
Abstract
Dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 1 A (DYRK1A) maps to the Down syndrome critical region; copy number increase of this gene is thought to have a major role in the neurocognitive deficits associated with Trisomy 21. Truncation of DYRK1A in patients with developmental delay (DD) and autism spectrum disorder (ASD) suggests a different pathology associated with loss-of-function mutations. To understand the phenotypic spectrum associated with DYRK1A mutations, we resequenced the gene in 7162 ASD/DD patients (2446 previously reported) and 2169 unaffected siblings and performed a detailed phenotypic assessment on nine patients. Comparison of our data and published cases with 8696 controls identified a significant enrichment of DYRK1A truncating mutations (P=0.00851) and an excess of de novo mutations (P=2.53 × 10−10) among ASD/intellectual disability (ID) patients. Phenotypic comparison of all novel (n=5) and recontacted (n=3) cases with previous case reports, including larger CNV and translocation events (n=7), identified a syndromal disorder among the 15 patients. It was characterized by ID, ASD, microcephaly, intrauterine growth retardation, febrile seizures in infancy, impaired speech, stereotypic behavior, hypertonia and a specific facial gestalt. We conclude that mutations in DYRK1A define a syndromic form of ASD and ID with neurodevelopmental defects consistent with murine and Drosophila knockout models.
Similar content being viewed by others
Introduction
Autism spectrum disorder (ASD) is a genetically and clinically heterogeneous neurodevelopmental disorder representing various subtypes of social communication dysfunction and unusually restricted interests or repetitive behavior.1 Rarely, monogenic ASD subtypes can be clinically recognized, such as in individuals with macrocephaly and a PTEN or CHD8 mutation, developmental regression in Rett syndrome, and typical dysmorphisms in Fragile X syndrome and tuberous sclerosis.2, 3 Recently, a potentially novel form of syndromic ASD caused by de novo mutations in ADNP (MIM 611386) was reported.4 Exome-sequencing studies of ASD have identified de novo mutations in candidate genes with a limited number of recurrent mutations owing to the extensive locus heterogeneity underlying this disease.5, 6 We recently applied a targeted resequencing approach of candidate genes to a large cohort of affected individuals to identify potentially monogenic subforms of ASD for further investigation.7, 8 Recurrent disruptive mutations in six genes were shown to contribute to 1% of sporadic ASD, including dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 1 A (DYRK1A; MIM 600855) in three individuals (3/2446) with ASD and microcephaly, suggesting that it might be one of the most common genes associated with de novo truncating mutations after CHD8.7
DYRK1A, a member of the dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase (DYRK) family, is a highly conserved gene located in the Down syndrome critical region on chromosome 21. When present in three copies, DYRK1A is one of the two genes thought to be primarily responsible for neurocognitive deficits associated with Down syndrome.9, 10 Previously, disruption due to translocation or deletion of DYRK1A was reported in four individuals with intellectual disability (ID), primary microcephaly and overlapping facial dysmorphisms (Supplementary Figure 1), pointing towards a possible emerging syndrome.11, 12, 13 Four individuals with DYRK1A variants have recently been concisely reported. The lack of microcephaly in one of these questioned the variability of the associated phenotype.13, 14, 15 To determine the core phenotype of the DYRK1A disruption, we targeted DYRK1A (NM_001396.3) for resequencing in a mixed cohort of individuals referred for ID, epilepsy and/or ASD.
Materials and methods
Patient and control samples
The resequencing cohort consisted of 3387 patients with ID/developmental delay (DD) and 1329 patients with DD or a clinical or psychiatric definition of autism. For the purpose of this study, ID was defined as individuals with an IQ<70. In addition, we resequenced 2193 unaffected siblings from the Simons Simplex Collection (SSC) as controls. The independent exome-sequencing cohort consisted of 20 sporadic patients displaying syndromic forms of microcephaly. For 17 out of 20 samples (including the one reported in this paper) parental DNA was available for library preparation, capturing and whole-exome sequencing, allowing trio-based variant filtering. This study was approved by the institutional review boards at the collaborating institutions (Commissie Mensgebonden Onderzoek Regio Arnhem-Nijmegen NL36191.091.11; University of Washington HSD #42744; for the exome sequencing cohort Commissie Medische Ethiek (MEC) UZ KU Leuven/ Onderzoek S-52853). Written informed consent was obtained for all individuals. We also included three patients identified in a resequencing screen of 2446 probands from the SSC initially sequenced in O’Roak et al.7 An additional 6503 controls were sourced from the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project.
Clinical assessment
We performed a detailed clinical assessment of all eight patients with sporadic DYRK1A mutations, including the five new patients described here as well as three individuals with ASD for whom there was previously only limited clinical information available.7 Additionally we assessed a ninth patient whose variant was determined to not likely lead to DYRK1A loss of function. Patients underwent a thorough assessment, including physical exam, dysmorphological assessment, a review of medical history, and comprehensive neurocognitive and diagnostic battery. The battery included clinician observation and parent reporting across the domains of cognition, memory, language, motor, executive functioning, social, repetitive and atypical behaviors, and adaptive ability. In addition, psychiatric presentation was evaluated and gold standard ASD assessments (Autism Diagnostic Observation Schedule, Autism Diagnostic Interview—Revised, clinical judgment) were completed for patients where possible.
Molecular inversion probe (MIP) resequencing
We designed 38 overlapping MIPs designed to cover the protein-coding sequence of the RefSeq DYRK1A isoforms (NM_101396.3, NM_130436.2, NM_101395.2 and NM_130438.2) and performed sequencing and analysis as described in O’Roak et al.7 Briefly, samples were barcoded and 192 samples were sequenced using a paired-end 101 -bp protocol per HiSeq 2000 (Illumina, San Diego, CA, USA) lane. Sequences were then trimmed to remove the MIP sequencing primer sites and to remove any overlap between the paired reads. Sequences were aligned using Burrows-Wheeler Aligner v0.5.6 and variants were identified using SAMtools v0.1.7. Annotation of predicted functions was performed using SeattleSeq, while Alamut (Interactive Biosoftware, Rouen, France) was used to predict splice effects. All the variants were validated using standard polymerase chain reaction (PCR) and Sanger sequencing. The majority of MIPs demonstrated high coverage (>20X) in >95% of QC-passing samples, covering 93.3% of the largest isoforms’ coding sequence. Four MIP designs failed to generate coverage (<8% of samples covered), representing a loss of 6.7% of the largest isoform, with 99% of the missing sequence at a low-complexity segment of exon 13: NM_101396.3:c.950_951 in exon 8 and NM_101396.3:c.1768_1921 in exon 13. The additional low-coverage region is specific to one short isoform in exon 12b NM_101395.2:c.1751toc.1755.
Whole-exome sequencing
Sequencing libraries were prepared with the TruSeq DNA Sample Prep Kit (Illumina, San Diego, CA, USA) and enriched with the SeqCap EZ Human Exome Library v3.0 kit (Roche NimbleGen, Madison, WI, USA). The samples were then sequenced on an HiSeq 2000 or 2500 machine (Illumina). Paired-end sequence reads were aligned to the human genome reference sequence (hg19) with the Burrows-Wheeler Aligner (v0.6.2). SAMtools (v0.1.18) was used for SAM to BAM files conversion, sorting and indexing alignments. Quality metrics were calculated using Picard tools (v1.78) and PCR-generated duplicates marked for downstream analysis. The Genome Analysis Toolkit (GATK v2.4.9) software package was used to perform local realignment, base call recalibration and single-nucleotide polymorphism (SNP) calling. For each called SNP, the GATK Unified Genotyper tool was used to estimate the most likely genotypes. Variant annotation was performed with ANNOVAR (v11-02-2013), including data sets from dbSNP137, the NHLBI 6500 Exome and 1000 genomes projects for variant frequencies, amino-acid-change functional predictions from SIFT, Polyphen2, LRT, MutationTaster and PhyloP, GERP++ conservation scores. Trio-based filtering of the annotated variants was manually conducted according to a de novo inheritance pattern for genotype prediction filtering; ‘knowngene’-based annotations were used for further filtering: only exonic, splicing, nonsilent and rare variants (MAF<1% or absent in all the aforementioned databases) were included. Candidate de novo variants were confirmed by standard Sanger sequencing using the BigDye Terminator v3.1 chemistry (Life Technologies). Sequencing traces were then visualized with Alamut (Interactive Biosoftware).
Results
To determine the frequency and core phenotype of the DYRK1A disruption, we targeted DYRK1A using MIP sequencing, which was applied to 4716 new cases (3387 cases of ID/ DD and 1329 cases of ASD).7 In addition, we incorporated 2446 cases of ASD previously assayed for DYRK1A mutations.7 The inheritance status of potentially disruptive mutations was assessed using standard Sanger sequencing approaches on DNA extracted from the peripheral blood of probands and parents where available.
In total, we observed eight putative truncating variants (three from a previous screen7 and five from the 4716 new cases) in a screen of 7162 probands and one additional patient based on whole-exome sequencing in a separate cohort of 20 individuals presenting with microcephaly. No truncating variants in DYRK1A were observed in the NHLBI Exome Sequencing Project in 6503 individuals. We also resequenced the gene in 2193 unaffected siblings from the SSC and found no occurrences of truncating mutations, indicating that recurrent truncating mutations were significant for ID/ASD (P=0.0017, Fisher’s exact test). In all but one case where parental DNA was available (8/9), the DYRK1A mutations were shown to be de novo. Applying a de novo rate of 1.2 nonsynonymous coding events per individual and a probabilistic model derived from human–chimpanzee fixed differences and sequence context,7 we calculated the probability of detecting seven de novo truncating events in DYRK1A within our cohort as P=2.53 × 10−10 (binomial test).
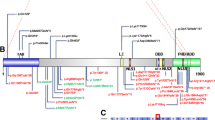
The de novo heterozygous disruptive mutations included two nonsense (c.798_799delinsTT and c.367C>T), two frameshift (c.143_144del and c.1491del) and four splice-site (c.1098+1G>A, c.1240-2A>G, c.516+2T>C and c.665-9_665-5delTTCTC) variants, which are predicted to disrupt the local splice site (Figure 1, Supplementary Table 1). The nonsense, frameshift and one splice-site variants all predict premature stop codons leading to a potential loss of function. The c.143_144del frameshift mutation leads to p.Ile48Lysfs*2 (the earliest truncation event identified in this study) and occurs before all annotated functional elements in the DYRK1A protein. The c.1491del leads to p.Ala498Profs*94, which occurs after the known functional elements. The c.1240-2A>G splice-site mutation likely results in the loss of exon 11 leading to p.Glu414Valfs*76 in the protein-kinase domain disrupting the helix–turn–helix functional element. The c.516+2T>C splice-site mutation is predicted to lead to an inframe deletion of exon 6, containing the nuclear localization sequence, DYRK homology box, a confirmed phosphorylation site and part of the kinase domain. The c.1098+1G>A splice-site mutation is predicted to result in an inframe deletion of exon 9, which is internal to the protein-kinase domain, eliminating several annotated helices and disrupting the critical activation segment that is likely to affect the function of the protein.16

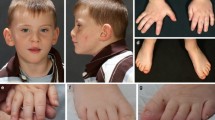
(a) Characteristic facial features can be noted in SSC12099, Troina1818, UMCN1, GF2852, UMCN2 and Leuven 306636. During infancy and childhood, the face is characterized by deep-set eyes, mild upslanting palpebral fissures, a short nose with a broad tip and a retrognathic but prominent chin. In adulthood, the nasal bridge becomes high and the alae nasi short, giving the nose a more prominent appearance. If the facial gestalt is not clearly visible in childhood, it may still develop in adulthood (Supplementary Figure 1B). (b) RefSeq DYRK1A splice isoforms examined in this study. Resequencing covered all coding exons (3 to 13). An asterisk indicates alternatively spliced exon variants. (c) Diagram of the largest protein isoform (RefSeq: NM_101396.3, UniProt: Q13627). Truncating variants are listed in purple. Splice variants are on the bottom, referenced by their cDNA effect for inframe exon loss (blue, or black if not likely pathogenic) or likely protein effect. Previously published variants from Redin et al.14 and Courcet et al.13 are indicated in italics. DYRK1A, dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 1A.
The sole inherited c.208-1G>A splice-site variant was detected in an individual who had been diagnosed with Asperger’s disorder during childhood. Alternative splicing events yield at least four isoforms of DYRK1A. Three utilize a longer version of exon 5 (exon 5a) and one uses a shorter variant (exon 5b) (Supplementary Figure 2). Both the long and short isoforms are expressed in various tissues, including the brain (Supplementary Figure 3). In contrast to the de novo splice variants, this DYRK1A variant does not affect all four isoforms. It is predicted to lead to loss of the inframe exon 5a, which occurs before any annotated functional or structural elements, and was also predicted to create a novel splice acceptor site one base into exon 5a, potentially generating a premature stop (p.Val70*) in the three RefSeq isoforms that utilize exon 5a but not in the isoform that utilizes exon 5b (Supplementary Table 1). qPCR and cloning of the splice junction from cDNA showed that the proband expresses intact copies of both isoforms (Supplementary Figure 4). It is, therefore, unlikely that this variant results in a loss of DYRK1A function. The clinical details of these and previously reported individuals, including translocation and deletion patients (n=15, age range 2–69 years), are presented in Table 1, Figure 1, Supplementary Note case descriptions, Supplementary Video, and Supplementary Figures 5 and 6.
Discussion
To determine the core phenotype of the DYRK1A disruption, we targeted DYRK1A for resequencing in a mixed cohort of individuals referred for ID, epilepsy and/or ASD. In total, we observed eight de novo truncating variants in a screen of 7162 probands and one additional patient based on whole-exome sequencing in a separate cohort of 20 individuals presenting with microcephaly. In all but one case where parents were available, the DYRK1A mutations were shown to be de novo. We estimated the probability of detecting seven de novo truncating events in DYRK1A within our cohort as P=2.53 × 10−10. Moreover, all other previously reported cases, which included parental testing, including two balanced translocations, two single-gene deletions and three mutations, were also shown to be de novo (Supplementary Figure 1).11, 12, 13, 14
Unlike the de novo splice variants, the inherited DYRK1A variant affects only three of the four isoforms. We showed that the proband expresses intact copies of both major isoforms (Supplementary Figure 4) despite the fact that five different algorithms predicted 100% loss of the splice site (Supplementary Table 1). It is interesting that the variant sequence observed in this patient and its mother is consistent with nine noncanonical human splice sites with a GT–AA sequence.17 This is consistent with the patient’s rather atypical phenotype when compared with the other patients. This individual did not meet the criteria for ASD at 18 years of age and showed a normal intelligence without microcephaly. Moreover, his carrier mother is a normal healthy individual. Although it is possible that this variant may still contribute to splicing during development and that the mother may be buffered for its effect, it is more likely that this variant has no consequence despite its prediction. Similar to this case, we excluded another recently published variant from this review due to uncertain pathogenicity.15 Not only was parental testing not described for this case, but the variant (c.1699C>T / p.Gln557*) is located late in the protein past all annotated functional domains and is unlikely to induce nonsense-mediated decay.18 These examples serve as a cautionary note in the interpretation of putative loss-of-function mutations and diagnostic interpretation.
Microcephaly was noted in all 15 individuals after birth or in the first months thereafter. One individual showed microcephaly in infancy (−4.5 s.d. at 4 years of age), which remarkably resolved to −1.6 s.d. at 17 years of age. Information on the presence or absence of ASD was not provided in six out of seven of the previously reported cases and it may be questioned whether ASD was formally assessed, especially considering the young age of assessment for three of these individuals (⩽5 years). In the remainder of the cohort, ASD was reported in the majority of cases (88%). Stereotypic behavior was present in 91% of individuals and anxious behavior in 56%. All individuals experienced apparent speech problems; notably, expressive language was more severely affected compared with receptive language as the majority did not speak or only used one- to two-word sentences (Supplementary Video).
In almost all individuals a specific facial gestalt could be recognized, especially at an older age. During infancy and childhood, the face is characterized by deep-set eyes, mild upslanting palpebral fissures, a short nose with a broad tip, and retrognathia with a broad chin. In adulthood, the nasal bridge becomes high and the alae nasi short, giving the nose a more prominent appearance. In case the facial gestalt is not clearly visible in childhood, it may still develop by adulthood (Supplementary Figure 6). Febrile seizures were present in the majority of individuals (77%), whereas at a later age, epilepsy was diagnosed in only five individuals (33%). Nine individuals showed clear motor disturbances characterized by an abnormal gait and eight individuals showed hypertonic musculature. In general, the level of ID was variable. Moderate to severe ID was noted in 12 individuals (80%). Mild ID was noted in three individuals (20%)—one with a translocation and two with a frameshift mutation. This may reflect a variable genotype–phenotype correlation based upon the type of DYRK1A truncation as, for instance, the p.Ala498Profs*94 mutation does not disrupt the main functional domain and is predicted to be less impactful than the alternative shorter isoforms. The previously reported translocation does not affect these DYRK1A isoforms either.
From our detailed clinical evaluation, we conclude that truncating mutations in DYRK1A lead to a well-recognizable syndromic form of ASD and ID, characterized by a specific facial gestalt, microcephaly, lack of speech, seizures, neonatal feeding problems, hypertonia and gait disturbances. Considering the core phenotype delineated within this study, DYRK1A testing should be considered when autism and/or ID is accompanied by microcephaly, especially when two or more of the additional characteristic key features are present, such as the typical facial gestalt, seizures, hypertonia, feeding problems or an abnormal gait. Cardiac and ophthalmologic evaluation should be performed in all the affected individuals as anomalies have been reported in 18% and 27% of cases, respectively (Table 1).
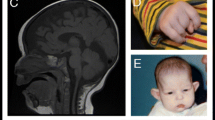
The clinical presentation of individuals with DYRK1A mutations demonstrates some neurodevelopmental features in common with Drosophila and mouse models where mutants show defects in cell proliferation, neurogenesis, neuronal differentiation, cell death and synaptic plasticity. Mutants of the Drosophila ortholog, Minibrain (Mnb) gene show greatly reduced adult brains (40–50%) with no gross changes in neuronal architecture reported.19, 20 Similarly, newborn Dyrk1a+/− mice show a decreased viability, pre- and postnatal growth retardation, DD and behavioral abnormalities, including increased anxiety and altered reactivity to stress.21 Loss of function of Dyrk1a in mice results in a 30% reduction in brain size, with a disproportionate reduction in the size of the mid- and hindbrain regions.19, 20 Brain architecture remains largely unchanged, which is in concordance with brain-imaging results in humans where, except for corpus callosum hypoplasia and cortical atrophy, no major structural anomalies were noted (Table 1). In humans, expression of DYRK1A is high in multiple tissues, including different areas in the developing fetal and adult brain (Supplementary Figures 3 and 7). In mice, Dyrk1a is highly expressed in the developing and adult nervous system, most abundantly in the cerebellar cortex and functionally related structures, the spinal cord and most of the motor nuclei of the midbrain and brain stem.22 These data are in line with the altered locomotor activity, including gait disturbances, observed in both adult Dyrk1a +/− mice and affected individuals. In addition, the presence of DYRK1A in the presynaptic terminal of the neuromuscular junction suggests a physiological role of DYRK1A in the function of the nervous system.23 DYRK1A has also been linked to retinal development in Drosophila and mouse models, which may relate to the observed retinal detachment in one of the individuals.24, 25, 26
Half of the individuals displayed sleep disturbances characterized by difficulties in falling asleep and nighttime awakenings, although this observation does not reach statistical significance when compared with its prevalence among ASD patients. Of note, DYRK1A has been proposed as a clock-related protein kinase, influencing the regulation of the protein level of CRY2 and shortening the period length of the circadian clock in Dyrk1a knockdown mice.27 ASD, stereotypic behavior and anxious behavior were frequently noted. Similarly, Dyrk1a+/− mice demonstrate an enhanced freezing response, suggesting either increased anxiety or changes in emotional behavior.19
Although the molecular function of DYRK1A is not completely understood, it is noteworthy that Mnb, the Drosophila orthologue of DYRK1A, has been shown to interact with Snr1, the INI1 orthologue, which is a member of the switch/sucrose nonfermentable complex involved in the morphogenesis of dendritic arbors in Drosophila sensory neurons.28, 29 Similarly, in mice, DYRK1A binds a switch/sucrose nonfermentable complex, suggesting a potential role in chromatin remodeling.30 In the light of the importance of germline mutations in chromatin remodeling complexes for neurodevelopmental disease,3, 31, 32, 33, 34, 35 detailed investigation into these complexes and networks may reveal additional pathogenic mutations and potential novel syndromes associated with ID and autism.3, 36 Our results, however, caution that diagnostic interpretation of mutations should not rely solely on the predicted impact based on gene annotation even for high-penetrant genes such as DYRK1A. Rather, it is critical to consider familial transmission, isoform differences and functional assessment of putative-truncating mutations in conjunction with the clinical assessment.
References
Lai MC, Lombardo MV, Baron-Cohen S . Autism. Lancet 2014; 383: 896–910.
Rosti RO, Sadek AA, Vaux KK, Gleeson JG . The genetic landscape of autism spectrum disorders. Dev Med Child Neurol 2014; 56: 12–18.
Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014; 158: 263–276.
Helsmoortel C, Vulto-van Silfhout AT, Coe BP, Vandeweyer G, Rooms L, van den Ende J et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat Genet 2014; 46: 380–384.
O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011; 43: 585–589.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012; 485: 237–241.
O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012; 338: 1619–1622.
Brett M, McPherson J, Zang ZJ, Lai A, Tan ES, Ng I et al. Massively parallel sequencing of patients with intellectual disability, congenital anomalies and/or autism spectrum disorders with a targeted gene panel. PLoS One 2014; 9: e93409.
Park J, Chung KC . New perspectives of Dyrk1A role in neurogenesis and neuropathologic features of Down syndrome. Exp Neurobiol 2013; 22: 244–248.
Guimera J, Casas C, Pucharcos C, Solans A, Domenech A, Planas AM et al. A human homologue of Drosophila minibrain (MNB) is expressed in the neuronal regions affected in Down syndrome and maps to the critical region. Hum Mol Genet 1996; 5: 1305–1310.
Moller RS, Kubart S, Hoeltzenbein M, Heye B, Vogel I, Hansen CP et al. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am J Hum Genet 2008; 82: 1165–1170.
van Bon BW, Hoischen A, Hehir-Kwa J, de Brouwer AP, Ruivenkamp C, Gijsbers AC et al. Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin Genet 2011; 79: 296–299.
Courcet JB, Faivre L, Malzac P, Masurel-Paulet A, Lopez E, Callier P et al. The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J Med Genet 2012; 49: 731–736.
Redin C, Gerard B, Lauer J, Herenger Y, Muller J, Quartier A et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet 2014; 51: 724–736.
Okamoto N, Miya F, Tsunoda T, Kato M, Saitoh S, Yamasaki M et al. Targeted next-generation sequencing in the diagnosis of neurodevelopmental disorders. Clin Genet, e-pub ahead of print 25 August 2014.doi:10.1111/cge.12492.
Soundararajan M, Roos AK, Savitsky P, Filippakopoulos P, Kettenbach AN, Olsen JV et al. Structures of Down syndrome kinases, DYRKs, reveal mechanisms of kinase activation and substrate recognition. Structure 2013; 21: 986–996.
Parada GE, Munita R, Cerda CA, Gysling K . A comprehensive survey of non-canonical splice sites in the human transcriptome. Nucleic Acids Res 2014; 42: 10564–10578.
Kervestin S, Jacobson A . NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol 2012; 13: 700–712.
Fotaki V, Dierssen M, Alcantara S, Martinez S, Marti E, Casas C et al. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol Cell Biol 2002; 22: 6636–6647.
Dierssen M, de Lagran MM . DYRK1A (dual-specificity tyrosine-phosphorylated and -regulated kinase 1A): a gene with dosage effect during development and neurogenesis. ScientificWorldJournal 2006; 6: 1911–1922.
Arque G, de Lagran MM, Arbones ML, Dierssen M . Age-associated motor and visuo-spatial learning phenotype in Dyrk1A heterozygous mutant mice. Neurobiol Dis 2009; 36: 312–319.
Marti E, Altafaj X, Dierssen M, de la LS, Fotaki V, Alvarez M et al. Dyrk1A expression pattern supports specific roles of this kinase in the adult central nervous system. Brain Res 2003; 964: 250–263.
Arque G, Casanovas A, Dierssen M . Dyrk1A is dynamically expressed on subsets of motor neurons and in the neuromuscular junction: possible role in Down syndrome. PLoS One 2013; 8: e54285.
Laguna A, Aranda S, Barallobre MJ, Barhoum R, Fernandez E, Fotaki V et al. The protein kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev Cell 2008; 15: 841–853.
Luebbering N, Charlton-Perkins M, Kumar JP, Lochead PA, Rollmann SM, Cook T et al. Drosophila Dyrk2 plays a role in the development of the visual system. PLoS One 2013; 8: e76775.
Laguna A, Barallobre MJ, Marchena MA, Mateus C, Ramirez E, Martinez-Cue C et al. Triplication of DYRK1A causes retinal structural and functional alterations in Down syndrome. Hum Mol Genet 2013; 22: 2775–2784.
Kurabayashi N, Hirota T, Sakai M, Sanada K, Fukada Y . DYRK1A and glycogen synthase kinase 3beta, a dual-kinase mechanism directing proteasomal degradation of CRY2 for circadian timekeeping. Mol Cell Biol 2010; 30: 1757–1768.
Kinstrie R, Lochhead PA, Sibbet G, Morrice N, Cleghon V . dDYRK2 and Minibrain interact with the chromatin remodelling factors SNR1 and TRX. Biochem J 2006; 398: 45–54.
Parrish JZ, Kim MD, Jan LY, Jan YN . Genome-wide analyses identify transcription factors required for proper morphogenesis of Drosophila sensory neuron dendrites. Genes Dev 2006; 20: 820–835.
Lepagnol-Bestel AM, Zvara A, Maussion G, Quignon F, Ngimbous B, Ramoz N et al. DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum Mol Genet 2009; 18: 1405–1414.
Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet 2012; 90: 565–572.
Kosho T, Okamoto N, Ohashi H, Tsurusaki Y, Imai Y, Hibi-Ko Y et al. Clinical correlations of mutations affecting six components of the SWI/SNF complex: detailed description of 21 patients and a review of the literature. Am J Med Genet A 2013; 161A: 1221–1237.
Santen GW, Aten E, Vulto-van Silfhout AT, Pottinger C, van Bon BW, van Minderhout IJ et al. Coffin-Siris syndrome and the BAF complex: genotype-phenotype study in 63 patients. Hum Mutat 2013; 34: 1519–1528.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014; 515: 209–215.
Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014; 515: 216–221.
Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013; 45: 825–830.
Acknowledgements
We thank the patients and their parents for participation. We are grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A Beaudet, R Bernier, J Constantino, E Cook, E Fombonne, D Geschwind, R Goin-Kochel, E Hanson, D Grice, A Klin, D Ledbetter, C Lord, C Martin, D Martin, R Maxim, J Miles, O Ousley, K Pelphrey, B Peterson, J Piggot, C Saulnier, M State, W Stone, J Sutcliffe, C Walsh, Z Warren, E Wijsman). We appreciate obtaining access to phenotypic data on the Simons Foundation Autism Research Initiative (SFARI) Base. Approved researchers can obtain the SSC population dataset described in this study (https://ordering.base.sfari.org/~browse_collection/archive[ssc_v13]/ui:view) by applying at https://base.sfari.org. This study was financially supported by (1) the Ter Meulen Fonds (stipendium to BvB), (2) the Dutch Organisation for Health Research and Development: ZON-MW grants 917-86-319 (BBAdV) and 912-12-109 (BBAdV), and (3) the Simons Foundation Autism Research Initiative (SFARI 303241) and National Institutes of Health (NIH) grant R01MH101221 to EEE. EEE is an Investigator of the Howard Hughes Medical Institute. FC is a PhD aspirant of the Research Foundation Flanders (FWO).
Web resources
The URLs for data presented herein are as follows (accessed September 2014): Database of Genomic Variants, http://projects.tcag.ca/variation/; Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle WA: http://evs.gs.washington.edu/EVS/); The Genotype-Tissue Expression project portal http://www.gtexportal.org/home/; Human protein reference database: http://www.hprd.org; Online Mendelian Inheritance in Man (OMIM), http://www.omim.org; UCSC genome browser: http://genome.ucsc.edu/; Universal Protein Resource: http://www.uniprot.org.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
EEE is on the scientific advisory board (SAB) of DNAnexus. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
PowerPoint slides
Rights and permissions
About this article

Cite this article
van Bon, B., Coe, B., Bernier, R. et al. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol Psychiatry 21, 126–132 (2016). https://doi.org/10.1038/mp.2015.5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2015.5
- Springer Nature Limited
This article is cited by
-
Single-nucleus gene and gene set expression-based similarity network fusion identifies autism molecular subtypes
BMC Bioinformatics (2023)
-
GenIDA: an international participatory database to gain knowledge on health issues related to genetic forms of neurodevelopmental disorders
Journal of Neural Transmission (2023)
-
Characterizing Sensory Phenotypes of Subgroups with a Known Genetic Etiology Pertaining to Diagnoses of Autism Spectrum Disorder and Intellectual Disability
Journal of Autism and Developmental Disorders (2023)
-
Impaired macroglial development and axonal conductivity contributes to the neuropathology of DYRK1A-related intellectual disability syndrome
Scientific Reports (2022)
-
Social motivation a relative strength in DYRK1A syndrome on a background of significant speech and language impairments
European Journal of Human Genetics (2022)