

Abstract
Permafrost contains about 50% of the global soil carbon1. It is thought that the thawing of permafrost can lead to a loss of soil carbon in the form of methane and carbon dioxide emissions2,3. The magnitude of the resulting positive climate feedback of such greenhouse gas emissions is still unknown3 and may to a large extent depend on the poorly understood role of microbial community composition in regulating the metabolic processes that drive such ecosystem-scale greenhouse gas fluxes. Here we show that changes in vegetation and increasing methane emissions with permafrost thaw are associated with a switch from hydrogenotrophic to partly acetoclastic methanogenesis, resulting in a large shift in the δ13C signature (10–15‰) of emitted methane. We used a natural landscape gradient of permafrost thaw in northern Sweden4,5 as a model to investigate the role of microbial communities in regulating methane cycling, and to test whether a knowledge of community dynamics could improve predictions of carbon emissions under loss of permafrost. Abundance of the methanogen Candidatus ‘Methanoflorens stordalenmirensis’6 is a key predictor of the shifts in methane isotopes, which in turn predicts the proportions of carbon emitted as methane and as carbon dioxide, an important factor for simulating the climate feedback associated with permafrost thaw in global models3,7. By showing that the abundance of key microbial lineages can be used to predict atmospherically relevant patterns in methane isotopes and the proportion of carbon metabolized to methane during permafrost thaw, we establish a basis for scaling changing microbial communities to ecosystem isotope dynamics. Our findings indicate that microbial ecology may be important in ecosystem-scale responses to global change.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Main
Multiple factors—including hydrology, vegetation, organic matter chemistry, pH and soil microclimate—are affected by permafrost loss5,8,9. Together these factors regulate microbial metabolisms that release carbon dioxide (CO2) and methane (CH4) from thawing permafrost10,11,12 and are the basis for Earth-system model predictions of future CH4 emissions7,13,14. However, the role of microbial community composition in regulating the metabolic processes that drive ecosystem-scale fluxes is unknown.
At our study site in Stordalen mire, as in other thawing permafrost peatlands8,15, permafrost loss causes hydrological and vegetation shifts: well-drained permafrost-supported palsas collapse into partly thawed bogs dominated by moss (Sphagnum spp.) and fully thawed fens dominated by sedges (such as Eriophorum angustifolium)4. Between 1970 and 2000, 10% of Stordalen’s palsa habitat thawed into such wetlands4. This transition drives an appreciable global warming impact because CO2-emitting palsa is converted to bogs and fens, which take up CO2 but emit CH4 (a more potent greenhouse gas3)4,5,16. The net effect is that the high-methane-emitting fen contributes sevenfold as much greenhouse impact per unit area as the palsa. This thaw progression is also associated with an increase in overall organic matter lability, including a decrease in C:N ratio and an increase in humification rates9. We speculated, consistent with previous studies of in situ bog and fen systems17,18,19, that thaw progression also facilitates a shift from hydrogenotrophic to acetoclastic CH4 production.
We used the distinct isotopic signatures of different microbial CH4 production and consumption pathways to directly relate changes in CH4 dynamics across the thaw gradient to underlying changes in the microbial community. Methane produced by hydrogenotrophic methanogens generally has lower δ13C and higher δD (δ13C = −110‰ to −60‰ and δD = −250‰ to −170‰) relative to that produced by acetoclastic methanogens (δ13C = −60‰ to −50‰ and δD = −400‰ to −250‰)19,20. If methanotrophic microbes then oxidize CH4, lighter molecules are preferentially consumed, leaving the remaining CH4 enriched in 13C and D relative to the original CH4 pool (expected patterns are shown in Extended Data Fig. 1)19,20.
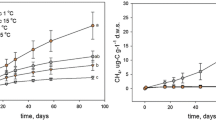
High-temporal-resolution measurements of the magnitude and isotopic composition of CH4 emissions, using a quantum cascade laser spectrometer (Aerodyne Research Inc.) connected to autochambers, showed that CH4 emissions and their 13C content increased with thaw. Average CH4 fluxes increased from effectively zero at the intact permafrost palsa site to 1.46 ± 0.37 mg CH4 m−2 h−1 (all errors are reported as s.e.m.) at the thawing Sphagnum site, and to 8.75 ± 0.50 mg CH4 m−2 h−1 at the fully thawed Eriophorum site (Fig. 1a; P < 0.001). The average δ13C of emitted CH4 also increased significantly, from −79.6 ± 0.9‰ in the Sphagnum site to −66.3 ± 1.6‰ in the Eriophorum site (Fig. 1b; P = 0.03). This consistent 10–15‰ divergence between sites was maintained through the growing season but overlain by parallel fluctuations in δ13C-CH4, suggesting that weather patterns exerted a common influence over the magnitude of isotopic fractionation. Porewater CH4 isotopes showed a similar pattern, with Eriophorum site porewater δ13C about 10‰ higher than that of Sphagnum (July and August; Fig. 1b and Extended Data Table 1). Porewater CH4 was 13C-enriched by 5–20‰ relative to emitted CH4, as expected, as a result of diffusive fractionation (Methods, equation (2))18,21.

a, Average daily CH4 emissions (error bars represent s.e.m.; n = 2–3). b, δ13C composition of emitted and porewater CH4 (error bars represent s.e.m.; flux n = 2–3, porewater n = 6–9). c, Relative abundance of methanogenic groups as inferred by taxonomic identity assigned from 16S rRNA amplicon sequencing (n = 3). For the intermediate-thaw Sphagnum site, aerobic communities were sampled above the water table; anaerobic communities were sampled below the water table.
The apparent fractionation factor for carbon in porewater CH4 relative to CO2, αC (Methods, equation (2), and Extended Data Table 1), is a related index of changes in CH4 production22. Greater fractionation is associated with hydrogenotrophic methanogenesis and was found in the thawing Sphagnum site (αC = 1.053 ± 0.002). Significantly less fractionation (P = 0.002) associated with more acetoclastic production or with consumption by oxidation was found in the fully thawed Eriophorum porewater (αC = 1.046 ± 0.001). Here, increases in acetoclastic production, not oxidation, best explain isotopic shifts because lower αC and higher δ13C-CH4 are accompanied by significantly lower δD-CH4 (Extended Data Fig. 1; P < 0.001)19. This is consistent with the pattern of isotopes in CH4 emissions, in incubations of Stordalen peat9 and in studies showing bog-to-fen shifts from hydrogenotrophic to acetoclastic methanogenesis17,18,19.
The CH4 flux and isotope results provide compelling but indirect evidence for changes in CH4-cycling microbial communities with permafrost thaw. These microbiological changes could be shifts in activity of particular community members or changes in community composition. We examined the role of community composition through 16S rRNA gene amplicon sequencing. All known methanogens belong to a small number of archaeal lineages within the Euryarchaeota23. As expected, the shift from CH4-neutral intact permafrost palsa to CH4-emitting wetland corresponded to a substantial increase in the relative abundance of methanogenic archaeal lineages (Fig. 1c and Extended Data Table 2, 3). In the aerobic palsa and surface Sphagnum habitats, methanogens were found in low relative abundance (average less than 0.6%), whereas the anaerobic environments of the Eriophorum and deeper (below the water table) Sphagnum habitats harboured communities with a substantially higher relative abundance of methanogens (20–30%).
More significantly, the abundance of specific methanogenic lineages varied across the thaw gradient (Fig. 1c and Extended Data Table 2) in a manner corresponding to shifts in CH4 production mechanism inferred from the isotope data (Fig. 1b). At the partly thawed Sphagnum site, where CH4 isotopes were more hydrogenotrophic, the methanogen community was dominated by hydrogenotrophic populations (at least 57% of sequences). Members of the genus Methanobacterium and close relatives of the recently described hydrogenotroph Candidatus ‘Methanoflorens stordalenmirensis’6 (a partial genome of which has also been identified in incubations of Alaskan permafrost12) were the most abundant phylotypes. Although present, the metabolically versatile Methanosarcina (capable of using a wide range of substrates, including acetate and hydrogen24), was much less abundant, averaging about 15% of the methanogen sequences. At the fully thawed Eriophorum site (where isotope signatures shifted towards acetoclastic), members of the obligately acetoclastic genus Methanosaeta increased in abundance, comprising roughly one-third of the methanogenic population. The remaining methanogenic community at the Eriophorum site was taxonomically diverse and included lineages also present at the Sphagnum site, as well as the hydrogenotrophic genus, Methanoregula (Extended Data Table 2). Differences in the functional (hydrogenotrophic versus acetoclastic) composition of the methanogen community between the sites were smallest in October, coinciding with a convergence in δ13C-CH4 (Fig. 1a and Extended Data Tables 2 and 3).
Taken together, the isotope and microbial sequence data suggest that shifts in microbial communities drive large, concordant variations in CH4 isotope biogeochemistry both seasonally and during permafrost thaw, a novel observation at the ecosystem scale. The early successional hydrogenotroph ‘M. stordalenmirensis’6 dominates methanogenic metabolism in the early stages of thaw, followed by the subsequent emergence of a more diverse methanogen community, including obligate acetoclastic methanogens. This microbial succession provides direct evidence for how changes in ecosystem structure during permafrost thaw (plant succession and increases in organic matter quality9) translate into altered CH4 biogeochemistry.
To quantify the effect of this shifting microbial community composition for CH4 isotopic patterns, we examined the relationships between isotope fractionation (αC), environmental conditions known or expected to impact methanogenesis, and the relative abundance of specific methanogenic lineages (Extended Data Table 4). Rather than a functional group (such as hydrogenotrophic methanogens), a single organism—the hydrogenotroph ‘M. stordalenmirensis’—was the best one-variable predictor of isotopic patterns in the field (Fig. 2a). Several variables that typically differentiate bogs from fens, including pH and water table depth18, were significant predictors of αC; however, it was the relative abundance of ‘M. stordalenmirensis’ that explained both the large range of αC observed at the Sphagnum site (R2 = 0.7, P < 0.001) and patterns across sites (R2 = 0.6, P < 0.001). This suggests, contrary to the current practice of focusing on the functional diversity of communities, that an individual microbial lineage can have a disproportionate influence on ecosystem biogeochemistry.

a, The relative abundance of a single methanogen, Candidatus ‘Methanoflorens stordalenmirensis’, in the field was significantly correlated (linear regression, P < 0.001, n = 41) with porewater effective fractionation (αC), an isotopic indicator of the methanogenic production pathway. b, Anaerobic incubations of peat collected from a related thaw sequence at Stordalen mire (see methods in ref. 9) show a significant correlation between αC and the CH4:CO2 production ratio (linear regression, P = 0.004, n = 9), suggesting that the abundance of ‘M. stordalenmirensis’ may be indicative of the proportion of organic matter metabolized to CH4.
Stepwise regression identified environmental variables (water table depth, peat C:N ratio and peat δ13C) that improved model predictions of αC (to R2 = 0.8, P < 0.001). Although confirming the central importance of ‘M. stordalenmirensis’ in explaining variation in αC (Extended Data Table 5), this model also supports the hypothesis that organic matter chemistry underlies shifts in CH4 metabolism9,25. The dependence on the abundance of this lineage was evident despite the relative rather than the absolute nature of the community composition analysis, and the measurement of abundance rather than activity. We speculate that direct measures of gene expression or metabolic activity (metatranscriptomics and metaproteomics) will have an even stronger association than community composition data with isotopic signatures.
Further analysis showed that αC is significantly correlated (R2 = 0.7, P = 0.004) with the large range in CH4:CO2 production ratio (0.13–0.84) measured in anaerobic incubations of Stordalen peat (Fig. 2b). It is therefore likely that changes in the proportion of anaerobically mineralized C that ends up as CH4—a key, but poorly constrained, parameter in global CH4 models26—tracks the abundance of ‘M. stordalenmirensis’, which acts as an index of the concerted changes in microbial community and organic matter chemistry that together control the efficiency of carbon metabolism.
Incorporating this understanding of the imprint of microbial communities could be crucial to both improved model prediction of future climate change CH4 feedbacks and accurate attribution of the portion of global atmospheric CH4 change that is derived from permafrost thaw. First, in simulating CH4 cycling, Earth-system models typically prescribe as fixed the fraction of anaerobically metabolized carbon that becomes CH4 (ref. 26). The lack of a basis for predicting this parameter across ecosystems and in response to climate change limits current modelling efforts3. Our finding that the CH4:CO2 production ratio is highly variable and predictable from isotopic indicators of methanogenic community composition (Fig. 2b) supports the need to improve the representation of microbial ecology in models17,27. Although simulating microbial population dynamics is beyond the scope of current global models, the identification of microbial lineages that predict key parameters, such as the CH4:CO2 ratio, provides insights that improve simulations of CH4 biogeochemistry used to estimate global emissions.
Second, atmospheric inversion studies that use CH4 mixing ratios and isotopes to infer global sources and sinks of atmospheric CH4 assume that wetland microbial sources are dominated by acetate fermentation (−58‰ to −65‰), and, critically, that isotopic signatures from biological sources are constant over time28,29. In contrast, we observed isotopic compositions that varied across a gradient of permafrost thaw: hydrogenotrophic methanogenesis was estimated to produce about 50–75% of total CH4 emission at Stordalen (Extended Data Table 6), with δ13C averaging −80‰ (Fig. 1b). The hydrogenotrophic δ13C observed at Stordalen and other Arctic wetlands30 may be a ubiquitous characteristic of thawing permafrost, particularly during thaw stages that generate recalcitrant organic matter9,25, such as that observed at Stordalen in the intermediate-thaw Sphagnum site.
To test whether these observed thaw-induced changes in microbial metabolism might be relevant for large-scale atmospheric methane dynamics, we used a simple box model of atmospheric mixing (Methods, equation (3)) to quantify the effect of different methanogen communities within recently constructed scenarios of CH4 emission from thawing permafrost2 (Extended Data Fig. 2a, b). We found that if hydrogenotrophic lineages regulated CH4 isotope patterns in permafrost thaw generally, as at Stordalen, then projected CH4 emissions (Fig. 3a) would produce larger decreases in δ13C of atmospheric CH4 than expected from current inversion model assumptions that acetoclasts dominate emissions (Fig. 3b and Extended Data Fig. 2c, d). This in turn would constrain our simple box model to substantially overestimate the amount of CH4 released from thawing permafrost and underestimate emissions from non-wetland sources, most notably fossil fuels (Fig. 3c). The greater the prevalence of hydrogenotrophic lineages in CH4 emissions, the larger will be the overestimate of fluxes from thaw (Fig. 3c). The numerical size of the mis-estimation error here is illustrative; state-of-the-art three-dimensional inversion models have spatially resolved constraints that would probably force smaller flux mis-estimations. However, the general implication is that microbial effects are sufficiently important that accurate global accounting of the different sources of CH4 under future climate change can be improved by understanding the microbial community dynamics underlying biological feedbacks in natural systems.

a, Modelled CH4 emissions under high (red bounding lines) and low (orange bounding lines) climate warming scenarios, and a range within each (grey tint) spanning high and low C-release scenarios2. b, Consequent decreases (simulated by the intermediate emissions scenario indicated by the red dashed line in a) in δ13C of atmospheric CH4 due to emissions dominated by hydrogenotrophic lineages, as in intermediate-thaw Sphagnum sites (green line, δ13C = −80‰), or more by acetoclasts, as in fully thawed Eriophorum sites (blue line, δ13C = −65‰). Atmospheric inversion models typically assume that emissions have δ13C ranging from −60‰ (black line) to −65‰ (blue line). (The dotted horizontal line indicates the current detection limit for atmospheric CH4 isotopes28). These imply an underestimate of the effect on atmospheric δ13C for the given emissions scenario (blue or green). c, To match observed atmospheric isotopes, the box model would then require a corresponding overestimate of CH4 flux attributed to permafrost thaw (vertical axis). The magnitude of the overestimate depends on the mismatch between model-assumed isotopic composition (upper line, −60‰; lower line, −65‰) and the actual isotopic composition produced by different communities, which ranges here along the horizontal axis from −80‰ (hydrogenotroph-dominated, as in the partly thawed Sphagnum sites) to −65‰ (acetoclastic, as in the fully thawed Eriophorum sites).
By showing that the abundance of key microbial lineages can be used to predict atmospherically relevant patterns in CH4 isotopes and the proportion of carbon metabolized to CH4 during permafrost thaw, this work establishes a basis for scaling changing microbial communities to ecosystem-scale and global-scale atmospheric isotope dynamics. It also highlights the central role of microbial ecology in ecosystem-scale responses to global change and the benefit of incorporating microbial dynamics into Earth-system models.
Methods
Site description and permafrost thaw
Stordalen is a sub-arctic palsa mire located 10 km east of Abisko in the discontinuous permafrost zone of northern Sweden (68° 21′ N, 18° 49′ E, altitude 363 m above sea level). This work focuses on three distinct subhabitats, common to northern wetlands and together covering about 98% of the mire’s surface: permafrost-dominated, well-drained palsas occupied by feather mosses and ericaceous and woody plants, covering 49% of the mire; intermediate permafrost sites with variable water table depth, dominated by Sphagnum spp., covering 37% of the mire; and full summer-thaw, wet sites with Eriophorum angustifolium, covering 12% of the mire. Between 1970 and 2000, as permafrost thawed and palsas collapsed, Sphagnum sites and Eriophorum sites expanded by 3% and 54%, respectively4.
The formation of wetlands after permafrost thaw, as observed at Stordalen, is a widespread characteristic of peatlands affected by permafrost loss8,31,32,33. Thawing of ice-rich features results in peatland collapse and the formation of bogs and fens. At Stordalen, thaw is associated with a progression from ombrotrophic bogs to minerotrophic fens due to thaw-induced subsidence increasing hydrological connectivity. A similar successional shift from bogs dominated by Sphagnum spp. to tall graminoid fens has been observed in other northern peatlands8,33,34,35. More generally, landscape features and hydrological conditions dictate the characteristics and trajectory of wetland communities formed after permafrost thaw36. For example, rapid fen development is observed at the subsiding margins of permafrost plateaux37, whereas collapse bogs and thermokarst lakes often form within large, thawing peatland complexes32. Large uncertainty in model predictions of the extent and characteristics of wetland formation arising from permafrost thaw is a critical limitation to current understanding of carbon–climate feedbacks7,14. As demonstrated in this study, improved characterization and modelling of peatland transformation during thaw will be essential for accurately predicting post-thaw microbial communities and the resultant magnitude and isotopic composition of CH4 emissions under climate change.
Methane isotope systematics
We use standard δ notation for quantifying the isotopic compositions of CH4 and CO2: the ratio R of 13C to 12C (or D to H) in the measured sample is expressed as a relative difference (denoted δ13C or δD) from the Vienna Pee Dee Belemnite (VPDB) international standard material. For example, for C:

δ13C is often expressed in parts per thousand (‰).
Isotopic fractionation in chemical reactions (including methanogenesis or methanotrophy) or due to diffusion may be quantified as

For diffusive fractionation, Rsource is taken to be the isotopic ratio in the concentrations of the gradient and Rproduct the ratio in the resultant net flux. Because diffusion discriminates against the heavy isotope, Rproduct < Rsource, which implies, for example, that the isotopic ratio of porewater (the ‘source’) will be greater than that of the flux of gas diffusing out, as we see here (Fig. 1a). Methanogenesis and methanotrophy also discriminate against the heavier isotopes, so that Rproduct< Rsource (and hence α > 1) for both C and H in methane. Note that α > 1 for methanotrophy implies that the products of CH4 oxidation (CO2 and H2O) are lighter (have lower R) in both C and H relative to the source CH4; however, mass balance then requires the residual methane not oxidized to become heavier in both C and H relative to the starting composition of the CH4 pool before oxidation.
The degree of C isotopic fractionation between CO2 and CH4 differs between the two main biochemical pathways of methanogenesis, namely acetoclastic (CH3COOH →CH4 + CO2) and hydrogenotrophic (CO2 + 4H2 → 2H2O + CH4). Carbon isotope fractionation (αC) is greater for hydrogenotrophic than for acetoclastic methanogenesis, but αH (hydrogen isotope fractionation) follows the opposite pattern: αH (hydrogenotrophic) < αH (acetoclastic) (Extended Data Fig. 1; ref. 19). Hence, variations in C and H isotopic compositions of CH4 that arise from variations in methanogenic pathway will be anti-correlated: shifts from hydrogenotrophic to acetoclastic production will cause C isotope ratios to increase but H isotope ratios to decline, moving along a negatively sloped ‘production line’ in H–C isotope space (Extended Data Fig. 1). Isotopic variations that arise from variations in the degree of methanotrophy, by contrast, will be positively correlated: shifts towards increasing methanotrophy will cause both C and H isotope ratios to increase along a positively sloped ‘oxidation line’ (Extended Data Fig. 1).
In a field study such as this, it is difficult to estimate fractionation factors directly; we therefore follow standard practice in the methane biogeochemistry literature (see, for example, refs 22, 38) and estimate the net or effective fractionation factor from in situ pore water data. For example, we estimate αC, the effective fractionation factor for C in CH4, by applying equation (2), setting δproduct =  and δsource =
and δsource =  , where
, where  and
and  are the observed C compositions of CH4 and CO2, respectively38. Using CO2 isotope composition for δsource follows directly for hydrogenotrophic methanogenesis (for which CO2 is the source C substrate) and has been found to work also in practice for acetoclastic methanogenesis, because porewater CO2 arises primarily from respiration of organic matter (a non-discriminatory reaction), and so is typically isotopically indistinguishable from organic matter20,39.
are the observed C compositions of CH4 and CO2, respectively38. Using CO2 isotope composition for δsource follows directly for hydrogenotrophic methanogenesis (for which CO2 is the source C substrate) and has been found to work also in practice for acetoclastic methanogenesis, because porewater CO2 arises primarily from respiration of organic matter (a non-discriminatory reaction), and so is typically isotopically indistinguishable from organic matter20,39.
Autochamber measurements
The autochamber system at Stordalen mire has previously been described in detail for measurements of CO2 and total hydrocarbons16,40. In brief, a system of eight automatic gas-sampling chambers made of transparent Lexan was installed in the three habitat types at Stordalen mire in 2001 (n = 3 each in the palsa and Sphagnum habitats, and n = 2 in the Eriophorum habitat). Each chamber covers an area of 0.14 m2 (38 cm × 38 cm), with a height of 25–45 cm, and is closed once every 3 h for a period of 5 min. The chambers are connected to the gas analysis system, located in an adjacent temperature controlled cabin, by 3/8-inch Dekoron tubing through which air is circulated at approximately 2.5 l min−1. During the 2011 season the system was updated with a new chamber design similar to that described in ref. 41. The new chambers each cover an area of 0.2 m2 (45 cm × 45 cm), with a height ranging from 15 to 75 cm depending on habitat vegetation. At the Palsa and Sphagnum site the chamber base is flush with the ground and the chamber lid (15 cm in height) lifts clear of the base between closures. At the Eriophorum site the chamber base is raised 50–60 cm on Lexan skirts to accommodate vegetation of large stature. In addition, each chamber is instrumented with thermocouples measuring air and surface ground temperature, and the depth of the water table is measured manually three to five times per week. The Palsa site chambers are located within the palsa site in ref. 6 and correspond to the hummock site class (I) described in ref. 4. The Sphagnum site chambers are located within the bog site in ref. 6 or site S in ref. 9 and correspond to the semi-wet and wet site classes (II and III) described in ref. 4. The Eriophorum site chambers are located within the fen site in ref. 6 or site E in ref. 9 and correspond to the tall graminoid site class (IV) described in ref. 4.
Quantum cascade laser spectrometer measurement and calibration
Methane fluxes and isotopes were measured with a quantum cascade laser spectrometer (QCLS; Aerodyne Research Inc.), deployed to Stordalen mire in June 2011. The QCLS instrument at Stordalen is a modification of the technology described in detail in ref. 42. In brief, the QCLS uses a room-temperature continuous-wave mid-infrared laser whose frequency was tuned to scan rapidly (900 kHz) across 12CH4 and 13CH4 absorption lines in the 7.5-μm region. The laser light enters a multipass sample cell (effective path length about 200 m) containing sample air at low pressure (about 5 kPa) and is detected by a thermoelectrically cooled detector (no cryogens are needed). Aerodyne Research’s custom TDL Wintel software averages high-frequency spectra to produce independent 12CH4 and 13CH4 mixing ratios in the sample airstream at 1-s intervals. The ratio R of 13CH4 to 12CH4 can then be expressed in standard notation as δ13C, the part-per-thousand (‰) deviation of the measured ratio from the VPDB standard 13C/12C ratio RVPDB, as in equation (1).
Instrument precision in the field at Stordalen mire was assessed by using time-series measurements of calibration tank air over 30–40 min. The precision of δ13C-CH4 measurements using a 1-s integration time was 1‰. The Allan variance technique (used to characterize the minimum possible measurement error and the averaging time required to achieve it43), showed that the minimum measurement error on δ13C-CH4 was less than 0.2‰, achieved with 60 s of averaging time. This approaches the precision of comparable measurements made with gas chromatography–isotope ratio mass spectrometry (GC–IRMS).
We connected the QCLS to the main autochamber circulation with ¼-inch Dekoron tubing and a solenoid manifold that enabled selection between the autochamber flow and an array of calibration tanks. During measurement periods, filtered (0.45 μm Teflon filter) and dried (Perma Pure PD-100T-24MSA) sample air flows at 1.4 standard litres per minute through the 2-litre QCLS sample cell volume at 5.6 kPa. A downstream solenoid controls the QCLS return flow so that air recirculates only during autochamber measurement periods; during calibration periods, exhaust air is vented to the room.
Calibrations were performed every 60 min with three calibration gases spanning the observed concentration range (1.5–10 p.p.m.). The CH4 concentration and δ13C composition of each calibration tank was determined by inter-calibration with a set of four well-characterized primary standard tanks. The primary tanks (Scott Marin, Inc.) were calibrated to the VPDB scale by means of flask samples, which were analysed by GC–IRMS at Florida State University (see porewater methods for GC–IRMS details). Each isotopologue, 12CH4 and 13CH4, was treated as an independent measurement and calibrated separately. For each calibration period a linear calibration curve was fitted for each isotopologue and the fit parameters were then linearly interpolated between calibration periods. The interpolated fit parameters were applied to the measured sample isotopologue mixing ratios to give calibrated measurements of 12CH4, 13CH4 and total CH4, from which δ13C-CH4, was calculated.
Autochamber data processing
For each autochamber closure we calculated the flux and δ13C signature of emitted CH4. Fluxes were calculated by using a method consistent with that detailed in ref. 44 for CO2 and total hydrocarbons, using a linear regression of changing headspace CH4 concentration over a period of 2.5 min. Eight 2.5-min regressions were calculated, staggered by 15 s, and the most linear fit (highest r2) was then used to calculate flux. Keeling plots45,46,47 using the entire closure period were used to estimate the isotopic composition of the emitted CH4. As demonstrated in ref. 42, negligible error in measurement of CH4 relative to that of δ13CH4 for this instrumentation meant that type I regression was sufficient for the Keeling plot analysis. When the total change in headspace CH4 was low45, there was high error in the Keeling intercept. We used a threshold of 3‰ uncertainty in the Keeling intercept as a cutoff for including isotopic values in the calculation of daily and annual averages, resulting in a total of 1,569 observations at the Sphagnum site and 1,168 at the Eriophorum site. No Palsa chamber closures had sufficient CH4 flux to calculate δ13CH4. Daily and whole-season average flux and isotopic composition for each habitat were calculated on the basis of individual chambers as the unit of replication (n = 3 for Palsa and Sphagnum, n = 2 for Eriophorum). Significant differences in the magnitude and isotopic composition of CH4 emissions were determined with Student’s t-test (isotopic composition) and analysis of variance (flux magnitude) in R48, with seasonal averages for each autochamber as the unit of replication. Statistical significance was determined at α = 0.05.
Porewater sampling and analysis
Porewater samples were collected on 12 July 2011, 15 August 2011 and 15 October 2011 at three locations adjacent to the Sphagnum and Eriophorum autochamber sites (Extended Data Table 1). Samples were collected by suction with a syringe through a stainless steel tube and filtered through 25-mm diameter Whatman Grade GF/D glass microfibre filters (2-μm particle retention). Porewater pH was measured in the field (Oakton Waterproof pHTestr 10; Eutech Instruments). Samples for the analysis of the concentration and δ13C of CH4 and CO2 were injected into 30-ml evacuated vials sealed with butyl rubber septa and frozen within 8 h of collection. The samples for δD-CH4 were injected into 120-ml evacuated vials sealed with butyl rubber septa and containing 0.5 g of KOH. For δD-H2O, water was filtered directly into 2-ml plastic screw-cap vials so that the vials were completely filled, then frozen within 8 h of collection. All samples were shipped frozen to Florida State University for analysis.
Samples collected for analysis of CH4 and CO2 concentrations and δ13C were thawed, acidified with 0.5 ml of 21% H3PO4, and brought to atmospheric pressure with helium. The sample headspace was analysed for concentrations and δ13C of CH4 and CO2 on a continuous-flow Hewlett-Packard 5890 gas chromatograph (Agilent Technologies) at 40 °C coupled to a Finnigan MAT Delta S isotope ratio mass spectrometer via a Conflo IV interface system (Thermo Scientific). The headspace gas concentrations were converted to porewater concentrations on the basis of their known extraction efficiencies, defined as the proportion of formerly dissolved gas in the headspace. An extraction efficiency of 0.95 (based on repeated extractions) was used for CH4, and the extraction efficiency for CO2 relative to dissolved inorganic carbon (DIC) was determined on the basis of CO2 extraction from dissolved bicarbonate standards49.
Samples collected for analysis of δD-CH4 were brought to atmospheric pressure with helium and measured on a gas chromatograph connected to a ThermoFinnegan Delta Plus continuous-flow isotope ratio mass spectrometer at the National High Magnetic Field Laboratory (Tallahassee, FL). δD of CH4 is affected by δD of H2O because CH4 exchanges H atoms with water during methanogenesis20,38,50, so measurement of δD-H2O is necessary for the correct assignment of CH4 production mechanisms and oxidation based on δD and δ13C of CH4. Samples collected for δD-H2O were measured on an LGR DT-100 liquid water stable isotope analyser at Florida Agricultural and Mechanical University (Tallahassee, FL). Data analysis for these samples was performed with an MS Excel template from the IAEA Water Resources Programme (http://www.iaea.org/water).
Significant differences in αC and δD and δ13C of porewater CH4 between the Sphagnum and Eriophorum sites were determined with Student’s t-test (αC, δD-CH4, δ13C-CH4) and Hotelling’s t-test (multivariate δD and δ13C of CH4) in R48. Statistical significance was determined at α = 0.05.
Peat sampling
Peat samples were collected on 12 July 2011, 16 August 2011 and 16 October 2011 at three locations adjacent to the Palsa, Sphagnum and Eriophorum autochamber sites. For the Sphagnum and Eriophorum sites, samples were collected at the same depths and locations as those used for porewater sampling (Extended Data Table 1); sample depths for the Palsa site are detailed in ref. 6. Peat cores were collected with a push corer 11 cm in diameter (Palsa and Sphagnum sites) or a 10 cm × 10 cm Wardenaar corer (Eriophorum site). Cores were subsampled by depth and were subdivided in the field for microbial and chemical analysis, avoiding the outer 1 cm of the core. Samples for microbial analysis were placed in cryotubes, saturated with about 3 volumes of LifeGuard solution (MoBio Laboratories) and stored at −80° C until processing. Samples for chemical analysis were placed in plastic bags and frozen until processing.
Peat chemical analysis
For peat %C, %N, C:N ratio and δ13C measurements, 5–10 g of peat was dried at 60 °C until completely dry (3–10 days) and ground to a fine powder. Subsamples of ground peat (80–100 μg for %C and δ13C analysis, and 5–6 mg for %N analysis) were wrapped in tin capsules and analysed by combustion to CO2 and N2 at 1,020 °C in an automated CHN elemental analyser coupled with a ThermoFinnegan Delta XP isotope ratio mass spectrometer at the National High Magnetic Field Laboratory. Samples were run in non-dilution mode for carbon analysis and dilution mode (×10) for nitrogen analysis. C:N was calculated as (%C)/(%N) (by weight) for corresponding pairs of subsamples.
Small-subunit ribosomal RNA gene amplicon analysis
Sampling and extraction was performed as described previously6. Several additional samples were analysed for this paper; multiplex identifiers for those runs not reported in ref. 6 are provided in Extended Data Table 7. Small-subunit rRNA gene sequences were processed with APP 3.0.3 (https://github.com/Ecogenomics/APP). Homopolymer errors were corrected with Acacia51 and the resulting reads were processed by using the CD-HIT-OTU 0.0.2 pipeline with minor adjustments52. All reads were trimmed to 250 base pairs, and reads of less than 250 base pairs were discarded. Sequences were clustered at 97% identity and each cluster was assigned a taxonomy using BLASTN 2.2.22 (ref. 53) through the QIIME script assign_taxonomy.py54 against the GreenGenes October 2012 database clustered at 99% identity (Supplementary Table 1). The taxonomy of each methanogenic cluster was confirmed by using parsimony insertion in ARB55. Amplicon sequence clusters were identified as potential hydrogenotrophic or acetoclastic methanogens based on taxonomic relationship to known methanogenic lineages (Extended Data Table 2)23,24,56. Within the order Methanosarcinales, lineages most closely related to Methanosaeta were classified as obligate acetoclasts, whereas those most closely related to Methanosarcina were considered facultative acetoclasts, having the potential for both acetoclastic or hydrogenotrophic production23.
Regression analysis
A stepwise regression approach with Akaike’s information criterion (AIC) as the model selection criterion was used to identify a subset of microbial and environmental predictor variables that best explained CH4 metabolism patterns quantified as porewater αC (Extended Data Table 5). Model selection was performed with the stepAIC package in R, and the relative importance of the predictor variables in the selected model was then calculated with the relaimpo R package48. Variables included in the model selection process included the relative abundances of the six most abundant methanogen operational taxonomic units (comprising more than 93% of the total methanogen sequences; see Extended Data Table 2) plus soil temperature, water table depth, pH, porewater CH4 and DIC concentration, and peat C:N, %C, %N and δ13C (Extended Data Table 1). Strong correlation between pH and both water-table depth and peat δ13C as well as peat %N and both %C and C:N meant that pH and %N were excluded from the regression analysis. Removal of non-significant predictor variables (DIC and relative abundance of an unidentified Methanobacterium spp. (otu-3636; Extended Data Table 2)) had a minimal effect on the model AIC value (less than 1); this simplified version was therefore selected as the optimal model (model 2 in Extended Data Table 5). Stepwise regression was also performed with δ13C-CH4 as the dependent variable. This analysis resulted in a similar model outcome, but with a lower R2 (model 1 in Extended Data Table 8). Stepwise regression analysis with environmental predictor variables and the relative abundance of the influential methanogen ‘M. stordalenmirensis’ (otu-10747) as the dependent variable showed that patterns in this methanogen’s abundance were influenced by environmental conditions, particularly water table depth and peat chemistry (model 2 in Extended Data Table 8). However, these environmental variables alone cannot fully replace microbial data when modelling αC. Stepwise regression analysis using only environmental variables to predict αC yielded a model with a lower AIC and R2 (model 3 in Extended Data Table 8). It is the combination of methanogen and environmental variables that yields a model that explains the most variability in αC (Extended Data Table 5).
Box model of atmospheric methane
The model used here was a one-box model simplified from the two-box model of ref. 57 (and also used in the methane inversion study28):


where M is the mixing ratio (in p.p.b.v.) of CH4 in the atmosphere,  is the source flux of CH4 to the atmosphere, λ is the atmospheric removal rate (1/9 yr−1, assumed for this illustration to be fixed), the R terms are the ratio of 13CH4 to 12CH4, as defined for equation (1), and αOH is the isotopic fractionation (0.994, or about −6‰) for the atmospheric oxidation of CH4 by OH (ref. 28). Baseline flux to the atmosphere (
is the source flux of CH4 to the atmosphere, λ is the atmospheric removal rate (1/9 yr−1, assumed for this illustration to be fixed), the R terms are the ratio of 13CH4 to 12CH4, as defined for equation (1), and αOH is the isotopic fractionation (0.994, or about −6‰) for the atmospheric oxidation of CH4 by OH (ref. 28). Baseline flux to the atmosphere ( ) was set to 559 Tg CH4, the 1980 value28. The isotopic composition of CH4 inputs to the atmosphere (
) was set to 559 Tg CH4, the 1980 value28. The isotopic composition of CH4 inputs to the atmosphere ( ) was set to the equivalent of −53‰ to allow steady-state modern atmospheric CH4 to have the observed value of about −47‰.
) was set to the equivalent of −53‰ to allow steady-state modern atmospheric CH4 to have the observed value of about −47‰.
We implemented this model numerically in the R software package48, simulating the effect on the atmosphere of CH4 emission due to permafrost thaw and partial decomposition of the 1,700 Pg C stock of permafrost C expected over the next 300 years, as summarized in refs 1, 2. High and low permafrost carbon release scenarios for both the high climate change scenario (Intergovernmental Panel on Climate Change (IPCC) scenario RCP8.5, leading to the release of 120–195 Pg C) and the low climate change scenario (IPCC scenario RCP2.6, approximated as one-third of the C release of the high scenario) (Extended Data Fig. 2a) generated CH4 emissions (Fig. 3a) (based on 2.3% of released permafrost carbon emerging as CH4 (ref. 2)) and corresponding impacts on the atmospheric concentrations of CH4 (Extended Data Fig. 2b). We simulated the impacts of these emissions on the isotopic composition of atmospheric CH4 by assuming that the δ13C of CH4 emitted was in the range of what we report here for Stordalen mire, from very light (−80‰, like that measured at the Sphagnum site) to only moderately light (−65‰, like that measured at the Eriophorum site), giving a range of isotopic perturbations to atmospheric CH4 under high climate change (Extended Data Fig. 2c) and under low climate change (Extended Data Fig. 2d). In all scenarios, the induced change in atmospheric δ13C is significantly larger than the atmospheric detection limit of 0.1‰ (reported in ref. 28 and shown as a dotted horizontal line in Extended Data Fig. 2c, d).
For the analysis shown in Fig. 3 we focused on a mid-range value of permafrost C release (high climate change scenario with low C release, 120 Pg total C by 2100), corresponding to emissions of 2.8 Pg C as CH4 by 2100 (the dashed black and red line in Fig. 3a). (By comparison, the IPCC estimates that up to 5 Pg C may be released as CH4 by 2100 (ref. 3).) We explored the misattribution of C release that would occur, by (mistakenly) assuming that the isotopic composition of emitted CH4 was in the range of assumptions used in previous atmospheric inversions, from −60‰ to −65‰ (ref. 28), instead of the range measured at Stordalen mire (−65‰ to −80‰). We estimated the magnitude of misattribution (or error flux; Fig. 3c) by simulating the amount of additional carbon that would need to be released (at nominally assumed isotopic composition values of −60 or −65) to have the same effect on atmospheric composition as the carbon released under scenarios with isotopic compositions like those observed in the field.
References
Tarnocai, C. et al. Soil organic carbon pools in the northern circumpolar permafrost region. Glob. Biogeochem. Cycles 23, GB2023 (2009)
Schuur, E. A. G. et al. Expert assessment of vulnerability of permafrost carbon to climate change. Clim. Change 119, 359–374 (2013)
Ciais, P. et al. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (Cambridge University Press, 2013)
Johansson, T. et al. Decadal vegetation changes in a northern peatland, greenhouse gas fluxes and net radiative forcing. Glob. Change Biol. 12, 2352–2369 (2006)
Christensen, T. R. et al. Thawing sub-arctic permafrost: effects on vegetation and methane emissions. Geophys. Res. Lett. 31, L04501 (2004)
Mondav, R. et al. Discovery of a novel methanogen in thawing permafrost. Nature Commun. 5, 3212, http://dx.doi.org/10.1038/ncomms4212 (14 February 2014)
Melton, J. R. et al. Present state of global wetland extent and wetland methane modelling: conclusions from a model inter-comparison project (WETCHIMP). Biogeosciences 10, 753–788 (2013)
Jorgenson, M. T., Racine, C. H., Walters, J. C. & Osterkamp, T. E. Permafrost degradation and ecological changes associated with a warming climate in central Alaska. Clim. Change 48, 551–579 (2001)
Hodgkins, S. B. et al. Changes in peat chemistry associated with permafrost thaw increase greenhouse gas production. Proc. Natl Acad. Sci. USA 111, 5819–5824 (2014)
Olefeldt, D., Turetsky, M. R., Crill, P. M. & McGuire, A. D. Environmental and physical controls on northern terrestrial methane emissions across permafrost zones. Glob. Change Biol. 19, 589–603 (2012)
Lee, H., Schuur, E. A. G., Inglett, K. S., Lavoie, M. & Chanton, J. P. The rate of permafrost carbon release under aerobic and anaerobic conditions and its potential effects on climate. Glob. Change Biol. 18, 515–527 (2012)
Mackelprang, R. et al. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480, 368–371 (2011)
Riley, W. J. et al. Barriers to predicting changes in global terrestrial methane fluxes: analyses using CLM4Me, a methane biogeochemistry model integrated in CESM. Biogeosciences 8, 1925–1953 (2011)
Koven, C. D. et al. Permafrost carbon-climate feedbacks accelerate global warming. Proc. Natl Acad. Sci. USA 108, 14769–14774 (2011)
Turetsky, M. R., Wieder, R. K. & Vitt, D. H. Boreal peatland C fluxes under varying permafrost regimes. Soil Biol. Biochem. 34, 907–912 (2002)
Bäckstrand, K. et al. Annual carbon gas budget for a subarctic peatland, northern Sweden. Biogeosciences 7, 95–108 (2010)
Bridgham, S. D., Cadillo-Quiroz, H., Keller, J. K. & Zhuang, Q. Methane emissions from wetlands: biogeochemical, microbial, and modeling perspectives from local to global scales. Glob. Change Biol. 19, 1325–1346 (2013)
Hornibrook, E. R. C. & Bowes, H. L. Trophic status impacts both the magnitude and stable carbon isotope composition of methane flux from peatlands. Geophys. Res. Lett. 34, 2–6 (2007)
Chanton, J. P., Chaser, L. C., Glaser, P. & Siegel, D. in Stable Isotopes and Biosphere–Atmosphere Interactions (eds Flanagan, L. B., Ehleringer, J. R. & Pataki, D. E. ) 85–105 (Elsevier, 2005)
Whiticar, M. J. Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane. Chem. Geol. 161, 291–314 (1999)
Popp, T. J., Chanton, J. P., Whiting, G. J. & Grant, N. Methane stable isotope distribution at a Carex dominated fen in North Central Alberta. Glob. Biogeochem. Cycles 13, 1063–1077 (1999)
Whiticar, M. J., Faber, E. & Schoel, M. Biogenic methane formation in marine and freshwater environments: CO2 reduction vs. acetate fermentation—isotope evidence. Geochim. Cosmochim. Acta 50, 693–709 (1986)
Ferry, J. G. How to make a living by exhaling methane. Annu. Rev. Microbiol. 64, 453–473 (2010)
Liu, Y. & Whitman, W. B. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. NY Acad. Sci. 1125, 171–189 (2008)
Hornibrook, E. R. C., Longstaffe, F. J. & Fyfe, W. S. Spatial distribution of microbial methane production pathways in temperate zone wetland soils: stable carbon and hydrogen isotope evidence. Geochim. Cosmochim. Acta 61, 745–753 (1997)
Wania, R. et al. Present state of global wetland extent and wetland methane modelling: methodology of a model inter-comparison project (WETCHIMP). Geoscient. Model Devel. 6, 617–641 (2013)
Wieder, W. R., Bonan, G. B. & Allison, S. D. Global soil carbon projections are improved by modelling microbial processes. Nature Clim. Change 3, 909–912 (2013)
Kai, F. M., Tyler, S. C., Randerson, J. T. & Blake, D. R. Reduced methane growth rate explained by decreased Northern Hemisphere microbial sources. Nature 476, 194–197 (2011)
Bousquet, P. et al. Contribution of anthropogenic and natural sources to atmospheric methane variability. Nature 443, 439–443 (2006)
Hines, M. E., Duddleston, K. N., Rooney-Varga, J. N., Fields, D. & Chanton, J. P. Uncoupling of acetate degradation from methane formation in Alaskan wetlands: connections to vegetation distribution. Glob. Biogeochem. Cycles 22, 1–12 (2008)
Payette, S. Accelerated thawing of subarctic peatland permafrost over the last 50 years. Geophys. Res. Lett. 31, 1–4 (2004)
O’Donnell, J. a. et al. The effects of permafrost thaw on soil hydrologic, thermal, and carbon dynamics in an Alaskan peatland. Ecosystems 15, 213–229 (2012)
Vitt, D. H., Halsey, L. A. & Zoltai, S. C. The changing landscape of Canada’s western boreal forest: the current dynamics of permafrost. Can. J. For. Res. 30, 283–287 (2000)
Quinton, W. L., Hayashi, M. & Chasmer, L. E. Permafrost-thaw-induced land-cover change in the Canadian subarctic: implications for water resources. Hydrol. Processes 25, 152–158 (2011)
Zoltai, S. C. Cyclic development of permafrost in the peatlands of Northwestern Alberta, Canada. Arct. Alp. Res. 25, 240–246 (1993)
Camill, P. & Clark, J. S. Climate change disequilibrium of boreal permafrost peatlands caused by local processes. Am. Nat. 151, 207–222 (1998)
Dyke, L. D. & Sladen, W. E. Permafrost and peatland evolution in the Northern Hudson Bay Lowland, Manitoba. Arctic 63, 429–441 (2010)
Whiticar, M. J. & Faber, E. Methane oxidation in sediment and water column environments—isotopic evidence. Org. Geochem. 10, 759–768 (1986)
Conrad, R. Quantification of methanogenic pathways using stable carbon isotopic signatures: a review and a proposal. Org. Geochem. 36, 739–752 (2005)
Bäckstrand, K., Crill, P. M., Mastepanov, M., Christensen, T. R. & Bastviken, D. Non-methane volatile organic compound flux from a subarctic mire in Northern Sweden. Tellus B Chem. Phys. Meterol. 60, 226–237 (2008)
Bubier, J. L., Crill, P. M., Mosedale, A., Frolking, S. & Linder, E. Peatland responses to varying interannual moisture conditions as measured by automatic CO2 chambers. Glob. Biogeochem. Cycles 17, 1066, http://dx.doi.org/10.1029/2002GB001946 (2003)
Santoni, G. W. et al. Mass fluxes and isofluxes of methane (CH4) at a New Hampshire fen measured by a continuous wave quantum cascade laser spectrometer. J. Geophys. Res. 117, D10301 (2012)
Werle, P., Mücke, R. & Slemr, F. The limits of signal averaging in atmospheric trace-gas monitoring by tunable diode-laser absorption spectroscopy (TDLAS). Appl. Phys. B 139, 131–139 (1993)
Bäckstrand, K., Crill, P. M., Mastepanov, M., Christensen, T. R. & Bastviken, D. Total hydrocarbon flux dynamics at a subarctic mire in northern Sweden. J. Geophys. Res. 113, G03026, http://dx.doi.org/10.1029/2008JG000703 (2008)
Pataki, D. E. The application and interpretation of Keeling plots in terrestrial carbon cycle research. Glob. Biogeochem. Cycles 17, 1022, http://dx.doi.org/10.1029/2001GB001850 (2003)
Keeling, C. D. The concentration and isotopic abundances of atmospheric carbon dioxide in rural areas. Geochim. Cosmochim. Acta 13, 322–334 (1958)
Keeling, C. D. The concentration and isotopic abundances of carbon dioxide in rural and marine air. Geochim. Cosmochim. Acta 24, 277–298 (1960)
R Development Core Team. A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2012)
Corbett, J. E. et al. Partitioning pathways of CO2 production in peatlands with stable carbon isotopes. Biogeochemistry 114, 327–340 (2013)
Chanton, J. P., Fields, D. & Hines, M. E. Controls on the hydrogen isotopic composition of biogenic methane from high-latitude terrestrial wetlands. J. Geophys. Res. 111, 1–9 (2006)
Bragg, L., Stone, G., Imelfort, M., Hugenholtz, P. & Tyson, G. W. Fast, accurate error-correction of amplicon pyrosequences. Nature Methods 9, 425–426 (2012)
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of proteins or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006)
Altschul, H. J. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997)
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7, 335–336 (2010)
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371 (2004)
Conrad, R. The global methane cycle: recent advances in understanding the microbial processes involved. Environ. Microbiol. Rep. 1, 285–292 (2009)
Tans, P. P. A note on isotopic ratios and the global atmospheric methane budget. Glob. Biogeochem. Cycles 11, 77–81 (1997)
Acknowledgements
We thank the Abisko Scientific Research Station for infrastructure and logistical support; T. Logan and N. Rakos for their assistance in the field; and S. Wofsy and S. Frolking for feedback on a draft of this paper. This work was supported by the US Department of Energy Office of Biological and Environmental Research (award DE-SC0004632), and by the University of Arizona Technology and Research Initiative Fund, through the Water, Environmental and Energy Solutions Initiative. R.M. was supported by an Australian Postgraduate Award Scholarship.
Author information
Authors and Affiliations
Contributions
S.R.S., V.I.R., P.M.C., J.C. and G.W.T. designed the study. C.K.M., S.B.H., R.A.W., P.M.C., J.C. and S.R.S. designed and/or performed flux/porewater/isotope measurements and laboratory incubations. C.K.M., B.J.W., R.M., E.-H.K., S.R.S., V.I.R. and G.W.T. designed and/or performed analyses integrating bioinformatics and biogeochemistry. C.K.M., V.I.R. and S.R.S. wrote the paper in consultation with B.J.W., S.B.H., J.C., P.M.C., E.-H.K., R.M. and G.W.T.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Expected and observed relationships between the δD and δ13C content of porewater CH4.
The thick grey arrow shows the expected pattern in H and C isotopes of CH4 when variations are caused by shifts between acetoclastic (lower right) and hydrogenotrophic (upper left) production. The thin black arrows pointing to the upper right indicate the expected pattern in H and C isotopes of CH4 when variations are caused by changes in CH4 oxidation19. The points are observed isotopic compositions of samples collected between July and October 2011 at the partly thawed Sphagnum and fully thawed Eriophorum sites; site averages are shown with error bars (error bars represent s.e.m.; n = 13 (Sphagnum) and 20 (Eriophorum)). Although the scatter allows for some variation in both production and oxidation, the average Eriophorum porewater CH4 had significantly more 13C and less D relative to Sphagnum porewater (Hotelling’s T2 test, P = 0.0001, n = 33), indicating that the overall inter-site isotopic differences were due mostly to differences in the CH4 production pathway rather than to differences in CH4 oxidation. Additionally, in August there was a significant negative relationship between δ13C-CH4 and δD-CH4 of porewater samples collected across sites (dashed line, linear regression, R2 = 0.5, P < 0.02, n = 12). Note that on the vertical axis δD-H2O has been subtracted from δD-CH4 to correct for the effect of δD exchange between H2O and CH4 (refs 20, 38, 50).
Extended Data Figure 2 Simulations, using high and low temperature and C release scenarios, of the effect of CH4 release from thawing permafrost on atmospheric δ13C-CH4.
a, Scenarios of permafrost C release due to thaw (red bounding lines, high temperature; orange bounding lines, low temperature; the range in each case is defined by high and low C release scenarios). b, Impact on atmospheric methane mixing ratios (assuming that 2.3% of released C is emitted as methane). c, Impact of the high climate change scenario on atmospheric methane isotopes, assuming Eriophorum-like emissions (blue bounding lines, δ13C ≈ −65‰), or assuming Sphagnum-like emissions (green bounding lines, δ13C ≈ −80‰). d, As in c, except for the low climate change scenario. In c and d, dotted horizontal lines indicate the detection limit for CH4 isotopes28.
Supplementary information
Supplementary Data
Operational taxonomic unit (OTU) table from 16S rRNA gene amplicon analysis. Each row represents an OTU. The first set of columns show the number of that 16S rRNA gene amplicon found in each sample. The rightmost columns show the taxonomy of that OTU predicted with BLAST. The samples presented in this study represent a subset of a larger sampling campaign (eg. Mondav et al 2014) therefore not all OTU's identified in the larger sample-set are present in this table. (XLS 9134 kb)
Rights and permissions
About this article

Cite this article
McCalley, C., Woodcroft, B., Hodgkins, S. et al. Methane dynamics regulated by microbial community response to permafrost thaw. Nature 514, 478–481 (2014). https://doi.org/10.1038/nature13798
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature13798
- Springer Nature Limited
This article is cited by
-
Climate warming restructures food webs and carbon flow in high-latitude ecosystems
Nature Climate Change (2024)
-
Multifarious Responses of Forest Soil Microbial Community Toward Climate Change
Microbial Ecology (2023)
-
Effects of microbial-converted ancient permafrost organic carbon on the growth and reproduction of Daphnia magna
Oecologia (2023)
-
Microbial iron cycling during palsa hillslope collapse promotes greenhouse gas emissions before complete permafrost thaw
Communications Earth & Environment (2022)
-
Improved global wetland carbon isotopic signatures support post-2006 microbial methane emission increase
Communications Earth & Environment (2022)