
Abstract
Despite therapeutic advances, multiple myeloma remains incurable, with limited options for patients with refractory disease. We conducted a large, multi-cohort clinical trial testing various doses and treatment schedules of pomalidomide and dexamethasone (Pom/dex) in patients with refractory multiple myeloma. Overall, 345 patients were enrolled to six cohorts based on number and type of prior lines of therapy, pomalidomide dose and schedule. Median prior lines of therapy were three with near universal prior exposure to proteasome inhibitors and/or immunomodulatory drugs. A confirmed response rate of 35% was noted for all cohorts (range 23–65%) with higher responses in cohorts with fewer prior lines of therapy. Median time to confirmed response was ⩽2 months and the longest progression-free survival and overall survival seen in any cohort were 13.1 and 47.9 months, respectively. Observed adverse reactions were as expected, with myelosuppression and fatigue being the most common hematologic and non-hematologic adverse events (AEs), respectively. Longer durations of treatment and response, higher response rates and fewer AEs were noted with the 2 mg pomalidomide dose. This is the longest follow-up data for Pom/dex in refractory multiple myeloma and will help shape the real-world utilization of this regimen.
Similar content being viewed by others
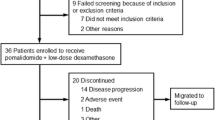

Introduction
Treatment of multiple myeloma has evolved significantly over the years. Immunomodulatory drugs (IMiDs) have emerged as a cornerstone therapeutic strategy in myeloma, with utilization in several different regimens and lines of therapy over the course of a patient’s treatment. Recent data from several randomized clinical trials have shown that a combination of three drugs (triplet) is superior to two drugs (doublet) in significantly improving progression-free survival (PFS) and in some cases overall survival (OS).1, 2, 3, 4, 5 With the improvement in efficacy and emergence of newer classes of drugs, many of these triplet regimens are being used relatively early on in disease management. At the same time, there is evidence to suggest that longer duration of therapy utilizing a maintenance treatment strategy is more beneficial in improving patient outcomes.6 As such, disease that is refractory to novel agents, especially IMiDs and proteasome inhibitors, which are the backbone of all established triplet regimens, is being encountered more frequently and earlier in the life of a myeloma patient. Multiple myeloma that is relapsed and/or refractory to proteasome inhibitors and IMiDs carries a poor prognosis even in the absence of conventional adverse risk prognostic markers and remains a challenge.7, 8, 9
The use of pomalidomide, an IMiD with higher in vitro potency than both thalidomide and lenalidomide, has been reported as a single agent10, 11 as well as in combination with dexamethasone11, 12, 13, 14, 15, 16, 17 in patients with relapsed and/or refractory myeloma. We report on a multi-cohort phase II clinical trial of different doses and schedules of pomalidomide in combination with low-dose dexamethasone (Pom/dex) in patients with refractory multiple myeloma and provide the longest follow-up provided in a prospective clinical trial utilizing this regimen.
Materials and methods
Eligibility
Patients were eligible to enter the study if they had relapsed or were refractory to therapy. Eligibility criteria based on prior treatment were defined depending on the treatment cohort the patients were accrued to, including: refractory myeloma with <3 prior regimens of treatment; myeloma refractory to prior lenalidomide; myeloma refractory to prior lenalidomide and bortezomib; and refractory myeloma without any limit on prior regimens of treatment. Induction therapy followed by autologous stem cell transplant and consolidation was considered one regimen. Patients were required to have measurable disease defined as by International Myeloma Working Group.18 Patients also needed a platelet count >75 × 109/l, absolute neutrophil count >1.0 × 109/l and creatinine <2.5 mg/dl. All previous cancer therapy must have been discontinued ⩾2 weeks before study registration. Patients with uncontrolled infection, another active malignancy, deep vein thrombosis that had not been therapeutically anticoagulated, Eastern Cooperative Oncology Group performance score of 3 or 4, grade 3 or 4 peripheral neuropathy, pregnant or nursing women, women of child-bearing potential who were unwilling to use a dual method of contraception and men who were unwilling to use a condom were excluded. The study was approved by the Mayo Clinic Institutional Review Board in accordance with federal regulations and the Declaration of Helsinki and was registered at http://ClinicalTrials.gov, number NCT00558896.
Treatment schedule
The trial included six treatment cohorts with pomalidomide administered orally at a dose of 2 mg daily (three treatment cohorts) or 4 mg daily (three treatment cohorts). Pomalidomide was given on days 1–28 of a 28-day cycle in five of the treatment cohorts and on days 1–21 of a 28-day cycle in one cohort. Dexamethasone was given orally at a dose of 40 mg daily on days 1, 8, 15 and 22 of each cycle. The different cohorts based on prior treatment regimens, starting dose and schedule of pomalidomide are shown in Table 1. Patients also received aspirin 325 mg once daily for thromboprophylaxis, but were allowed to substitute full-dose anticoagulation with either low-molecular-weight heparin or warfarin at physician discretion.
Dose adjustments were permitted based on toxicity as follows: pomalidomide was to be permanently discontinued in the event of a grade 4 rash, neuropathy or hypersensitivity, and grade 3 or higher bradycardia or cardiac arrhythmia. It was progressively reduced for other related grade 3 or higher adverse events (AEs) to dose levels of 2 or 4 mg for 21 days of each 28-day cycle. Subsequent dose reductions were done in 1 mg increments for the 4 mg cohorts until a dose of 2 mg for 21 days of each 28-day cycle was reached. Dose reductions required subsequent to a 2 mg dose for 21 days of each 28-day cycle were then done in 0.5 mg increments. When grade 3 or 4 AEs occurred prior to day 15 of a cycle and resolved to grade 2 or lower before day 28 of the cycle, pomalidomide was resumed at the next lower dose level, with the next cycle continuing at the reduced dose level. For grade 3 or 4 AEs occurring on or after day 15 of a given cycle, pomalidomide was held for the remainder of the cycle and reduced by one dose level beginning with the next cycle. Dose reductions were permitted for dexamethasone-related toxicity, by lowering the dose of dexamethasone progressively to 20, 12, 8 and 4 mg once weekly. Therapy was discontinued permanently in patients unable to tolerate the lowest doses of pomalidomide or dexamethasone. In the absence of grade 3 or higher toxicity, the daily dose of pomalidomide could be increased at physician discretion to 4 mg in patients who had not achieved a 25% reduction in serum or urine monoclonal protein levels after two cycles of therapy or who had previously responded and had rising serum or urine monoclonal protein levels. Among patients who had a previous dose reduction, escalation was allowed as long as there was no current grade 3 or 4 toxicity.
Response and toxicity criteria
Responses were assessed according to the then-published criteria of the International Myeloma Working Group.18
The National Cancer Institute Common Terminology Criteria for Adverse Events, version 3, was used to grade AEs as well as to assign perceived attribution to the study treatment regimen. Toxicity was defined as AEs at least possibly related to treatment.
We were interested in specifically looking at responses among high-risk patients. High risk was defined, according to the published criteria at the time of primary analysis of the trial,18 as cytogenetic studies (hypodiploidy or karyotypic deletion of chromosome 13), fluorescent in situ hybridization (presence of translocations t(4;14) or t(14;16) or deletion 17p) or plasma cell labeling index ⩾3%.
Statistical design and analysis
The primary end point for all the cohorts was the confirmed overall response rate (ORR=stringent complete response, very good partial response or partial response). Cohorts 1–5 used a one-stage binomial design or a one-stage design with an interim analysis based on a Simon design. Accrual within any cohort did not halt while waiting for interim analysis and patient accrual to the cohorts was done in a sequential manner, with patients accrued to a subsequent cohort after the previous one was complete. For cohort 6, a total of 120 evaluable patients were enrolled to show that a 90% confidence interval (CI) for confirmed response rate would lie above 26% if 24 or more responses were observed. Secondary end points included OS, PFS, duration of response (DOR) and AE profile. Exact binomial CIs were constructed for the primary end point of confirmed response. The distributions of (1) OS time (time from study entry to death), (2) PFS time (time from study entry to the earlier of disease progression or death) and (3) DOR (time from first documentation of response until disease progression in patients who achieved a response), were estimated using the method of Kaplan–Meier. Simple descriptive statistics were used to summarize the adverse event (AE) profile and baseline patient characteristics. The relationship between prognostic factors and response (responder vs non-responder) was evaluated by Fisher’s exact tests. Cox proportional hazards models were used to evaluate the impact of multiple factors simultaneously on time to progression, where the branch and bound algorithm of Furnival and Wilson was used to identify factors for the final multivariable model.
Results
Patient population
Overall, 345 patients were accrued to the study from 1 November 2007 to 31 March 2012. These included 60, 34, 35, 35, 61 and 120 patients accrued sequentially to the 6 cohorts, respectively. The cohort treatment characteristics and accrual periods are detailed in Table 1. Two of the patients were inevaluable (one each from cohorts 5 and 6), leading to 343 patients in the final analysis. Patient characteristics are presented in Table 2. The median time from diagnosis to enrollment on study was variable among the cohorts, with the shortest time since diagnosis noted in patients on cohort 5 (refractory to <3 prior regimens, 4 mg; 38.4 months) and the longest in patients on cohort 4 (refractory to lenalidomide and bortezomib, 4 mg; 71.6 months). The median number of prior therapies across all cohorts was 3 (range 1–14) with the highest number in cohorts with patients refractory to bortezomib and lenalidomide (dual-refractory). More than 90% of patients in every cohort had prior exposure to proteasome inhibitors and/or IMiDs while more than two-thirds of patients had prior stem cell transplant. High-risk disease19 was noted in 43.7% patients overall (n=146), with as high as 60% in cohort 4.
Follow-up
Treatment characteristics and patient follow-up data on study are summarized in Supplementary Table 1. The median number of cycles administered was 5 (range 1–89), with the highest, 11.5 cycles in cohort 1 (2 mg, refractory and <3 prior regimens) and the lowest, 3 cycles in cohort 4 (4 mg, dual-refractory). The most common cause for treatment discontinuation (69.7%) was disease progression. The median follow-up for all living patients was 64.4 months (range 1.2–104.1 months). Overall, 47% patients had treatment delays. Of the 388 total events of treatment delays, 62% were secondary to AEs, primarily grade 3 or 4 neutropenia. Thromboprophylaxis was given with 325 mg of aspirin in 85% of treatment cycles and full-dose anticoagulation was administered with heparin or Coumadin in the rest.
Efficacy
ORR (partial response or better) for all cohorts analyzed together was 35% (95% CI: 30–40%). In various cohorts it ranged from 23% (cohort 5; 2 mg, dual-refractory) to 65% (cohort 1; 2 mg, refractory with <3 prior regimens; Supplementary Figure 1). In general, higher response rates were seen in cohorts 1 and 5, both of which had patients with <3 prior regimens of treatment. complete response or stringent complete response were seen in at least 1 patient in all of the cohorts except cohort 3 (2 mg, dual-refractory). Among all cohorts, response rates were evaluated separately for patients with high-risk disease, with the highest rate (74%) seen in cohort 1 (2 mg, refractory with <3 prior regimens). Details of patient outcomes along with response rates are shown in Table 3.
Median time to a confirmed partial response or better was 2 months or less in all cohorts (range 0.8–14.8 months). The largest cohort (cohort 6) where pomalidomide was administered in accordance with its current Food and Drug Administration approval had a median time to response of 1.1 months (range 0.8–8.8 months). Median DOR varied from 3.9 months (95% CI: 1.8–14.5) in cohort 4 (4 mg, dual-refractory) to 36.5 months (95% CI: 9.4–72.2) in cohort 5 (4 mg, refractory with <3 prior regimens). The cohorts that were less heavily pre-treated (cohorts 1 and 5) had a longer median DOR than the others.
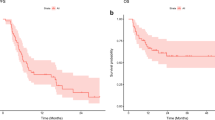
The median PFS was 3.5 months (95% CI: 2.2–6.7) in cohort 4 (4 mg, dual-refractory) and as high as 13 months (95% CI: 7.8–22) in cohort 1 (2 mg, refractory with <3 prior regimens). In cohort 6 the median PFS was 4.3 months (95% CI: 2.9–5.1; Figure 1a). The median OS ranged from 9.2 months (95% CI: 5.5–21.3) in cohort 4 (4 mg, dual-refractory) to 47.9 months (95% CI: 35.4–66.7) in cohort 1 (2 mg, refractory with <3 prior regimens). Cohort 6 had a median OS of 13.4 months (95% CI: 10.4–16.2; Figure 1b).

(a) PFS by study cohort. (b) OS by study cohort.
We performed an analysis by prior lines of therapy across all cohorts, which showed better responses in less heavily pre-treated patients, ranging from 24% in those with ⩾4 to 66% in those with 1 prior lines of therapy. Another subgroup analysis was done by dose, comparing all patients treated with 2 mg (n=129) to those treated with 4 mg of pomalidomide every 21 days (n=119) or continuously (n=95) (Supplementary Table 2). The ORR was highest in the 2 mg group (45%) and lowest in 4 mg/21-day group (24%). We noted that the median duration of treatment was also longest in the 2 mg group (7.4 months, range: 0.6–82.6 months) and shortest in the 4 mg/21-day group (3.3 months, range: 0.3–45.8 months). Similarly, the median DOR was longest in the 2 mg group (17.8 months, 95% CI: 12–30.6) and shortest in the 4 mg/21-day group (10.2 months, 95% CI: 6.4–36.7). Other predictors of response across all groups analyzed together included absence of 17p− abnormality (P=0.02), absence of t(4;14) abnormality (P=0.02), low beta-2-microglobulin (P=0.004), lactate dehydrogenase within normal limit (P=0.001), plasma cell labeling index 1<(P+0.01), no prior lenalidomide exposure (P<0.001), no prior bortezomib exposure (P<0.001) and hemoglobin ⩾10 g/dl (P=0.003). In a multivariate analysis looking at factors associated with a longer time to progression in all patients grouped together, we noted that absence of 12q− on fluorescent in situ hybridization (hazard ratio (HR) 1.44, 95% CI: 1.04–2.0; P=0.03), ⩽3 prior regimens of therapy (HR 1.48, 95% CI: 1.12–1.97; P=0.006). Lactate dehydrogenase within normal limits (HR 1.73, 95% CI: 1.31–2.29, P<0.001) and no prior bortezomib exposure (HR 1.66, 95% CI: 1.22–2.29; P=0.001) were significant.
Specifically comparing outcomes in patients with high-risk (N=146) and standard-risk (N=188) disease, the median PFS for the high-risk group was 4.3 months (95% CI: 2.8–5.5) as compared to 7.4 months (95% CI: 6.2–9.3) in the standard-risk group (Figure 2a). A similar effect was seen for OS, where the high-risk group had a median OS of 12.8 months (95% CI: 11–16.3) as compared to 35.1 months (95% CI: 26.4–43.5) for the standard-risk group (Figure 2b). We further performed a subset analysis of patients with del17p within the high-risk group across all cohorts (n=56). The ORR for these patients was 21% with a median OS of 11.8 months (95% CI: 9.2–18.5) and median PFS of 3.0 months (95% CI: 1.9–5.3).In an attempt to afford comparison with other large, current data sets, we attempted an analysis by Revised International Staging System (R-ISS) but some patients did not have all the variables available as the study did not collect the data specifically and spanned a period when all the components of the R-ISS were not necessarily assessed. Available information on R-ISS by treatment cohort is presented in Table 2. PFS by R-ISS ranged from median 10.2 months (95% CI: 6.9–22.2) for stage I to median 2.8 months (95% CI: 1.9–3.2) for stage III. Similarly, median OS ranged from median 50.1 months (95% CI: 32.6–not attained) for stage I to median 4.8 months (95% CI: 2.8–10.3) for stage III.

(a) PFS by myeloma risk category. (b) OS by myeloma risk category
Adverse events
The treatment was overall well tolerated across all cohorts. AEs consisted primarily of myelosuppression. Grade 3 or 4 hematologic AEs regardless of attribution occurred in 71% of patients in all the cohorts (range 53–83%). In general, the cohorts utilizing 4 mg pomalidomide had a higher incidence of hematological AEs with the exception of cohort 3 (2 mg, refractory to lenalidomide and bortezomib) where they were seen in 83% of patients; highest among all cohorts. Grade 3+ non-hematological AEs regardless of attribution were seen overall in 59% of the patients and were less frequent than hematologic AEs except in cohort 1, where they were slightly more frequent (72 versus 63%). When considering at least possible attribution of AEs to pomalidomide, overall 65% patients had grade 3 or 4 hematologic and 36% patients had grade 3+ non-hematologic toxicities. The most common of all grade 3+ hematologic toxicities was neutropenia, seen more frequently in the cohorts utilizing 4 mg dose of pomalidomide (57%) than those using the 2 mg dose (45%). Similar trends were seen for anemia, lymphopenia, leukopenia and thrombocytopenia, although these were much less frequent than neutropenia and were not seen consistently in all the treatment cohorts. The most common non-hematologic toxicity was fatigue with grade 3 or 4 fatigue occurring in 9% of patients among all cohorts. Pomalidomide dose was modified in 195 patients (57%) while dexamethasone dose was modified in 136 patients (40%) in all the cohorts. The majority of these were secondary to AEs, most commonly due to neutropenia. There were a total of 12 episodes of deep vein thrombosis noted in all cohorts (regardless of attribution), with patients given therapeutic anticoagulation using low-molecular-weight heparin or Coumadin after the thrombosis was diagnosed. Among patients on all cohorts, a total of 11 cases (regardless of attribution), of second primary malignancies were noted. These included seven cases of non-melanoma skin cancer (two squamous cell carcinomas and five basal cell carcinomas) and one case each of cholangiocarcinoma, malignant melanoma, myelodysplastic syndrome and ductal carcinoma in situ of the breast. There were a total of 12 deaths reported in patients while on treatment and of these 3 were considered at least possibly associated with study treatment. The cause of death for all the 3 cases was reported as febrile neutropenia and infection, source not otherwise specified. AEs for all treatment cohorts are outlined in Table 4 and Figure 3.

Adverse Events Occurring in at least 10% of Patients on the Study.
Discussion
This multi-cohort clinical trial explores various schedules and doses of Pom/dex in multiple myeloma patients with varying disease and prior treatment characteristics, and demonstrates the safety and efficacy of this regimen across all cohorts evaluated. We have previously reported early results from cohorts 112 and 2,17 and combined results from cohorts 3 and 4 (2 mg/4 mg dose in patients dual-refractory),13 but this report provides updated results for these cohorts as well. This clinical trial with all its treatment cohorts presents one of the largest experiences with Pom/dex and certainly the longest follow-up reported so far in patients with refractory multiple myeloma.
Evaluating various treatment cohorts included in the trial, we noted the highest response rates (partial response or better) as well as clinical benefit (stable disease or better) in patients who were less heavily pre-treated (cohorts 1 and 5), suggesting increased benefit from pomalidomide, a more potent IMiD, when used earlier in the patient’s treatment paradigm for myeloma. This was true despite the fact that ~80% of patients in cohort 1 and all the patients in cohort 5 had been treated with an IMiD previously, some of them with both, thalidomide and lenalidomide. This suggests the better efficacy of pomalidomide even in IMiD-refractory patients when used relatively earlier in the treatment. These results replicate findings from previously reported trials11, 14, 15, 20 and also help establish the efficacy of pomalidomide in thalidomide and lenalidomide refractory patient. Similar to the higher response rates, the median OS and PFS were also noted to be highest in these cohorts with patients who had <3 prior regimens of treatment. Considering all patients, we noted a longer median duration of treatment, a higher DOR and higher ORR in the 2 mg cohorts, potentially due to better tolerability of the smaller dose, especially in the continuous regimen.6 This is suggested by the lower AE rate, especially the hematological AEs in the 2 mg treatment cohorts (Table 4) and is supported by other trials with longer duration of therapy providing better patient outcomes.6 Grouping the patients by pomalidomide dose did show that patients in the 2 mg group had lesser prior exposure to IMiDs, especially lenalidomide, and patients in the 4 mg/21-day group had a slightly higher prevalence of high-risk disease with other characteristics being rather similar (Supplementary Table 2). Nevertheless, this finding is intriguing as the 4 mg dose of pomalidomide is approved by the Food and Drug Administration for treatment of multiple myeloma in the United States, and questions the current paradigm of drug development, where typically the maximally tolerated dose is selected rather than the minimally effective dose.
We have previously reported on the bortezomib and lenalidomide refractory patients (cohorts 3 and 4) from this trial.13 A longer follow-up in this report shows 37% survival at 18 months in both cohorts, which is expectedly inferior to the other cohorts in this trial, nevertheless, it shows the efficacy of Pom/dex in this double-refractory population, which has traditionally had an extremely poor outcome.9 Previous trials that have specifically reported on the outcomes of these double-refractory patients showed a median PFS of 3.8 months and a median OS of 13–14 months for these patients.11, 14 We noted a superior median PFS (6.3 months) and OS (14.7 months) for the double-refractory patients in cohort 3 (2 mg dose) while cohort 4 (4 mg dose) had findings closer to the previously reported trials (median PFS: 3.5 months and median OS: 9.2 months). Results from either of our treatment schedules are superior to a large analysis by the International Myeloma Working Group, which showed a median OS of 9 months only in patients with disease refractory to bortezomib and who had relapsed following, were refractory to or were ineligible to receive an IMiD.9 Our results show Pom/dex to have an ORR of 29% in double-refractory patients treated with the currently Food and Drug Administration-approved dose of pomalidomide (4 mg), which is close to that reported by previous clinical trials of Pom/dex20 and close to that seen with newer agents, including monotherapy with daratumumab, an anti-CD38 antibody.21
Several clinical trials have now shown that Pom/dex is an effective regimen for patients with relapsed and/or refractory multiple myeloma (Table 5). Our large, multi-cohort clinical trial with the longest reported follow-up helps understand the positioning of Pom/dex in the continuum of management of myeloma patients, especially with a larger selection of agents and classes of drugs available at present, as well as in special populations that may no longer be candidates for some of the more commonly used agents like bortezomib or lenalidomide. Previous reports that Pom/dex may improve health-related quality of life in patients with relapsed and/or refractory myeloma22, 23 and our report of the efficacy of this regimen when used in relatively less heavily pre-treated patients despite refractory disease, supports using the convenient, all-oral Pom/dex as a preferred regimen for refractory myeloma so that other regimens or drugs may remain options for subsequent therapy in this so far incurable disease.
References
Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015; 373: 621–631.
Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Spicka I, Oriol A et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015; 372: 142–152.
Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016; 375: 1319–1331.
Moreau P, Masszi T, Grazsko N, Bahlis NJ, Hansson M, Pour L et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016; 374: 1621–1634.
Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med 2016; 375: 754–766.
Palumbo A, Gay F, Cavallo F, Di Raimondo F, Larocca A, Hardan I et al. Continuous therapy versus fixed duration of therapy in patients with newly diagnosed multiple myeloma. J Clin Oncol 2015; 33: 3459–3466.
Meadows JP, Mark TM . Management of double-refractory multiple myeloma. Curr Hematol Malig Rep 2013; 8: 253–260.
Sonneveld P, Broijl A . Treatment of relapsed and refractory multiple myeloma. Haematologica 2016; 101: 995.
Kumar SK, Lee JH, Lahuerta JJ, Morgan G, Richardson PG, Crowley J et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia 2012; 26: 149–157.
Streetly MJ, Gyertson K, Daniel Y, Zeldis JB, Kazmi M, Schey SA . Alternate day pomalidomide retains anti-myeloma effect with reduced adverse events and evidence of in vivo immunomodulation. Br J Haematol 2008; 141: 41–51.
Richardson PG, Siegel DS, Vij R, Hofmeister CC, Baz R, Jagannath S et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood 2014; 123: 1826–1832.
Lacy MQ, Hayman SR, Gertz MA, Dispenzieri A, Buadi F, Kumar S et al. Pomalidomide (CC4047) plus low-dose dexamethasone as therapy for relapsed multiple myeloma. J Clin Oncol 2009; 27: 5008–5014.
Lacy MQ, Alfred JB, Gertz MA, Hayman, Short KD, Buadi F et al. Pomalidomide plus low-dose dexamethasone in myeloma refractory to both bortezomib and lenalidomide: comparison of 2 dosing strategies in dual-refractory disease. Blood 2011; 118: 2970–2975.
Leleu X, Attal M, Arnulf B, Moreau P, Traulie C, Marit G et al. Pomalidomide plus low-dose dexamethasone is active and well tolerated in bortezomib and lenalidomide-refractory multiple myeloma: Intergroupe Francophone du Myelome 2009-02. Blood 2013; 121: 1968–1975.
Dimopoulos MA, Palumbo A, Corradini P, Cavo M, Delforge M, Di Ramonodo F et al. Safety and efficacy of pomalidomide plus low-dose dexamethasone in STRATUS (MM-010): a phase 3b study in refractory multiple myeloma. Blood 2016; 128: 497–503.
Siegel DS, Weisel KC, Dimopoulos MS, Baz R, Richardson P, Delforge M et al. Pomalidomide plus low-dose dexamethasone in patients with relapsed/refractory multiple myeloma and moderate renal impairment: a pooled analysis of three clinical trials. Leuk Lymphoma 2016; 57: 2833–2838.
Lacy MQ, Hayman DR, Gertz MA, Short KD, Dispenzieri A, Kumar S et al. Pomalidomide (CC4047) plus low dose dexamethasone (Pom/dex) is active and well tolerated in lenalidomide refractory multiple myeloma (MM). Leukemia 2010; 24: 1934–1939.
Kyle RA, Rajkumar SV . Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009; 23: 3–9.
Mikhael JR, Dingli D, Roy V, Reeder CB, Duadi FK, Hayman SR et al. Management of newly diagnosed symptomatic multiple myeloma: updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) consensus guidelines 2013. Mayo Clin Proc 2013; 88: 360–376.
San Miguel J, Weisel K, Moreau P, Lacy M, Song K, Delforge M et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol 2013; 14: 1055–1066.
Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016; 387: 1551–1560.
Song KW, Dimopoulas MA, Weisel KC, Moreau P, Palumbo A, Belch A et al. Health-related quality of life from the MM-003 trial of pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone in relapsed and/or refractory multiple myeloma. Haematologica 2015; 100: e63–e67.
Weisel K, Dimopoulos M, Song KW, Moreau P, Palumbo A, Belch A et al. Pomalidomide and low-dose dexamethasone improves health-related quality of life and prolongs time to worsening in relapsed/refractory patients with multiple myeloma enrolled in the MM-003 Randomized Phase III Trial. Clin Lymphoma Myeloma Leuk 2015; 15: 519–530.
Acknowledgements
Author contributions
BRL, SK, SVR and MQL designed the clinical trial; SA, JRM, SK, VR, DD, PLB, FKB, SVR, RF, MAG, PK, TS, SRH, AKS, AD, WIG, CBR, YL, RSG, NL, TK, JAL, SJR, AAC-K and MQL conducted the clinical trial; BRL and KML performed the analysis; SA, BRL, KML and MQL wrote the manuscript; and all authors revised and approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
SA has served on advisory board for Amgen, Novartis, Takeda and Pharmacyclics, and receives research funding from Pharmacyclics; JRM receives research funding from Abbvie, Celgene and Sanofi; SK receives research funding from Takeda, Celgene, Novartis, Abbvie, Janssen and Roche; DD receives research funding from Karyopharm, Amgen and Takeda; PLB receives research funding from Novartis, Constellation and Mundipharma, and has served on advisory board for Incyte, Janssen, Adaptive Bioscience and Juno Therapeutics; MAG receives research funding from Celgene, Janssen, Prothena, Ionis, Alnylam and Sandoz; PK has served on advisory board for Sanofi-Genzyme and receives research funding from Takeda, Celgene and Amgen; AKS receives research funding from Celgene, Amgen, Bristol Myers Squibb and Janssen; AD receives research funding from Celgene, Millennium, Pfizer and Janssen, and has received travel grant from Pfizer and Prothena; CBR receives research funding from Celgene, Novartis, Takeda and BMS; YL receives research funding from Janssen; ACK receives research funding from Pharmacyclics; MQL receives research funding from Celgene. The remaining authors declare no conflict of interest. Funding from CA 186781 was utilized to support investigator effort in data analysis and manuscript preparation for this trial.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Rights and permissions
About this article

Cite this article
Ailawadhi, S., Mikhael, J., LaPlant, B. et al. Pomalidomide–dexamethasone in refractory multiple myeloma: long-term follow-up of a multi-cohort phase II clinical trial. Leukemia 32, 719–728 (2018). https://doi.org/10.1038/leu.2017.258
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.258
- Springer Nature Limited
This article is cited by
-
Pomalidomide and dexamethasone combination with additional cyclophosphamide in relapsed/refractory multiple myeloma (AMN001)—a trial by the Asian Myeloma Network
Blood Cancer Journal (2019)
-
CD26 is a potential therapeutic target by humanized monoclonal antibody for the treatment of multiple myeloma
Blood Cancer Journal (2018)