
Abstract
Recent data suggest that intraneuronal accumulation of metabolites of the amyloid-β-precursor protein (APP) is neurotoxic. We observed that transgenic mice overexpressing in neurons a human APP gene harboring the APPE693Q (Dutch) mutation have intraneuronal lysosomal accumulation of APP carboxylterminal fragments (APP-CTFs) and oligomeric amyloid β (oAβ) but no histological evidence of amyloid deposition. Morphometric quantification using the lysosomal marker protein 2 (LAMP-2) immunolabeling showed higher neuronal lysosomal counts in brain of 12-months-old APPE693Q as compared with age-matched non-transgenic littermates, and western blots showed increased lysosomal proteins including LAMP-2, cathepsin D and LC3. At 24 months of age, these mice also exhibited an accumulation of α-synuclein in the brain, along with increased conversion of LC3-I to LC3-II, an autophagosomal/autolysosomal marker. In addition to lysosomal changes at 12 months of age, these mice developed cholinergic neuronal loss in the basal forebrain, GABAergic neuronal loss in the cortex, hippocampus and basal forebrain and gliosis and microgliosis in the hippocampus. These findings suggest a role for the intraneuronal accumulation of oAβ and APP-CTFs and resultant lysosomal pathology at early stages of Alzheimer’s disease-related pathology.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder that typically begins with mild short-term memory deficits and culminates in total loss of cognition and executive functions. The neuropathological features of AD include brain parenchymal senile plaques, cerebral amyloid angiopathy and neurofibrillary tangles.1 Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D), a familial form of AD,2, 3 is caused by an E693Q substitution in the amyloid-β-precursor protein (APP).4 The disease is characterized by cerebral amyloid angiopathy leading to hemorrhagic strokes,5, 6 as well as cerebral lesions caused by the hemorrhages,7 non-fibrillar parenchymal deposits of amyloid-β (Aβ)6, 8, 9, 10 and dementia.11, 12 Aβ harboring the Dutch substitution at position 22 has been associated with enhanced aggregation and oligomerization13, 14 and reduced clearance as compared with wild-type Aβ.15, 16
Two mouse lines overexpressing the E693Q-substituted human APP under the neuron-specific Thy1.1 expression cassette were generated.16, 17 Similar to the brains of HCHWA-D patients,18 higher Aβ40 to Aβ42 ratio was observed in the brains of the APPE693Q transgenic mice as compared with non-transgenic littermates.16, 17 It was shown that neurons in the brain of 12-month-old APPE693Q mice contain vesicles immunopositive for antibodies that interact with full-length APP (flAPP), amyloid precursor protein (APP-CTFs) and Aβ.17 Aβ has been shown to exist in different forms, including soluble Aβ monomers and oligomers, as well as insoluble amyloid fibrils and multiple studies have documented the correlation between Aβ pathology and cognitive decline in AD.19, 20, 21 Synaptic dysfunction and neuronal degeneration have been demonstrated in the brains lacking amyloid plaques and cerebral amyloid angiopathy during the early stages of AD, and several lines of evidence have suggested that soluble oligomers of Aβ, but not monomers, cause the synaptic dysfunction and neurodegeneration in the brains of AD patients and animal models.19, 20, 21 In addition to Aβ, APP-CTFs have been shown to exhibit cytotoxicity in the absence of amyloid deposition in the brain.22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38 APPE693Q mice have behavioral deficits at ages when APP metabolites accumulate within neurons but no fibrillar amyloid deposits are detectable in either brain vasculature or parenchyma.17, 39, 40, 41
Although the cellular events triggered by neurotoxic oAβ accumulation that lead to synaptic deficits may vary in different forms of AD, data suggest that neuronal endocytic-lysosomal alterations are either an underlying pathological mechanism in the disease or a maladaptive response of neurons to other disease stresses in AD.42, 43, 44, 45, 46 Given that endosomal abnormalities were reported in cerebral endothelia in the brain of HCHWA-D patients43 and that we have documented intraneuronal punctate accumulation of APP metabolites in cortical and hippocampal brain regions of APPE93Q transgenic mice,17 we investigated the endosomal-lysosomal pathway in the brain of these APPE93Q transgenic mice. Here we demonstrate the intraneuronal lysosomal accumulation of APP-CTFs associated with intraneuronal lysosomal abnormalities and neuronal loss in the absence of fibrillar Aβ.
Materials and methods
Animals
A breeding colony of APPE693Q mice17 on C57BL/6J background was maintained at Nathan S. Kline Institute for Psychiatric Research. Both male and female APPE693Q transgenic mice and littermate controls were studied at 12 and 24 months of age. No gender differences between the genotypes were found. All animal procedures were performed following the National Institutes of Health guidelines with approval from the Institutional Animal Care and Use Committee at the Nathan S. Kline Institute for Psychiatric Research. All the experiments were performed with a sample size of 4–5 mice per genotype per age. The sample size was determined on the basis of findings from our previous APPE693Q transgenic mouse studies.17
Thioflavin S staining and immunocytochemistry
For all histological procedures, identification cards were coded to render the experimenter blind to the sex and genotype of each subject. Mice were anesthetized with ketamine (50 g kg−1)/xylazine (5 mg kg−1) and transcardially perfusion-fixed with 4% paraformaldehyde in 0.1 m sodium cacodylate buffer.47 To detect fibrillar amyloid deposits, brain sections from 12- and 24-month-old mice were slide-mounted and immersed in a solution of filtered 1% thioflavin S in ddH2O for 10 min (Sigma Aldrich, St. Louis, MO, USA), rinsed (× 3) for 1 min in ddH2O and subsequently cover-slipped with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA). Immunolabeling was performed using 4G8 antibody (which recognizes flAPP/APP-CTF/Aβ; 1:200; Covance ImmunoTechnologies, Denver, PA, USA; Cat # SIG-39220), C1/6.1 to the carboxyl-terminal cytoplasmic domain of APP (1:100; provided by Dr Paul M Mathews). 22C11 antibody (which recognizes full-length and soluble amino terminal fragment of APP, sAPP) (1:200; EMD Millipore, Billerica, MA, USA; Cat # MAB 348), anti-glial fibrillary acidic protein (GFAP) (1:500; Dako, Carpinteria, CA, USA; Cat # Z0334), anti-Iba1 (1:500, Wako Chemicals, Richmond, VA, USA; Cat # 019-1974), anti-rab5a (1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA; Cat # SC-309), anti-cathepsin D (Cat D) (1:500; Scripps, San Diego, CA, USA; Cat # 90236), anti-LC3 (1:500; Novus Biologicals, Littleton, CO, USA; Cat # NB600-1384), anti-lysosomal marker protein 2 (LAMP-2) (1:100; Developmental Studies Hybridoma Bank, Iowa City, IA, USA; Cat # 93 S), and anti-ChAT antibody (1:500; Millipore; Cat # AB144P) antibodies. Double immunofluorescent labeling was performed to identify coincidence of localization of flAPP/APP-CTFs/Aβ using 4G8 (1:200) or flAPP/APP-CTFs using antibody to the carboxyl-terminal cytoplasmic domain of APP using C1/6.148 (1:100) with antibody to the lysosomal marker LAMP-2 (1:100). Anti-parvalbumin (PV) (1:5000; Swant Technologies, Marly, Switzerland; Cat # PV25) staining was performed to detect GABAergic interneurons. Secondary antibodies used were: fluorescence-conjugated anti-rabbit, anti-sheep, and anti-mouse antibodies (Invitrogen, Carlsbad, CA, USA).
PV-positive GABAergic interneurons were quantified in the hippocampus of four individual brain sections per mouse, five mice per genotype, at the age of 12 months using ImageJ (NIH, Bethesda, MD, USA). For lysosomal quantification, approximately 30 neurons per mouse, four mice per genotype, were analyzed and LAMP-2 signal was measured using ImageJ randomly by a genotype-blinded observer.47 LAMP-2 labeled brain sections were further stained with Nissl (Neurotrace) (1:100; Molecular Probes, Eugene, OR, USA) to quantify lysosomes in the large neurons of the medial septal nucleus (MSN), counting ~30 neurons per mouse, four mice per genotype. LAMP-2 signal was measured using ImageJ. Qualitative analysis of GFAP- or Iba1-positive staining was performed on four individual brain sections per mouse, five mice per genotype, at the ages of 12 and 24 months.
Optical fractionator procedure
The number of immunofluorescence-labeled ChAT cells in the MSN was estimated using the optical fractionator method,49 utilizing ImageJ software as previously described.50 Briefly, the MSN was sampled in every 3rd consecutive 40- μm-thick vibratome section rostral to the anterior commissure. A grid of optical disector sampling sites with 0.25 mm spacing was placed on each section. At each sampling site, a × 40 oil-immersion objective with 1.3 numerical aperture was used to collect a z-stack of 15 3-μm-spaced images starting at a focal plane just beneath the tissue surface. Cell counting was done on 198 × 143 μm counting boxes drawn onto each z-stack, using the top two slices as the upper guard zone, the next 10 slices as a 30 μm deep counting box and the subsequent slices as the lower guard zone. Section thickness (40.5±1.7 μm mean±s.d., n=8 animals), measured in triplicate on each section, showed no evidence of shrinkage from the original 40 μm section thickness. On average, 191±69 cells were counted per MSN, and the coefficient of error51 was 0.08±0.01 (mean±s.d.).
Western blot analysis
Mice were anesthetized with ketamine (50 g kg−1)/xylazine (5 mg kg−1) and brains were rapidly removed over ice. Brains of 12- (n=4) and 24- (n=5) month-old mice were snap frozen and stored at −80 °C before biochemical analyses. Entorhinal cortices were dissected out from fresh brains of 12- (n=4) and 24- (n=4) month-old mice and stored at −80 °C. Western blot immunolabeling was performed as previously described.52 Primary antibodies used were: anti-LAMP-2 (1:1000), anti-rab4a (1:1000; Santa Cruz Biotechnology; Cat # SC-312), anti-rab5a (1:1000), anti-rab7 (1:1000; Sigma; Cat # R8779), anti-rabaptin5 (1:1000; Santa Cruz Biotechnology; Cat # SC-6162), anti-LC3 (1:1000), anti-Cat D (1:1000), C1/6.1 (1:1000),48 anti-mTOR and anti-AKT (phosphorylated and total protein) (1:1000, Cell Signaling Technology, Danvers, MA, USA; Cat # 2983 S and Cat # 9272 S respectively), anti-α-synuclein (1:1000; Sigma; Cat # S3062) and anti-β-tubulin (1:10 000; Sigma; Cat # T8535). Secondary antibodies used were: horseradish peroxidase-conjugated anti-rabbit and mouse antibodies (1:5000; GE Healthcare, Pittsburgh, PA, USA). The protein bands were scanned, optical density was calculated using the Image J, and the ratio of protein intensity to β-tubulin in the same lane was calculated. Western blot analyses were repeated three times.
Statistical analyses
Results were evaluated using one-way analysis of variance followed by post hoc multiple-comparison Bonferroni’s tests to determine the disparity among different age groups between the genotypes. Unpaired, two-tailed Student’s t-test was used to analyze results among genotypes. A P-value of P<0.05 was considered statistically significant. None of the animals were excluded from analysis. No randomization was used in this study. The data from each group showed similar variance, presented in the study as mean±s.e.m.
Results
Lysosomal localization of APP metabolites in the brain of APPE693Q mice
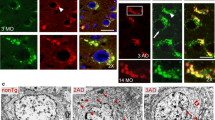
Immunocytochemistry revealed brain region-specific high levels of staining by antibodies to APP and its metabolites in APPE693Q mice as compared with their wild-type (WT) littermate controls. Strong intraneuronal, punctate, 4G8-immuno-positive staining that identifies flAPP, APP-CTFs and Aβ was detected in the entorhinal and frontoparietal cortices of 12-month-old APPE693Q mice as compared with WT mice (Figure 1a). Similarly, robust intraneuronal, punctate staining was observed with C1/6.1, an antibody that binds flAPP and APP-CTFs, in 12-month-old APPE693Q mice as compared with WT mice (Figure 1b). Staining with an antibody against the amino-terminus of APP (22C11) that binds both flAPP and amino terminal soluble fragments of APP (sAPP) showed a lower difference between APPE693Q and WT mice as compared with the staining with 4G8 and C1/6.1 in the brain of the APPE693Q mice (Figure 1c). Dual immunostaining with an antibody to the lysosomal protein LAMP-2 showed that staining with both 4G8 and C1/6.1 was largely localized to lysosomes in the brains of APPE693Q transgenic mice (Figures 1a and b). Compared with the staining with these two antibodies, fewer LAMP-2 lysosomes were stained with 22C11 (Figure 1c). These data indicate the accumulation of APP-CTFs, but not of flAPP or sAPP, in lysosomes in the brain of APPE693Q transgenic mice.

Age-dependent intraneuronal lysosomal accumulation of APP-CTFs and lysosomal abnormality in the brain of APPE693Q mice. Double immunostaining of the entorhinal cortices of 12-month-old APPE693Q and WT mice with antibodies to the lysosomal marker LAMP-2 (red) and 4G8 (green), which reacts with flAPP, APP-CTFs and Aβ (a), with C1/6.1, which reacts with flAPP and APP-CTFs (green) (b) or 22C11 which reacts with flAPP and sAPP (green) (c), (n=4) (scale bars, 10 μm), revealing intra-lysosomal accumulation of APP-CTFs. (d) Western blot analysis of cerebral homogenates of brains of 12- and 24-month-old APPE693Q mice and WT littermate controls using the C1/6.1 antibody revealed an increase in APP-CTFs levels in 24-month-old as compared with 12-month-old mice. Different exposure times of the same membrane are shown. β-tubulin reactivity is shown as a loading control. Densitometric analysis of the ratio of APP-CTFs signals to β-tubulin. Measurements are presented as the mean±s.e.m. (n=4/5). The difference among different age groups in mice were significant at *P<0.05. (e) Thioflavin-S (Thio-S) staining showing no fibrillar Aβ deposits in the frontoparietal cortex of APPE693Q mice at 24 months of age as compared with fibrillar Aβ-positive 24-month-old APP23 mouse brain (scale bar, 100 μm). Aβ, amyloid-β; APP, amyloid-β-precursor protein; CTF, APP carboxyl-terminal fragment; flAPP, full-length APP; WT, wild type.
Western blot analysis with the antibody C1/6.1 demonstrated that while there was no change in the expression level of flAPP, APP-CTFs levels were significantly elevated in the brains of 24-month-old compared with 12-month-old APPE693Q mice (Figure 1d). This reveals accumulation of APP-CTFs in the brains of APPE693Q mice with age, in addition to the previous demonstration of accumulation of oAβ.17, 41 No fibrillar amyloid deposits were observed in the brains of APPE693Q mice up to 24 months of age (Figure 1e).
Lysosomal pathology in the brain of APPE693Q mice
Immunocytochemical and western blot analyses were conducted in order to investigate whether accumulation of APP metabolites in neuronal lysosomes affects the endocytic pathway in APPE693Q mouse brains. Immunostaining with an antibody to the early endosomal protein rab5 showed no difference in endosomal size between APPE693Q mice and WT littermate control mice (Figure 2a). Furthermore, no differences were observed in 12- and 24-month-old APPE693Q mice by western blot analysis of hemibrain homogenates with antibodies to the early endosomal marker, rab5a, the rab5 effector protein, rabaptin 5, the late endosomal marker, rab7 and the recycling endosomal marker, rab4a (Figure 2b), suggesting that the endosomal pathway is unaffected.

Higher LAMP-2 and Cat D levels in the brain of APPE693Q mice compared with WT control mice. (a) No difference was observed between APPE693Q mice and littermate control neurons immunostained for the early endosomal marker, rab5, shown here in the frontoparietal cortex (scale bar, 5 μm). (b) Western blot analysis using anti-rab4a, rab5a, rab7 and rabaptin 5 antibodies did not show differences in the levels of these proteins in hemibrain homogenates of APPE693Q as compared with WT mice (n=4). (c) Higher signal for LAMP-2 immunoreactivity was observed in the frontoparietal cortex of 12-month-old APPE693Q mice compared with age-matched WT controls as found in the entorhinal cortex (Figures 1a–c). (d) Morphometric measurements of LAMP-2-labeled lysosomes revealed higher numbers of lysosomes per neuron in APPE693Q as compared with WT mice. The ratio of LAMP-2-positive lysosomes per neuron in APPE693Q mice was normalized to control mice. (e) Western blot of proteins in the entorhinal cortex of 12- and 24-month-old WT and APPE693Q mice, using anti-LAMP-2 antibody. β-tubulin reactivity is shown as a loading control. (f) The ratio of LAMP-2 to β-tubulin reactivity in APPE693Q mice was normalized to control mice. (g) Immunocytochemical staining of the entorhinal cortex of mice at 12 months of age using anti-Cat D antibody (scale bar, 50 μm). (h) Western blot analysis of homogenates of the entorhinal cortex confirmed higher active Cat D level in 12- and 24-month-old APPE693Q mice compared with WT mice. β-tubulin reactivity is shown as a loading control. (i) The ratio of Cat D to β-tubulin reactivity in APPE693Q mice was normalized to control mice. Measurements are presented as the mean±s.e.m. (n=4/5). The difference from WT mice was significant at *P<0.05. APP, amyloid-β-precursor protein; EC, entorhinal cortex; FC, frontoparietal cortex; LAMP-2, lysosomal marker protein 2; WT, wild type.
Immunocytochemistry of lysosomal proteins have shown higher intensity staining and higher number of LAMP-2-positive vesicles per neuron in the entorhinal and frontoparietal cortices in the brains of 12-month-old APPE693Q compared with WT mice (Figures 1a–c and 2c). Quantification of LAMP-2 positive lysosomes in the frontoparietal and entorhinal cortices revealed higher lysosomal counts (Figure 2d) in brain neurons of APPE693Q mice as compared with WT controls mice. Similar to LAMP-2, higher intensity staining and higher number of stained vesicles were observed with the antibody to the lysosomal hydrolase Cat D in the entorhinal cortices of 12-month-old APPE693Q as compared with WT mice (Figure 2g). While western blot analysis of entorhinal cortical homogenates of 12-month-old mice with antibodies to LAMP-2 and Cat D showed a non-significant trend toward greater levels in APPE693Q compared with WT mice, western blot analysis of entorhinal cortical homogenates of 24-months-old mice showed significantly higher levels of LAMP-2, and active Cat D in APPE693Q as compared with WT mice (Figures 2e, f, h and i). These data show that immunocytochemical staining revealed lysosomal changes in specific neuronal populations, apparent already in the brain of 12-month-old transgenic mice, whereas western blot analysis of brain tissue that contains multiple cellular populations, including astrocytes and microglia, did not show lysosomal changes at 12 months of old. Significantly higher levels of lysosomal proteins were observed by western blot analysis of entorhinal cortices of APPE693Q compared with WT mice at 24 months of age. These data reveal an age-dependent increase in lysosomal pathology in entorhinal cortical neurons of APPE693Q mice. Although similar changes in the lysosomal markers LAMP-2 and Cat D were observed in multiple brain regions, the entorhinal cortex was the region most affected by lysosomal pathology in the brain of APPE693Q mice. Furthermore, immunostaining and western blots showed higher LC3 expression levels in the entorhinal cortex of APPE693Q as compared with WT mice at 24 months of age (Figures 3a and c). In addition, higher levels of conversion from LC3-I to LC3-II were observed in APPE693Q mice as compared with WT littermate controls (Figure 3d). Although higher levels of LC3 and conversion from LC3-I to LC3-II were observed in brain homogenate of APPE693Q mice as compared with WT littermate controls, study of the mammalian target of rapamycin (mTOR) signaling pathway to test its involvement in autophagic abnormalities in APPE693Q mice did not show differences in the phosphorylation of proteins such as P70S6K, AKT, and mTOR between APPE693Q mice and littermate controls (Figure 3e), suggesting lysosomal dysfunction, but not autophagic pathology, that leads to defective clearance of neuronal autophagic substrates in APPE693Q mice.

Higher LC3 levels and conversion to LC3-II in the brain of 24-month-old APPE693Q mice compared with WT controls. (a) Brain sections immunostained with anti-LC3 antibody showed higher intensity in the entorhinal cortices of APPE693Q as compared with WT mice (scale bar, 10 μm). (b) Western blot analysis of entorhinal cortical homogenates of APPE693Q as compared with WT mice using an anti-LC3 antibody showed higher intensity of both the LC3-I and LC3-II bands. (c) Quantification of the ratio of total LC3 (LC3-I+LC3-II) to β-tubulin bands in APPE693Q normalized to WT mice, (n=4). (d) Quantification of the ratio of the LC3-II to total LC3 bands in APPE693Q normalized to WT mice, (n=4). (e) Western blot analysis of hemibrain homogenates using anti-LC3, mTOR and AKT antibodies (total and phosphorylated protein) showed higher intensity of both the LC3-I and LC3-II bands in APPE693Q as compared with WT mice whereas no differences were observed in mTOR and AKT expression and phosphorylation levels (n=4). (f) Western blot analysis of brain homogenates showed higher α-synuclein protein levels in hemibrain homogenates of APPE693Q compared with WT mice. (g) Quantification of the ratio of α-synuclein to β-tubulin bands (n=5). All data are presented as the mean±s.e.m. The differences from WT mice were significant at *P<0.05 and **P<0.01. α-Syn, α-synuclein; APP, amyloid-β-precursor protein; mTOR, mammalian target of rapamycin; WT, wild type.
On the basis of recent indications that impaired lysosomal clearance may also be associated with accumulation of neurodegeneration-related proteins, including α-synuclein, in an APP transgenic mouse model,53 we sought to determine the levels of α-synuclein in the brains of APPE693Q mice. Western blot analysis of 24-month-old APPE693Q mouse brains exhibited higher levels of α-synuclein as compared with WT mice (Figures 3f and g), supporting impaired lysosomal function.
Cholinergic neuronal loss in the MSN of APPE693Q mice
Basal forebrain cholinergic neurons are particularly vulnerable to degeneration in AD.54 We undertook to investigate whether lysosomal abnormality in neurons affects basal forebrain cholinergic neuron in the MSN of APPE693Q mice. Comparison of cholinergic neurons in the MSN of APPE693Q and WT mice by immunostaining with antibody to ChAT revealed lower number of immunopositive cells in APPE693Q mice at 12 months of age as compared with littermate controls (Figure 4a). Using quantitative unbiased stereology, we found that APPE693Q mice had significantly fewer ChAT-positive cells in the MSN as compared with WT mice (Figures 4b). We further investigated the lysosomal abnormalities in large neurons of the MSN immunostained with LAMP-2, counter-stained with Nissl (Figure 4c). Quantification of LAMP-2-positive lysosomes in these large septal neurons showed bigger lysosomal size (Figure 4d) in brains of APPE693Q mice as compared with WT controls mice.

Loss of cholinergic neurons, GABAergic interneurons and gliosis in the brain of APPE693Q mice at 12 months of age. (a) Loss of ChAT-immuno-positive neurons in the MSN of APPE693Q mice compared with age-matched WT controls (scale bar, 50 μm). (b) Unbiased stereological quantitation of ChAT-immunopositive cells in the MSN of WT and APPE693Q mice is depicted as cell number±s.e.m. (n=5). (c) Nissl counter-stained neurons in the MSN of APPE693Q mice immunostained for LAMP-2 showed higher signal for LAMP-2 compared with age-matched WT controls (scale bars, 20 μm). (d) Morphometric measurements of LAMP-2 labeled lysosomes revealed higher lysosomal area per neuronal area in the Nissl stained large neurons of the MSN (arrowheads) in APPE693Q normalized to WT mice (n=4). (e) Representative pictures of PV-positive GABAergic cells in the MSN (scale bar, 50 μm), cingulate cortex (scale bar, 100 μm) and hippocampus (scale bar, 200 μm) in WT and APPE693Q mice. (f) Quantification of PV-positive cells in the hippocampus (n=5). (g) Immunocytochemical staining with anti-GFAP antibody of sections of APPE693Q mouse brains revealed astrogliosis in the hippocampal region as compared with WT mice (scale bar, 200 μm). (h) APPE693Q mice also exhibited microgliosis in the hippocampal region when immunostained with anti-Iba1 antibody (Scale bar, 50 μm). Differences from WT were significant at *P<0.05 and **P<0.01. APP, amyloid-β-precursor protein; CCx, cingulate cortex; GFAP, glial fibrillary acidic protein; Hippo, hippocampus; LAMP-2, parvalbumin; MSN, medial septal nucleus; PV, parvalbumin; WT, wild type.
Loss of GABAergic interneurons in the brain of APPE693Q mice
Immunostaining of GABAergic interneurons with an antibody to PV has shown that the GABAergic cell population was decreased in the cortex, hippocampus, and MSN areas in 12 months-old APPE693Q as compared with WT mice (Figure 4e). PV-positive cells in the hippocampi of APPE693Q and WT mice were quantified revealing a lower number of these inhibitory interneurons in the hippocampus of APPE693Q mice as compared with WT controls (Figure 4f).
Activation of astrocytes and microglia in the brain of APPE693Q mice
In 29-month-old mice of the model described by Herzig et al.,16 microglial activation demonstrated by Iba-1 immunostaining was observed in the immediate vicinity of amyloid-laden vessels and adjacent areas, and GFAP-positive-activated astrocytes were observed in neocortical areas affected by cerebral amyloid angiopathy. In the APPE693Q mice studied here we observed higher intensity of GFAP and Iba1 immunostaining, suggesting activated glial and microglial cells in the hippocampus, particularly in the hilus of dentate gyrus and extended to the granular cell layer and corpus callosum (Figures 4g and h) in the absence of amyloid deposition.
Discussion
This study revealed age-dependent intraneuronal accumulation of APP-CTFs in the brains of APPE693Q mice in the absence of cerebral amyloid deposition, in addition to the previously demonstrated intraneuronal accumulation of oAβ.17, 39, 40, 41 Characterization of cellular APP/CTFs accumulation in the absence of parenchymal Aβ deposits and resultant neuropathology are valuable for understanding the pathological determinants of cognitive dysfunction in AD. Our extensive study of the intraneuronal vesicular localization of APP metabolites revealed their accumulation within lysosomes in the brain of 12-month-old APPE693Q mice. Dysfunction of lysosomes containing APP-CTFs and/or oAβ aggregates was demonstrated by the observation of higher staining intensity of LAMP-2 and increased numbers of enlarged LAMP-2-positive lysosomes in neurons dually stained with antibodies to flAPP/APP-CTFs/Aβ of 12-month-old APPE693Q as compared with WT controls. Neurons in the brains of APPE693Q mice also had higher levels of the lysosomal hydrolase Cat D. A previous study showed no accumulation of soluble LC3, p62 or α-synuclein in the brains of APPE693Q mice at 6 months of age,41 suggesting an age-dependent lysosomal dysfunction due to the ineffective clearance of APP metabolites. The lysosomal abnormality became more severe at an older age, as revealed by the intraneuronal accumulation of the autophagosomal/autolysosomal marker LC3 as well as by the increased conversion of LC3-I into LC3-II in entorhinal cortical neurons at 24 months of age. These data suggest early mobilization of the lysosomal system, followed by delayed turnover of intra-lysosomal proteins such as LC3-II that is normally rapidly degraded after fusion of autophagosomes with lysosomes. In addition, we found increased α-synuclein in lysosomes of 24-month-old APPE693Q mice. α-synuclein forms insoluble fibrils under pathological conditions in Parkinson's disease, dementia with Lewy bodies,55, 56 and accumulates in the cortex of 30–50% AD patients.57 These data show that APP-CTFs accumulation is involved in the age-related changes in the brains of aged mice, and could be a cause of AD-relevant lysosomal dysfunction.
Evidence is mounting from transgenic mice and human patients, indicating that Aβ as well as APP-CTFs accumulate intraneuronally and contribute to AD progression.22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38 An association between elevated APP-CTFs levels, abnormal endosomal morphology, and basal forebrain cholinergic neuron degenerative changes have also been observed in mouse models of familial AD due to other APP mutations.47 Earlier studies using isotropic fractionator showed no evidence for total neuronal loss in the hippocampus, neocortex, and cerebellum of APPE693Q mice,58 but reduced dendritic arborization of apical dendrites and a decrease in postsynaptic density length in individual hippocampal neurons of 12 months-old APPE693Q mice was observed.59 However, our data show loss of cholinergic neurons in the MSN of our APPE693Q mice. Dysregulated cholinergic signaling is an early hallmark of AD.60 The severity and magnitude of cholinergic dysfunction appears to depend, at least in part, on age and brain region, in many APP-overexpressing mouse models, including APP23,47, 61, 62 TgCRND8,63 Tg257664 and APP/PS1K1.65 The cholinergic fiber loss was attributed to higher Aβ load.66 However, the observed shrinkage of cholinergic neurons61 and reduced enzymatic activity in the basal forebrain in 6–8-months-old APP23 transgenic mice62 and the cholinergic degenerative pathology in PDAPP mice,67 indicate the presence of an early cholinergic dysfunction prior to Aβ amyloid deposition. Oligomeric forms of Aβ have been implicated in the reduction in ChAT activity60 and loss of cholinergic innervation in the neocortex,68 preceding the loss of cholinergic neurons commonly observed in AD brains. However, the data presented here suggest that the neuronal lysosomal dysfunction in MSN cholinergic neurons and cortical regions of APPE693Q mice is due at least in part to the accumulation of APP-CTFs.
Besides showing significant loss of cholinergic cells in the MSN, 12-month-old APPE693Q mice also showed GABAergic neurotransmission deficits, similar to other APP transgenic mice, including TgCRND8,69 AbPPdE970 and J20 transgenic mice.71 Both cholinergic and GABAergic cell loss in the brain of these transgenic mice, in the absence of parenchymal and vascular β amyloid deposition, implies a role for intracellular accumulation of APP-CTFs/oAβ in neuronal degeneration and subsequent impaired spatial learning and memory, novel object recognition and increased anxiety in APPE693Q transgenic mice.17, 39, 40, 41
In conclusion, our results suggest that an age-dependent intraneuronal accumulation of APP-CTFs and oAβ initiates a cascade of pathogenic events, including lysosomal dysfunction in vulnerable brain regions and leads to inflammatory activation, cholinergic and GABAergic neuronal loss in transgenic mice overexpressing human APP gene harboring the E693Q mutation, leading to memory deficits.
References
Hardy J, Selkoe DJ . The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 297: 353–356.
Gudmundsson G, Hallgrimsson J, Jonasson TA, Bjarnason O . Hereditary cerebral haemorrhage with amyloidosis. Brain 1972; 95: 387–404.
Haan J, Roos RA, Briet PE, Herpers MJ, Luyendijk W, Bots GT . Hereditary cerebral hemorrhage with amyloidosis—Dutch type. Clin Neurol Neurosurg 1989; 91: 285–290.
Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG et al. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990; 248: 1124–1126.
Wattendorff AR, Bots GT, Went LN, Endtz LJ . Familial cerebral amyloid angiopathy presenting as recurrent cerebral haemorrhage. J Neurol Sci 1982; 55: 121–135.
Luyendijk W, Bots GT, Vegter-van der Vlis M, Went LN, Frangione B . Hereditary cerebral haemorrhage caused by cortical amyloid angiopathy. J Neurol Sci 1988; 85: 267–280.
Haan J, Algra PR, Roos RA . Hereditary cerebral hemorrhage with amyloidosis-Dutch type. Clinical and computed tomographic analysis of 24 cases. Arch Neurol 1990; 47: 649–653.
van Duinen SG, Castano EM, Prelli F, Bots GT, Luyendijk W, Frangione B . Hereditary cerebral hemorrhage with amyloidosis in patients of Dutch origin is related to Alzheimer disease. Proc Natl Acad Sci USA 1987; 84: 5991–5994.
Haan J, Roos RA, Algra PR, Lanser JB, Bots GT, Vegter-Van der Vlis M . Hereditary cerebral haemorrhage with amyloidosis—Dutch type. Magnetic resonance imaging findings in 7 cases. Brain 1990; 113: 1251–1267.
Coria F, Castano EM, Frangione B . Brain amyloid in normal aging and cerebral amyloid angiopathy is antigenically related to Alzheimer's disease β-protein. Am J Pathol 1987; 129: 422–428.
Haan J, Lanser JB, Zijderveld I, van der Does IG, Roos RA . Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type. Arch Neurol 1990; 47: 965–967.
Cohn-Hokke PE, Wong TH, Rizzu P, Breedveld G, van der Flier WM, Scheltens P et al. Mutation frequency of PRKAR1B and the major familial dementia genes in a Dutch early onset dementia cohort. J Neurol 2014; 261: 2085–2092.
Wisniewski T, Ghiso J, Frangione B . Peptides homologous to the amyloid protein of Alzheimer's disease containing a glutamine for glutamic acid substitution have accelerated amyloid fibril formation. Biochem Biophys Res Commun 1991; 179: 1247–1254.
Miravalle L, Tokuda T, Chiarle R, Giaccone G, Bugiani O, Tagliavini F et al. Substitutions at codon 22 of Alzheimer's Aβ peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J Biol Chem 2000; 275: 27110–27116.
Fraser PE, Nguyen JT, Inouye H, Surewicz WK, Selkoe DJ, Podlisny MB et al. Fibril formation by primate, rodent, and Dutch-hemorrhagic analogues of Alzheimer amyloid β-protein. Biochemistry 1992; 31: 10716–10723.
Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD et al. Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 2004; 7: 954–960.
Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC et al. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-β oligomers. Ann Neurol 2010; 68: 220–230.
Prelli F, Castano E, Glenner GG, Frangione B . Differences between vascular and plaque core amyloid in Alzheimer's disease. J Neurochem 1988; 51: 648–651.
Walsh DM, Selkoe DJ . Aβ oligomers - a decade of discovery. J Neurochem 2007; 101: 1172–1184.
Haass C, Selkoe DJ . Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat Rev Mol Cell Biol 2007; 8: 101–112.
Sengupta U, Nilson AN, Kayed R . The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016; 6: 42–49.
Bertrand E, Brouillet E, Caille I, Bouillot C, Cole GM, Prochiantz A et al. A short cytoplasmic domain of the amyloid precursor protein induces apoptosis in vitro and in vivo. Mol Cell Neurosci 2001; 18: 503–511.
McPhie DL, Golde T, Eckman CB, Yager D, Brant JB, Neve RL . β-Secretase cleavage of the amyloid precursor protein mediates neuronal apoptosis caused by familial Alzheimer's disease mutations. Brain Res Mol Brain Res 2001; 97: 103–113.
Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A et al. Alzheimer's-related endosome dysfunction in Down syndrome is Aβ-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci USA 2010; 107: 1630–1635.
Lauritzen I, Pardossi-Piquard R, Bauer C, Brigham E, Abraham JD, Ranaldi S et al. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J Neurosci 2012; 32: 16243–16255a.
Tamayev R, Matsuda S, Arancio O, D'Adamio L . β- but not γ-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol Med 2012; 4: 171–179.
Nhan HS, Chiang K, Koo EH . The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes. Acta Neuropathol 2015; 129: 1–19.
Xu W, Fitzgerald S, Nixon RA, Levy E, Wilson DA . Early hyperactivity in lateral entorhinal cortex is associated with elevated levels of AβPP metabolites in the Tg2576 mouse model of Alzheimer's disease. Exp Neurol 2015; 264: 82–91.
Kim S, Sato Y, Mohan PS, Peterhoff C, Pensalfini A, Rigoglioso A et al. Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer's disease. Mol Psychiatry 2016; 21: 707–716.
Cavanagh C, Colby-Milley J, Bouvier D, Farso M, Chabot JG, Quirion R et al. βCTF-correlated burst of hippocampal TNFα occurs at a very early, pre-plaque stage in the TgCRND8 mouse model of Alzheimer's disease. J Alzheimers Dis 2013; 36: 233–238.
Jiang Y, Rigoglioso A, Peterhoff CM, Pawlik M, Sato Y, Bleiwas C et al. Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: role of APP-CTF. Neurobiol Aging 2016; 39: 90–98.
Neve RL, Robakis NK . Alzheimer's disease: a re-examination of the amyloid hypothesis. Trends Neurosci 1998; 21: 15–19.
Tamayev R, D'Adamio L . Inhibition of γ-secretase worsens memory deficits in a genetically congruous mouse model of Danish dementia. Mol Neurodegener 2012; 7: 19.
Bach JH, Chae HS, Rah JC, Lee MW, Park CH, Choi SH et al. C-terminal fragment of amyloid precursor protein induces astrocytosis. J Neurochem 2001; 78: 109–120.
Devi L, Ohno M . Mitochondrial dysfunction and accumulation of the β-secretase-cleaved C-terminal fragment of APP in Alzheimer's disease transgenic mice. Neurobiol Dis 2012; 45: 417–424.
Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X et al. A second cytotoxic proteolytic peptide derived from amyloid β-protein precursor. Nat Med 2000; 6: 397–404.
Sopher BL, Fukuchi K, Smith AC, Leppig KA, Furlong CE, Martin GM . Cytotoxicity mediated by conditional expression of a carboxyl-terminal derivative of the β-amyloid precursor protein. Brain Res Mol Brain Res 1994; 26: 207–217.
Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL . Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science 1989; 245: 417–420.
Kim SH, Steele JW, Lee SW, Clemenson GD, Carter TA, Treuner K et al. Proneurogenic Group II mGluR antagonist improves learning and reduces anxiety in Alzheimer Aβ oligomer mouse. Mol Psychiatry 2014; 19: 1235–1242.
Knight EM, Williams HN, Stevens AC, Kim SH, Kottwitz JC, Morant AD et al. Evidence that small molecule enhancement of β-hexosaminidase activity corrects the behavioral phenotype in Dutch APP(E693Q) mice through reduction of ganglioside-bound Aβ. Mol Psychiatry 2015; 20: 109–117.
Knight EM, Kim SH, Kottwitz JC, Hatami A, Albay R, Suzuki A et al. Effective anti-Alzheimer Aβ therapy involves depletion of specific Aβ oligomer subtypes. Neurol Neuroimmunol Neuroinflamm 2016; 3: e237.
Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA . Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol 2000; 157: 277–286.
Cataldo A, Rebeck GW, Ghetri B, Hulette C, Lippa C, Van Broeckhoven C et al. Endocytic disturbances distinguish among subtypes of Alzheimer's disease and related disorders. Ann Neurol 2001; 50: 661–665.
Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biol Psychiatry 2010; 68: 885–893.
Ling D, Magallanes M, Salvaterra PM . Accumulation of amyloid-like Aβ1-42 in AEL (autophagy-endosomal-lysosomal) vesicles: potential implications for plaque biogenesis. ASN Neuro 2014; 6: e00139.
Perez SE, He B, Nadeem M, Wuu J, Ginsberg SD, Ikonomovic MD et al. Hippocampal endosomal, lysosomal, and autophagic dysregulation in mild cognitive impairment: correlation with Aβ and tau pathology. J Neuropathol Exp Neurol 2015; 74: 345–358.
Choi JH, Kaur G, Mazzella MJ, Morales-Corraliza J, Levy E, Mathews PM . Early endosomal abnormalities and cholinergic neuron degeneration in amyloid-β protein precursor transgenic mice. J Alzheimers Dis 2013; 34: 691–700.
Mathews PM, Jiang Y, Schmidt SD, Grbovic OM, Mercken M, Nixon RA . Calpain activity regulates the cell surface distribution of amyloid precursor protein. Inhibition of calpains enhances endosomal generation of β-cleaved C-terminal APP fragments. J Biol Chem 2002; 277: 36415–36424.
West MJ, Slomianka L, Gundersen HJ . Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec 1991; 231: 482–497.
Smiley JF, Konnova K, Bleiwas C . Cortical thickness, neuron density and size in the inferior parietal lobe in schizophrenia. Schizophr Res 2012; 136: 43–50.
Dorph-Petersen KA, Nyengaard JR, Gundersen HJ . Tissue shrinkage and unbiased stereological estimation of particle number and size. J Microsc 2001; 204: 232–246.
Kaur G, Sharma A, Xu W, Gerum S, Alldred MJ, Subbanna S et al. Glutamatergic transmission aberration: a major cause of behavioral deficits in a murine model of Down's syndrome. J Neurosci 2014; 34: 5099–5106.
Steele JW, Ju S, Lachenmayer ML, Liken J, Stock A, Kim SH et al. Latrepirdine stimulates autophagy and reduces accumulation of α-synuclein in cells and in mouse brain. Mol Psychiatry 2013; 18: 882–888.
Mufson EJ, Ginsberg SD, Ikonomovic MD, DeKosky ST . Human cholinergic basal forebrain: chemoanatomy and neurologic dysfunction. J Chem Neuroanat 2003; 26: 233–242.
Mezey E, Dehejia A, Harta G, Papp MI, Polymeropoulos MH, Brownstein MJ . α synuclein in neurodegenerative disorders: murderer or accomplice? Nat Med 1998; 4: 755–757.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M . α-synuclein in Lewy bodies. Nature 1997; 388: 839–840.
Yue Z, Friedman L, Komatsu M, Tanaka K . The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim Biophys Acta 2009; 1793: 1496–1507.
Brautigam H, Steele JW, Westaway D, Fraser PE St, George-Hyslop PH, Gandy S et al. The isotropic fractionator provides evidence for differential loss of hippocampal neurons in two mouse models of Alzheimer's disease. Mol Neurodegener 2012; 7: 58.
Price KA, Varghese M, Sowa A, Yuk F, Brautigam H, Ehrlich ME et al. Altered synaptic structure in the hippocampus in a mouse model of Alzheimer's disease with soluble amyloid-β oligomers and no plaque pathology. Mol Neurodegener 2014; 9: 41.
Nunes-Tavares N, Santos LE, Stutz B, Brito-Moreira J, Klein WL, Ferreira ST et al. Inhibition of choline acetyltransferase as a mechanism for cholinergic dysfunction induced by amyloid-β peptide oligomers. J Biol Chem 2012; 287: 19377–19385.
Boncristiano S, Calhoun ME, Kelly PH, Pfeifer M, Bondolfi L, Stalder M et al. Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J Neurosci 2002; 22: 3234–3243.
Van Dam D, Marescau B, Engelborghs S, Cremers T, Mulder J, Staufenbiel M et al. Analysis of cholinergic markers, biogenic amines, and amino acids in the CNS of two APP overexpression mouse models. Neurochem Int 2005; 46: 409–422.
Bellucci A, Luccarini I, Scali C, Prosperi C, Giovannini MG, Pepeu G et al. Cholinergic dysfunction, neuronal damage and axonal loss in TgCRND8 mice. Neurobiol Dis 2006; 23: 260–272.
Apelt J, Kumar A, Schliebs R . Impairment of cholinergic neurotransmission in adult and aged transgenic Tg2576 mouse brain expressing the Swedish mutation of human β-amyloid precursor protein. Brain Res 2002; 953: 17–30.
Christensen DZ, Bayer TA, Wirths O . Intracellular Aβ triggers neuron loss in the cholinergic system of the APP/PS1KI mouse model of Alzheimer's disease. Neurobiol Aging 2010; 31: 1153–1163.
Kar S, Quirion R . Amyloid β peptides and central cholinergic neurons: functional interrelationship and relevance to Alzheimer's disease pathology. Prog Brain Res 2004; 145: 261–274.
German DC, Yazdani U, Speciale SG, Pasbakhsh P, Games D, Liang CL . Cholinergic neuropathology in a mouse model of Alzheimer's disease. J Comp Neurol 2003; 462: 371–381.
Nyakas C, Granic I, Halmy LG, Banerjee P, Luiten PG . The basal forebrain cholinergic system in aging and dementia. Rescuing cholinergic neurons from neurotoxic amyloid-beta42 with memantine. Behav Brain Res 2011; 221: 594–603.
Albuquerque MS, Mahar I, Davoli MA, Chabot JG, Mechawar N, Quirion R et al. Regional and sub-regional differences in hippocampal GABAergic neuronalvulnerability in the TgCRND8 mouse model of Alzheimer's disease. Front Aging Neurosci 2015; 7: 30.
Ramos B, Baglietto-Vargas D, del Rio JC, Moreno-Gonzalez I, Santa-Maria C, Jimenez S et al. Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer's disease. Neurobiol Aging 2006; 27: 1658–1672.
Rubio SE, Vega-Flores G, Martinez A, Bosch C, Perez-Mediavilla A, del Rio J et al. Accelerated aging of the GABAergic septohippocampal pathway and decreased hippocampal rhythms in a mouse model of Alzheimer's disease. FASEB J 2012; 26: 4458–4467.
Acknowledgements
National Institutes of Health, Alzheimer’s association.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Kaur, G., Pawlik, M., Gandy, S. et al. Lysosomal dysfunction in the brain of a mouse model with intraneuronal accumulation of carboxyl terminal fragments of the amyloid precursor protein. Mol Psychiatry 22, 981–989 (2017). https://doi.org/10.1038/mp.2016.189
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2016.189
- Springer Nature Limited
This article is cited by
-
APP dyshomeostasis in the pathogenesis of Alzheimer’s disease: implications for current drug targets
Alzheimer's Research & Therapy (2024)
-
Cellular depletion of major cathepsin proteases reveals their concerted activities for lysosomal proteolysis
Cellular and Molecular Life Sciences (2024)
-
Autophagy–lysosomal-associated neuronal death in neurodegenerative disease
Acta Neuropathologica (2024)
-
Moschus ameliorates glutamate-induced cellular damage by regulating autophagy and apoptosis pathway
Scientific Reports (2023)
-
Accumulation of APP-CTF induces mitophagy dysfunction in the iNSCs model of Alzheimer’s disease
Cell Death Discovery (2022)