 20 μm. Images were acquired using a ×40 objective lens. f–h, The expression levels of ges-1, myo-3 and unc-119 gene are relatively stable throughout ageing. Inhibition of autophagy or proteasome system does not alter the expression levels of ges-1, myo-3 and unc-119 genes during ageing (NS, P > 0.05, **P < 0.01; one-way ANOVA). Error bars, s.e.m.
Extended Data Figure 7 Gene ced-9 acts in the same genetic pathway with dct-1, pink-1 and pdr-1 to mediate mitophagy.
a, CED-9 depletion does not affect the localization of DCT-1. Scale bars, 20 μm. Images were acquired using a ×63 objective lens. b, Knockdown of either CED-9, DCT-1, PINK-1 or PDR-1 increases mitochondrial mass in transgenic worms expressing a mitochondria-targeted GFP in the intestine. Simultaneous RNAi against ced-9/dct-1, pink-1/ced-9 and ced-9/pdr-1 does not increase further mitochondrial mass (n = 100; NS, P > 0.05, ***P < 0.001; one-way ANOVA). c, Transgenic animals expressing the mtRosella biosensor in body wall muscle cells, were treated with paraquat. Mitophagy induction is signified by the reduction of the ratio between pH-sensitive GFP to pH-insensitive DsRed. CED-9 is required for mitophagy. Mitophagy inhibition under stress conditions when transgenic animals are subjected to RNAi against ced-9 (n = 120; NS, P > 0.05, ***P < 0.001; one-way ANOVA). Error bars, s.e.m. d, Mitophagy inhibition under stress, in transgenic animals subjected to RNAi against ced-9. Scale bars, 20 μm. Images were acquired using a ×40 objective lens.
Extended Data Figure 8 Inhibition of mitophagy affects longevity in a genetic-background-specific manner.
a, Depletion of PDR-1, or PINK-1. b, c, Depletion of both DCT-1 and PINK-1. d, e, Depletion of both PDR-1 and PINK-1, f, Depletion of both DCT-1 and PDR-1, Knockdown of DCT-1 shortens the lifespan of long-lived clk-1(e2519) (g), and eat-2(ad465) (h) mutants. i, j, Mitochormesis is not engaged in mitophagy-deficient animals to influence lifespan. Depletion of either DCT-1, PINK-1 or PDR-1, with, or without NAC or BHA treatment. k, Depletion of DCT-1, PINK-1 or PDR-1, with, or without EGTA treatment. l, DCT-1 overexpression does not influence the lifespan of otherwise wild-type animals or pink-1(tm1779) mutants under normal conditions. DCT-1 overexpression extends only the maximum lifespan of DAF-2-depleted worms. However, PINK-1 deficiency abolishes the extended lifespan of both daf-2(RNAi) and daf-2(RNAi);Ex[pdct-1DCT-1::GFP] animals. Survival curves depict the percentage of animals remaining alive, plotted against animal age. Lifespan values are given in Supplementary Table 1; assays were performed at 20 °C.
Extended Data Figure 9 Mitophagy deficiency impairs mitochondrial homeostasis and stress resistance.
a, b, Fifteen-day-old dct-1(tm376), pink-1(tm1779), pdr-1(gk448) and wild-type animals incubated (a) at 37 °C for 7 h (n = 150; ***P < 0.001; one-way ANOVA) and (b) treated with 8 mM paraquat (n = 150; ***P < 0.001; one-way ANOVA). c, Transgenic animals expressing the H2O2-biosensor HyPer subjected to either dct-1 or pink-1 RNAi. The graph depicts pixel intensity ratios of oxidized to reduced HyPer (n = 70; **P < 0.01; one-way ANOVA). d, Mitophagy-depleted animals display increased ratios of mitochondrial ROS formation (MitoTracker Red CM-H2X ROS) to total mitochondrial content (intestinal mtGFP) (n = 150; ***P < 0.001; one-way ANOVA). e, TMRE staining of mitophagy-depleted adults at 20 °C or 25 °C (n = 150; *P < 0.05, ***P < 0.001; one-way ANOVA). f, The percentage of mitochondrial DNA relative to wild type is shown. Quantitative PCR was performed in wild-type, dct-1(tm376), pink-1(tm1779) and pdr-1(gk448) mutant animals. The percentage of mtDNA is not affected in mitophagy-deficient animals (NS, P > 0.05; one-way ANOVA). g, The genetic locus of dct-1 gene is depicted; dct-1(tm376) is homozygous deletion. The dct-1(tm376) mutant carries a deletion of 900 base pairs at the promoter region of dct-1 gene. h, i, Transgenic animals were generated carrying extra copies of wild-type dct-1 gene and dct-1 gene carrying deletions for the corresponding sequences of WXXL motif, MER domain and BH3 domain, and were exposed to heat stress (37 °C) or treated with paraquat. WXXL motif of DCT-1 is required for mitophagy process since WXXL-deleted DCT-1 fails to rescue resistance of dct-1(tm376) animals under stress conditions (n = 150; NS, P > 0.05, ***P < 0.001; one-way ANOVA). j, k, Wild-type, pink-1(tm1779) and pdr-1(gk448) transgenic animals expressing a full-length pdct-1DCT-1::GFP reporter fusion were exposed to heat stress (37 °C) or treated with paraquat. DCT-1 overexpression is sufficient to promote stress resistance in wild-type worms. However, the stress resistance of DCT-1 overexpressing animals is abolished in pink-1 and pdr-1 mutant background (n = 150; ***P < 0.001; one-way ANOVA). Error bars, s.e.m. l–o, DCT-1 is ubiquitinated in a PINK-1-dependent manner upon oxidative stress. DTC-1::GFP was immunoprecipitated from total lysates of wild-type and pink-1(tm1779) transgenic animals expressing a full-length pdct-1DCT-1::GFP reporter fusion. The immunoprecipitated material was trypsinized and subjected to nLC-MS/MS analysis. l, DCT-1 was identified by mass spectrometry with high confidence and a sequence coverage of ∼60% (underlined on DCT-1 sequence). The two most prominent tryptic peptides of DCT-1 (highlighted in blue) were used for the relative quantification of the ubiquitination within the different samples. Ubiquitin-modified lysine is highlighted in red. m, Numbers represent the following samples: untreated wild-type worms (sample 1), untreated wild-type transgenic worms expressing a full-length pdct-1DCT-1::GFP reporter fusion (sample 2), paraquat-treated wild-type transgenic worms expressing a full-length pdct-1DCT-1::GFP (sample 3) and paraquat-treated pink-1(tm1779) mutants expressing a full-length pdct-1DCT-1::GFP (sample 4). The most prominently identified tryptic peptides 19–50 (peptide 1) and 101–121 (peptide 2) were used to determine the relative abundance of modified and unmodified DCT-1 in the four samples. The maximum intensities of the peptide ions of each peptide in the different samples were normalized and used for the comparative analysis and K26-GG abundance. The unmodified peptides showed similar pattern and abundance in the different samples (not identified in control sample 1, which did not express DCT-1::GFP, as expected), while the tryptic peptide with the K26-GG modification (19–50) was significantly enriched in sample 3 where mitophagy was induced in the presence of PINK-1. n, o, K26-GG modification on the tryptic peptide 19–50 of DCT-1 was manually validated on the MS/MS spectra of the unmodified (n) and modified (o) peptide. Both peptides were identified by high amino-acid coverage, as is shown on their corresponding assigned spectra. The intact peptide masses after the loss of H2O and NH3 molecules are shown in green, and the characteristic b and y fragment ions used for sequencing the amino-acid residues of the peptides are shown in red and blue, respectively. The characteristic fragments indicating the K26-GG modification are marked by blue circles and underlined fragment numbers (o).
Extended Data Figure 10 SKN-1 regulates the transcription of mitochondrial genes and modulates mitochondrial homeostasis and integrity.
a, mRNA level analysis of the SKN-1 target genes gst-4 and gst-10 in wild-type animals and mitophagy-deficient animals (**P < 0.01; one-way ANOVA). b, Expression of the DAF-16-regulated, psod-3GFP transgene in animals subjected to dct-1 or pink-1 RNAi (n = 70; NS, P > 0.05, ***P < 0.001; one-way ANOVA). c, Expression of the mitochondrial stress reporter phsp-60GFP, in animals subjected to RNAi against dct-1 or pink-1 (n = 70; NS, P > 0.05; one-way ANOVA). d, Expression of the ER stress reporter phsp-4GFP, in animals subjected to RNAi against dct-1 or pink-1 (n = 70; NS, P > 0.05; one-way ANOVA). e, mRNA levels of atp-5, gas-1, hmg-5, tim-17, W09C5.8 and dct-1 in wild-type and SKN-1-depleted animals (skn-1(zu67) and skn-1(zu135) animals) (*P < 0.01; one-way ANOVA). f, The percentage of mitochondrial DNA relative to wild type is shown. The percentage of mtDNA is reduced in SKN-1-depleted worms (**P < 0.01; one-way ANOVA). g, Knockdown of SKN-1 alters mitochondrial network morphology in the intestine of young adult animals. Scale bar, 20 μm. Images were acquired using a ×40 objective lens. h, mRNA levels of pink-1 and pdr-1 in wild-type animals, DAF-16- and SKN-1-depleted animals (NS, P > 0.05; one-way ANOVA). i, Mitophagy is not stimulated under oxidative stress, in transgenic animals expressing mtRosella subjected to RNAi against skn-1 (n = 120; NS, P > 0.05, ***P < 0.001; one-way ANOVA). j, Monitoring mitophagy under oxidative stress, in transgenic animals subjected to RNAi against skn-1, skn-1 and dct-1, daf-16, daf-16 and dct-1 (n = 120; NS, P > 0.05, ***P < 0.001; one-way ANOVA). k, SKN-1 compensates for inhibition of mitophagy in otherwise wild-type animals. Simultaneous depletion of DCT-1 and SKN-1, or PINK-1 and SKN-1. Survival curves depict the percentage of animals remaining alive, plotted against animal age. Lifespan values are given in Supplementary Table 1; assays were performed at 20 °C. l, Transgenic animals co-expressing a mitochondria-tagged GFP (mtGFP) in body wall muscle cells and the autophagosomal protein LGG-1 fused to DsRed were exposed to heat stress (37 °C) or treated with CCCP. Induction of mitophagy, as indicated by co-localization of GFP and DsRed (for each group of images, mitochondria are shown in green on top, autophagosomes in red below, with a merged image at the bottom). Mitophagy inhibition under stress, in transgenic animals subjected to RNAi against skn-1. Scale bars, 20 μm. Images were acquired using a ×40 objective lens. m, Cytoplasmic calcium elevation upon mitophagy depletion is abolished upon treatment with EGTA (n = 100; NS, P > 0.05, ***P < 0.001; one-way ANOVA). n, Transgenic animals expressing pgst-4GFP reporter subjected to RNAi against dct-1, pink-1 and pdr-1, with or without EGTA treatment (n = 120; NS, P > 0.05, ***P < 0.001; one-way ANOVA). o, UNC-43 regulates the transcriptional activity of SKN-1 upon mitophagy depletion (n = 90; ***P < 0.001; one-way ANOVA). Error bars, s.e.m.
Supplementary information
Supplementary Information
This file contains Supplementary Table 1. (PDF 203 kb)
Rights and permissions
About this article

Cite this article
Palikaras, K., Lionaki, E. & Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528 (2015). https://doi.org/10.1038/nature14300
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14300
- Springer Nature Limited
This article is cited by
-
Autophagy-related 7 protein-dependent autophagy mediates resveratrol-caused upregulation of mitochondrial biogenesis and steroidogenesis in aged Leydig cell
Molecular Biology Reports (2024)
-
Impact of visceral adipose tissue on longevity and metabolic health: a comparative study of gene expression in perirenal and epididymal fat of Ames dwarf mice
GeroScience (2024)
-
Cholic and deoxycholic acids induce mitochondrial dysfunction, impaired biogenesis and autophagic flux in skeletal muscle cells
Biological Research (2023)
-
Quinolinic acid impairs mitophagy promoting microglia senescence and poor healthspan in C. elegans: a mechanism of impaired aging process
Biology Direct (2023)
-
Crosstalk between mitochondrial biogenesis and mitophagy to maintain mitochondrial homeostasis
Journal of Biomedical Science (2023)