
Abstract
Resistance to cytotoxic chemotherapy drugs remains as the major cause of treatment failure in acute myeloid leukemia. Histone deacetylases (HDAC) are important regulators to maintain chromatin structure and control DNA damage; nevertheless, how each HDAC regulates genome stability remains unclear, especially under genome stress conditions. Here, we identified a mechanism by which HDAC3 regulates DNA damage repair and mediates resistance to chemotherapy drugs. In addition to inducing DNA damage, chemotherapy drugs trigger upregulation of HDAC3 expression in leukemia cells. Using genetic and pharmacological approaches, we show that HDAC3 contributes to chemotherapy resistance by regulating the activation of AKT, a well-documented factor in drug resistance development. HDAC3 binds to AKT and deacetylates it at the site Lys20, thereby promoting the phosphorylation of AKT. Chemotherapy drug exposure enhances the interaction between HDAC3 and AKT, resulting in decrease in AKT acetylation and increase in AKT phosphorylation. Whereas HDAC3 depletion or inhibition abrogates these responses and meanwhile sensitizes leukemia cells to chemotoxicity-induced apoptosis. Importantly, in vivo HDAC3 suppression reduces leukemia progression and sensitizes MLL-AF9+ leukemia to chemotherapy. Our findings suggest that combination therapy with HDAC3 inhibitor and genotoxic agents may constitute a successful strategy for overcoming chemotherapy resistance.
Similar content being viewed by others

Introduction
Acute myeloid leukemia (AML) is a clonal disorder of hematopoietic stem cells characterized by the inhibition of differentiation and excessive proliferation of cells at various stages of incomplete maturation.1 The general treatment strategy for AML patients has not changed substantially during the past decades, with the combination of cytarabine (Ara-C) and an anthracycline as the chemotherapy backbone. The cytotoxicity of these chemotherapy agents is primarily mediated by the inhibition of DNA synthesis and subsequent generation of DNA strand breaks. The extent of DNA damage induced by these drugs exceeds the DNA repair capacity of cancer cells, resulting in cell cycle arrest and cell death. Although the initial complete remission rate can be achieved in around 70% for patients under the age of 60, drug resistance developed in a large proportion of these patients, which died of relapsed and refractory disease eventually.1, 2 The clinical efficacy of chemotherapy agents is severely impaired by intrinsic and acquired drug resistance. Therefore, characterization of mechanisms that contribute to drug resistance and identification of strategies to tackle the problem may improve the treatment outcome in AML.
The phosphoinositide 3-kinase (PI3K)/AKT pathway regulates the fundamental cellular functions and plays a crucial role in virtually all aspects of leukemia biology, such as cell survival and proliferation. PI3K/AKT pathway is frequently activated in AML and associated with a poor prognosis.3, 4, 5 In a wide range of cancer cells, elevated PI3K/AKT signaling has been involved in reduced sensitivity to conventional chemotherapy agents.6 O’Gorman et al.7 first demonstrated the important role of the PI3K/AKT pathway in causing resistance to drugs commonly used for AML treatment. A clinical study also showed that inhibition of AKT activation correlates with complete response in c-kit-positive relapsed/refractory AML.8 AKT is the center effector of the PI3K pathway. In response to growth factor signaling, various forms of cellular stress or oncogenic PI3K pathway mutations, AKT becomes phosphorylated at two sites Thr308 and Ser473. Besides, AKT can be regulated by the PI3K-related kinase family member DNA-dependent protein kinase (DNA-PK), which phosphorylates AKT at Ser473 in response to DNA damage,9 indicating AKT involves in the DNA damage response. Since altered DNA damage response is an important factor for drug resistance development,10, 11 AKT activation may promote cell survival through enhancing the DNA repair capacity of cancer cells. Therefore, manipulating AKT activity in AML chemotherapy might be a good strategy to increase the chemosensitivity of leukemia cells and reduce the likelihood of relapsed/refractory AML.
In a variety of cancers, histone deacetylases (HDACs) function and/or expression is perturbed and is often associated with poor prognosis.12 In AML, oncogenic fusion proteins can recruit HDACs to specific gene promoters to drive leukemogenesis.13 Besides, HDACs can regulate the genome stability by facilitating DNA damage repair,14 and overexpressed HDACs in cancer cells have been implicated in protecting cells from genotoxic insults.15 For example, HDAC3 was essential for DNA damage control and genome stability maintenance, while deletion of Hdac3 greatly impaired DNA repair.16 In consistent with this, HDAC inhibitors have been developed to interfere with DNA damage repair mechanisms and trigger apoptosis of cancer cells.12, 17 However, the mechanisms by which each HDAC regulates genome stability remain poorly understood.
Here we demonstrate that common chemotherapy drugs–Ara-C and doxorubicin (Doxo)–induces HDAC3 upregulation and AKT activation in AML cell lines, and that HDAC inhibitors dramatically enhance the cytotoxicity of both drugs. Mechanistically, we identify that HDAC3 binds to and deacetylates AKT at the site of Lys20, leading to enhanced activity of AKT, which in turn promotes the resolution of DNA damage. Following the HDAC inhibitors exposure, AKT dissociates with HDAC3, and becomes highly acetylated and less phosphorylated. We propose that the inhibition of AKT phosphorylation and disruption of DNA damage repair contributes to the efficacy of combination therapy with HDAC inhibitors and chemotherapy agents.
Materials and methods
Cell lines and reagents
K562, THP-1 and HEK293T cells were obtained from Shanghai Institute of Hematology (Shanghai, China), and K562 Doxo-resistant cell line K562/Doxo was obtained from Institute of Hematology, Chinese Academy of Medical Sciences (Tianjin, China). THP-1 cells were cultured with 0.1 μM Ara-C for 3 months for promoting Ara-C resistance. K562/Doxo cells were established by continuous culture of parental K562 cells with increasing concentrations of Doxo until cells readily grew in the presence of 0.5 μg/ml Doxo.18 K562, K562/Doxo and THP-1 were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine and antibiotics. K562/Doxo was cultured in complete medium with 0.5 μg/ml Doxo for drug resistance maintenance, and was re-cultured in complete medium without Doxo for 2 weeks before experiments. HEK293T cells were cultured in DMEM with 10% fetal bovine serum, 2 mM L-glutamine and antibiotics. Cells were grown at 37 °C in an atmosphere containing 5% CO2. Cell lines were authenticated by short tandom repeat profiling before conducting the experiment and were regularly monitored for the absence of mycoplasma.
Ara-C was from Pzifer (Shanghai, China). Doxo was from Shenzhen Main-Luck Pharmaceuticals (Shenzhen, China). The class I HDAC inhibitor valproic acid (VPA; #P4543) and HDAC3- (#S7229) specific inhibitor RGFP966 were purchased from Sigma-Aldrich (Shanghai, China) and Selleck (Shanghai, China), respectively. Cell Counting Kit-8 (CCK-8; #CK04) was from Dojindo (Shanghai, China). Annexin-V–FITC (#556420), Annexin-V–APC (#550474) and 7-AAD (#559925) were obtained from BD Biosciences (Shanghai, China). The origins of different antibodies were as follows: acetylated lysine (#9814), AKT (#4685), p-AKT308 (#13038), p-AKT473 (#4060), γH2AX (#9718), HDAC1 (#2062), HDAC2 (#2540), Flag (#14793), GFP (#2956) were from Cell Signaling Technology (Shanghai, China); HDAC3 (ab32369), HDAC8 (ab187139), p-DNAKcs (ab32566), DNA-PKcs (ab124918), Ku70 (ab92450), Ku80 (ab80592) were from Abcam (Shanghai, China); Actin (A5441) was from Sigma-Aldrich.
Patient samples
Peripheral blood and bone marrow samples were collected from patients with de novo or relapsed AML after informed consent. Mononuclear blasts were isolated through Ficoll (Axis-Shield, Oslo, Norway) density centrifugation, and cell viability was assessed by Trypan Blue Exclusion Assay. Protocol of sample handling and data analysis was approved by Ruijin Hospital Ethics Committee, and was performed in compliance with the Helsinki Declaration. Patients information was shown in Supplementary Table.
Mice and leukemia model
Female C57BL/6 mice were obtained from the Shanghai Laboratory Animal Center, Chinese Academy of Sciences, Shanghai. The mice were housed in specific pathogen-free conditions at the Research Center for Experimental Medicine of Rui-Jin Hospital. Mice were between 6 and 8 weeks of age at the start of the experiments. Sample sizes of more than six animals per group were sufficient for detecting differences in survival of 20–30% with statistical significance. Animals were assigned to different groups randomly. Blinding was not applied in the analysis. All experiments were carried out according to the National Institutes of Health Guide for Care and Use of Laboratory Animals.
For leukemia mice model, bone marrow GFP+ (MLL-AF9+) cells were harvested from primary MLL-AF9 leukemia mice and injected into secondary sublethally irradiated syngeneic normal mice via tail vein. Chemotherapy was initiated 1 week after transplantation and consisted of five daily doses of Ara-C (100 mg/kg) together with three doses of Doxo (3 mg/kg).19 VPA (300 mg/kg) or RGFP966 (20 mg/kg) was also administered once daily 1 week post transplantation for 5 days. Leukemia progression was monitored weekly by peripheral blood samples using an automated blood counter and flow cytometry for the percent of GFP+ cells. Mice were killed 21 days post transplantation and subjected to complete necropsy; Blood, bone marrow and spleen were analyzed by cell counts and flow cytometry.
Plasmid construction and lentiviral transduction
AKT construct was obtained from Genechem (Shanghai, China), and was used to generate domain deletion mutants. K562/Doxo cells were transduced with lentiviral particles carrying human HDAC3-directed short hairpin RNA (shRNA) (pLVX-shRNA2, Clontech, Beijing, China), and tranfected cells were selected from fluorescence activated cell sorting. For murine Hdac3 knockdown in mice MLL-AF9+ leukemia cells, pLVX-shRNA1 (Clontech) was used, and transduced cells were selected with 1.0 μg/ml puromycin for 24 h. A scramble shRNA was used as a control for off target effects of shRNA. The sequences were as follows: human shHDAC3-1: 5′-ACCTAGTGTCCAGATTCAT-3′; human shHDAC3-2: 5′-ACCCAATGAGTTCTATGAT-3′; murine shHdac3: 5′-ACCCGGTGTTGGACATATGAAA-3′; scramble shRNA: 5′-GCGCGCTTTGTAGGATTCG-3′.
Statistics
Data analysis was performed using GraphPad Prism software (Graphpad Software, La Jolla, CA, USA). Survival comparisons were performed using the Kaplan–Meier/log-rank test. Other differences between experimental groups were analyzed using a two-tailed unpaired Student’s t test. A P-value <0.05 was considered to be significant in all experiments.
Results
HDAC inhibitor VPA sensitizes leukemia cells to chemotherapy drugs-induced death by inhibiting AKT
We previously found that the HDAC inhibitor VPA could inhibit the phosphorylation of Akt in murine CD4+ T cells.20 Since enhanced AKT activity has been implicated as a crucial factor for drug resistance development in cancer cells, we reasoned that inhibition of AKT activity by HDAC inhibitors would sensitize AML cells to chemotherapy drugs. Therefore, in our present study, we chose K562/Doxo and THP-1 cells, which are resistant to Doxo or Ara-C, respectively. CCK-8 assays showed that VPA had remarkable synergistic effect with Doxo or Ara-C, especially when VPA was used at 1 mM, an achievable concentration in clinical settings (Supplementary Figure 1a). Annexin-V/7-AAD staining also showed VPA combination with Doxo or Ara-C and had more significant apoptosis-inducing effect than single agent (Figure 1a).

VPA enhances chemotherapy-induced DNA damage by inhibiting AKT activation and sensitizes cells to chemotherapy both in vitro and in vivo. (a) K562/Doxo and THP-1 cells were treated with indicated drugs, and cell apoptosis was analyzed 24 h later by flow cytometry. (b) K562/Doxo cells were treated with different concentrations of Doxo and/or VPA for 24 h, and then subjected to western blot to detect indicated proteins (left); THP-1 cells were treated with different concentrations of Ara-C and/or VPA for 24 h, and then subjected to western blot to detect indicated proteins (right). (c) Immunofluorescence staining of p-AKT473 and γH2AX in K562/Doxo cells after treating with Doxo (2 μg/ml) alone or in combination with VPA (1 mM) for 24 h. Results are representative of three independent experiments. Data are expressed as mean±s.d. **P<0.01; ****P<0.0001.
We then tried to examine whether AKT indeed played a role in drug resistance in AML cells. Comparison between K562 cells (Doxo sensitive) and K562/Doxo cells found that the protein level of both total AKT and phosphorylated AKT was much higher in K562/Doxo cells (Supplementary Figure 1b). Besides, treatment of K562/Doxo with different concentrations of Doxo showed a dose-dependent escalation of AKT phosphorylation and a correlated phosphorylation of histone H2AX (γH2AX) (Supplementary Figure 1c). Treatment of K562/Doxo with VPA had no obvious effect on γH2AX, but inhibited the phosphorylation of AKT (Supplementary Figure 1d). Moreover, combination of Doxo with VPA blocked the AKT phosphorylation induction, and dose dependently increased the level of γH2AX (Figure 1b), with no effect on the intracellular concentration of Doxo (Supplementary Figure 1e), suggesting that VPA combination enhanced Doxo-induced DNA damage in K562/Doxo cells. VPA co-treatment also inhibited Doxo-induced phosphorylation of DNA-PKcs (Supplementary Figure 1f), a key factor in non-homologous end joining and a phosphorylation target of AKT.21 Next, we examined the effect of Ara-C and VPA on THP-1 cells. Treating THP-1 with Ara-C increased the phosphorylation of AKT (Supplementary Figure 1g), while co-treatment with VPA inhibited AKT phosphorylation and significantly upregulated the level of γH2AX (Figure 1b). In consistent with previous research, immunofluorescence assay showed that Ser473-phosphorylated AKT co-localized with γH2AX (Figure 1c; Supplementary Figures 1h and i), suggesting AKT directly contributing to DNA damage repair. Moreover, co-treatment with VPA almost completely abrogated the co-localization between p-AKT473 and γH2AX, while markedly increased the number of γH2AX foci (Figure 1c). Taken together, these data demonstrate that AKT desensitizes cells to the toxicity of chemotherapy drugs by promoting DNA damage repair, whereas co-treatment with HDAC inhibitor VPA inhibits the pro-survival effect of AKT and reverses drug resistance in AML cells.
VPA enhances anti-leukemia effect in vivo in combination with chemotherapy
The MLL-rearranged leukemia represents a subset of poor prognosis leukemia characterized as chemotherapy-refractory and high relapse rate.22 Mice leukemia model induced by MLL fusion protein recapitulates these features.19, 23 Therefore, we used MLL-AF9-induced AML to study the ability of HDAC inhibitor to sensitize the chemo-resistant leukemia to conventional chemotherapy. Syngeneic mice were transplanted with primary MLL-AF9+ bone marrow cells and received different regimens of chemotherapy and/or VPA. Although VPA alone had no significant survival benefit, VPA combination with chemotherapy markedly prolonged the overall survival of secondary MLL-AF9+ leukemia mice (Supplementary Figure 2a). Besides, VPA co-treatment delayed the reappearance of circulating leukemia cells after chemotherapy (Supplementary Figures 2b and c). VPA co-treated mice also showed decreased infiltration of leukemia cells in bone marrow and spleen (Supplementary Figures 2d and e). Therefore, these data suggest VPA have in vivo chemo-sensitizing effect.
HDAC3 interacts with AKT and is upregulated in leukemia cells upon chemotherapy drugs exposure while downregulated by VPA
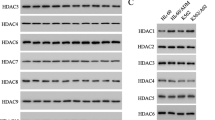
We next investigated the mechanism by which VPA modulated the phosphorylation of AKT. As VPA is a class I HDACs selective inhibitor,24 we looked for the regulation of VPA on these HDACs. As shown in Figure 2a, K562 and K562/Doxo cells expressed comparable level of HDAC2, while K562/Doxo cells had a higher protein level of HDAC3 and 8, but a lower protein level of HDAC1. VPA dose dependently decreased the expression of HDAC1, 3 and 8 in K562/Doxo cells (Figure 2a). We then looked for changes in class I HDACs expression upon treatment of K562/Doxo cells with Doxo. Western blot assay showed that only the expression of HDAC3 was remarkably increased by Doxo, but not other three HDACs (Figure 2a). Previous studies had shown HDAC3 as a critical factor in DNA damage control and genome stability maintenance.16, 25 Therefore, the upregulation of HDAC3 might be a mechanism used by K562/Doxo cells to maintain DNA stability upon Doxo treatment. Interestingly, the alteration of HDAC3 in THP-1 cells to Ara-C or VPA was similar to that in K562/Doxo cells, with an upregulation of HDAC3 in the presence of Ara-C but a downregulation upon VPA treatment (Figure 2b). Moreover, combined treatment of VPA with Doxo or Ara-C abrogated the induction of HDAC3 in K562/Doxo or THP-1 cells (Figure 2c). Taken together, these data suggest changes in HDAC3 upon VPA or chemotherapy drugs and the treatment might play a crucial role in modulating the genome stability.

HDAC3 interacts with AKT, and is regulated by chemotherapy drugs and HDAC inhibitors. (a) The protein level of class I HDACs in K562 cells and K562/Doxo cells treated for 24 h with different concentrations of VPA (left) or Doxo (right). (b) The protein level of class I HDACs in THP-1 cells after treatment with VPA (left) and Ara-C (right) for 24 h. (c) K562/Doxo cells and THP-1 cells were subjected to indicated treatment for 24 h, and were analyzed by western blot for the protein level of HDAC3. (d) AKT was immunoprecipitated from K562/Doxo cells and THP-1 cells. The immunoprecipitates were immunoblotted with class I HDACs antibodies. Results are representative of three independent experiments.
Previous study demonstrated the class III HDAC SIRT1 interacted with AKT and promoted AKT activation.26 Since our data showed a positive correlation between the expression of HDAC3 and the phosphorylation of AKT, we thus asked whether HDAC3 also interacted with AKT. Co-immunoprecipitation assay showed that AKT selectively interacted with HDAC3 in both K562/Doxo and THP-1 cells, but not with other class I HDACs (Figure 2f). Immunofluorescence staining further demonstrated that AKT interacted with HDAC3 in the nucleus (Supplementary Figure 3). Therefore, these data indicate that HDAC3 might regulate the genome stability through AKT.
HDAC3 deacetylates AKT at Lys20 and activates AKT
We then evaluated whether and how HDAC3 maintained genome stability through AKT. AKT immunoprecipitates from HEK293T cells contained HDAC3, and conversely, HDAC3 co-immunoprecipitated with AKT (Supplementary Figure 4a). Besides, expression of HDAC3 reduced the acetylation of endogenous AKT, while promoted the phosphorylation at both Thr308 and Ser473 sites (Supplementary Figure 4b). The phosphorylation promoting effect of HDAC3 depended on its deacetylase activity, since only wild-type HDAC3, but not a catalytically inactive mutant (Arg265→Pro; R265P)27, 28 increased AKT phosphorylation (Supplementary Figure 4c). We further confirmed these results by overexpressing Flag-tagged AKT and GFP-tagged wild-type or inactive mutant HDAC3 in HEK293T cells (Figure 3a).

HDAC3-mediated deacetylation at Lys20 activates AKT. (a) HEK293T cells transfected with Flag-AKT or empty vector were co-transfected with GFP vector control, GFP-HDAC3 or GFP-HDAC3 mutant, and lysates immunoprecipitated with Flag were immunoblotted for acetylated-lysine, GFP, p-AKT308, p-AKT473 and Flag. (b) HEK293T cells transfected with empty vector or HDAC3 were co-transfected with vector control, Flag-AKT, Flag-AKT-ΔPH, Flag-AKT-ΔKD, Flag-AKT-ΔC or Flag-AKT-KD, and lysates immunoprecipitated with Flag were immunoblotted for HDAC3 and Flag. (c) HEK293T cells transfected with empty vector or HDAC3 were co-transfected with vector control, Flag-AKT, Flag-AKT-ΔPH, Flag-AKT-ΔC or Flag-AKT-KD. Lysates immunoprecipitated with Flag were immunoblotted for HDAC3, p-AKT308 and Flag. (d) HEK293T cells transfected with empty vector or HDAC3 were co-transfected with vector control, wild-type Flag-AKT, Flag-AKT K20R mutant or Flag-AKT K20Q mutant, and lysates immunoprecipitated with Flag were immunoblotted for p-AKT308, p-AKT473 and Flag. (e) K562/Doxo cells stably transduced with scrambled shRNA or shHDAC3 were treated with 2 μg/ml Doxo for 24 h, and lysates were immunoprecipitated with AKT and immunoblotted for acetylated-lysine, p-AKT473, HDAC3 and AKT (left panel); THP-1 cells were treated with Ara-C (5 μM) or Ara-C (5 μM) plus RGFP966 (5 μM) for 24 h, and lysates were immunoprecipitated with AKT and immunoblotted for acetylated-lysine, p-AKT473, HDAC3 and AKT (right panel). Results are representative of three independent experiments.
AKT can be divided into three major domains: the N-terminal PH domain, the middle kinase domain and the C-terminal substrate binding domain (Supplementary Figure 4d). To investigate which domain mediated the interaction with HDAC3 and how HDAC3 regulated AKT activity, we generated several mutants of AKT (Supplementary Figure 4d). Co-expression of HDAC3 with full-length AKT or each mutant in HEK293T cells showed that the kinase domain of AKT mediated the interaction with HDAC3 (Figure 3b). However, the expression of HDAC3 could not increase the phosphorylation of mutants lacking the PH domain (the ΔPH mutant and the KD mutant; Figure 3c), indicating the PH domain is indispensable for HDAC3-mediated AKT activation. Combining these results, we hypothesized that HDAC3 might deacetylate AKT at its PH domain and thereby promote AKT phosphorylation. We next examined the possible lysine target sites of HDAC3. Previous study had demonstrated that Lys20 was a target of SIRT1 to mediate AKT activation.26 We therefore asked whether Lys20 is also needed for HDAC3-mediated activation of AKT. AKT mutant harboring Lys20→Arg (K20R), which resembled deacetylated lysine in terms of charge, showed remarkably increased phosphorylation (Figure 3d). Whereas AKT mutant harboring Lys20→Gln (K20Q), which resembled acetylated lysine, showed reduced phosphorylation (Figure 3d). Moreover, co-expression of HDAC3 only increased the phosphorylation of wild-type AKT, but not K20R or K20Q mutants, suggesting that deacetylation of Lys20 was essential for HDAC3-mediated activation of AKT (Figure 3d).
Since our data demonstrated HDAC3 as an important regulator for AKT activation, we then asked whether inhibition of HDAC3 increased AKT acetylation and thereby reduced its phosphorylation. Treatment of K562/Doxo or THP-1 cells with Doxo or Ara-C increased the phosphorylation of AKT, which was accompanied with reduced AKT acetylation and increased HDAC3 interaction with AKT (Figure 3e). HDAC3 knockdown (Supplementary Figure 4e) or inhibition by specific inhibitor RGFP966 reversed these effects of chemotherapy drugs, with an increase in AKT acetylation, a decrease in AKT phosphorylation and a dissociation of HDAC3 with AKT (Figure 3e). These responses were also mimicked by co-treatment with VPA (Supplementary Figures 4f and g).
Inhibition of HDAC3 downregulates AKT activity and sensitizes leukemia cells to the cytotoxicity of chemotherapy drugs
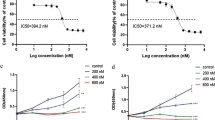
Since HDAC3 could regulate the activity of AKT and meanwhile AKT was a critical factor in DNA damage control and chemoresistance development, we hypothesized that the HDAC3/AKT axis maintained genome stability through AKT activity upregulation, while inhibition of HDAC3 downregulated AKT activity and thus sensitized AML cells to chemotoxic drugs. HDAC3 knockdown or inhibition sensitized K562/Doxo or THP-1 cells to Doxo- or Ara-C-induced apoptosis, respectively (Figure 4a; Supplementary Figure 5a). AKT-specific inhibitor MK2206 also sensitized cells to both cytotoxic agents (Supplementary Figure 5b). CCK-8 assays showed that cells depleted with HDAC3 or treated with RGFP966 exhibited dramatically lower resistance to chemotherapy drugs, as demonstrated by significantly decreased IC50 value of both Doxo and Ara-C in K562/Doxo and THP-1 cells, respectively (Figure 4b). Importantly, this response could be rescued by overexpression of AKT K20R mutant, but not AKT K20Q mutant (Figure 4b). To further reveal the functional significance of HDAC3/AKT axis on genome stability maintenance, we first investigated the influence of AKT on DNA damage response. Inhibition of AKT activity by MK2206 enhanced chemotherapy drugs-induced DNA damage in leukemia cells, as demonstrated by a significant increase in γH2AX level (Supplementary Figure 5c). Next, HDAC3 knockdown or inhibition reduced AKT phosphorylation and remarkably enhanced chemotherapy drugs-induced γH2AX (Figure 4c; Supplementary Figure 5d). Taken together, these data reveal HDAC3 regulated AKT phosphorylation, promotes leukemia cells to resist chemotherapy drugs and contributes to cell survival following drug exposure.

HDAC3 suppression sensitizes leukemia cells to chemotherapy through downregulating AKT activity. (a) K562/Doxo cells stably transduced with shHDAC3 were treated with indicated concentrations of Doxo for 24 and 48 h, and cell apoptosis was analyzed by Annexin V staining (left); THP-1 cells were treated with Ara-C (5 μM) and/or RGFP966 (5 μM) for 24 and 48 h, and cell apoptosis was analyzed by Annexin V/7-AAD staining (right). (b) The IC50 values of Doxo in K562/Doxo cells, and K562/Doxo cells stably transduced with scrambled shRNA or shHDAC3 were determined by CCK-8 assays (left); K562/Doxo cells stably transduced with shHDAC3 were co-transduced with AKT K20R or AKT K20Q, and were subjected to CCK-8 assays to determine the IC50 values of Doxo (middle); THP-1 cells were stably transduced with empty vector, AKT K20R or AKT K20Q, and were exposed to RGFP966 (5 μM). The IC50 values of Ara-C were determined by CCK-8 assays (right). (c) K562/Doxo cells stably transduced with shHDAC3 were treated with indicated concentrations of Doxo for 24 h, and lysates were immunoblotted for p-AKT473, AKT and γH2AX (left); THP-1 cells were treated with indicated concentrations of Ara-C and/or RGFP966 for 24 h, and lysates were immunoblotted for p-AKT473, AKT and γH2AX (right). Results are representative of three independent experiments. Data are expressed as mean±s.d. NS, P>0.05; *P<0.05; **P<0.01; ****P<0.0001.
We then isolated leukemia blasts from relapsed/refractory AML patients as well as de novo AML patients who responded well to conventional induction chemotherapy. Comparing the intracellular levels of HDAC3 and phosphorylated AKT between the two groups, we found a trend of elevated levels of HDAC3 and phosphorylated AKT in leukemia blasts from patients with relapsed/refractory AML. Further analysis also showed that the phosphorylation levels of AKT correlated with the protein levels of HDAC3 (Figure 5a). We also isolated paired samples from the AML patients before and 1 day after induction chemotherapy. Analysis of the paired samples revealed that HDAC3 levels as well as phosphorylated AKT levels were upregulated after chemotherapy, while the acetylated AKT levels were downregulated (Figure 5b). We further treated the freshly leukemia blasts with Doxo or Ara-C alone, or in combination with VPA or RGFP966. Both HDAC inhibitors sensitized leukemia blasts to the conventional cytotoxic agents (Supplementary Figure 6a). Besides, immunoblot assay on HDAC3 levels and AKT modifications revealed consistent changes in patient samples as in K562/Doxo and THP-1 cells (Figure 5c; Supplementary Figure 6b).

HDAC3 regulates the acetylation and phosphorylation of AKT in primary leukemia blasts, and inhibition of HDAC3 sensitizes primary leukemia to chemotherapy. (a) Primary AML blasts were isolated from relapsed/refractory AML patients as well as de novo AML patients who responded well to conventional induction chemotherapy. The levels of indicated proteins were analyzed by immunoblot. (b) Paired AML samples before and 1 day after chemotherapy were collected and analyzed for indicated proteins. (c) AML blasts from relapsed/refractory or de novo AML patients were treated as indicated and analyzed by immunoblot for p-AKT473, AKT, HDAC3, γH2AX and acetylated-lysine. (Doxo 0.5 μg/ml, Ara-C 1 μM, VPA 1 mM, RGFP966 10 μM).
HDAC3 inhibition synergizes with chemotherapy drugs to reduce MLL-AF9 leukemia in vivo
Because our results indicated the important role of HDAC3/AKT axis in DNA damage control and genome stability maintenance, we further evaluated whether HDAC3 suppression had synergistic anti-leukemia effect with chemotherapy in vivo. Syngeneic mice were inoculated with MLL-AF9+ leukemia cells transduced with shHdac3 or scrambled shRNA. Remarkably, depletion of Hdac3 readily provided significant survival benefit in mice bearing MLL-AF9+ leukemia (Figure 6a). Moreover, this survival benefit was also observed in AML mice treated with RGFP966 (Figure 6a). Chemotherapy combination further prolonged the median survival time in mice bearing Hdac3-depleted AML or being treated with RGFP966 (Figure 6a). Peripheral blood monitoring revealed that Hdac3 knockdown or inhibition reduced the leukemia burden, which was further enhanced by chemotherapy combination (Supplementary Figures 7a and b). Necropsy on Day 21 post transplantation also demonstrated that Hdac3 disruption resulted in fewer leukemia cells in bone marrow and spleen (Figures 6b and c; Supplementary Figure 7c).

C57BL/6 mice were sublethally irradiated and transplanted with primary murine MLL-AF9+ leukemia cells stably transduced with scramble shRNA or shHdac3, and subjected to indicated treatments as described in the Materials and Methods section. (a) The survival was monitored over time (n=15). (b, c) Leukemia burden BM (b) and spleen (c) of mice treated as in a, killed on Day 21 post transplantation (n=3). Results are representative of two independent experiments. Data are expressed as mean±s.d. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
Discussion
Resistance to cytotoxic chemotherapy drugs is a common event in AML treatment and has a severe impact on patient survival. Thus, it is urgent to develop new approaches to overcome drug resistance. One strategy is to design drug regimens combining conventional chemotherapy with targeted therapy that blocks intracellular mechanisms leading to chemotherapy evasion and leukemia progression. The expression of HDACs is frequently dysregulated in many kinds of cancers,29, 30 and is often associated with poor prognosis.31, 32, 33 Elevated expression of HDACs in cancer cells may protect cells from chemotoxic insults by enhancing the DNA damage repair ability, thereby contributing to drug resistance occurrence. Various HDAC inhibitors have been developed and are currently in clinical trials. However, modest anti-leukemia effect has been reported following HDAC inhibitor monotherapy and substantial leukemia regression is rarely observed.34 In order to improve the clinical responses rate to HDAC inhibitors, incorporation of these inhibitors into conventional chemotherapy regimens might be a reasonable strategy. Our study provides evidence that HDAC inhibitors significantly sensitize drug-resistant AML cells to the DNA damage chemotherapy agents Doxo and Ara-C. The frequency of HDACs alterations in AML combined with the ability of HDACs to maintain genome stability against cytotoxicity provides rationale to evaluate the combination of HDAC inhibitors with conventional chemotherapy in the clinic.
In chemotherapy-sensitive cancer cells, DNA damage resulting from chemo exposure exceeds the DNA repair ability, leading to cell apoptosis. However, cancer cells may develop various mechanisms to survive and evade from chemotherapy-induced cytotoxicity, such as enhanced DNA damage repair capacity.10, 11 The ability of HDACs to control DNA damage and maintain chromatin structure and genome stability has been widely documented. For example, HDAC1 and HDAC2 promote non-homologous end joining, and HDAC1/2-depleted cells exhibited defective double-strand break repair.35 HDAC3-null cells displayed a reduction in both homologous recombination and non-homologous recombination pathways.16 Although a number of elements in DNA repair pathways have been reported to be influenced by HDACs and their inhibitors, how each HDAC regulates DNA damage repair remains poorly characterized.15, 36 Our study finds that HDAC3 controls DNA damage by regulating the activity of AKT. The ability of AKT to influence DNA repair and promote cancer cells resistance to DNA damage has been well established.37 We find that AKT is highly phosphorylated in leukemia cell lines resistant to chemotherapy drugs, and AKT signaling becomes further activated following chemotherapy drugs exposure. Phosphorylated AKT localized at DNA damage induced γH2AX foci. AKT activation can phosphorylate DNA-PK and regulate its accumulation at DNA damage sites to improve the efficiency of double-strand break re-joining by non-homologous end joining.38 In consistent with this, we demonstrate that HDAC inhibitor or AKT inhibitor suppresses the phosphorylation of DNA-PK. AKT can also promote double-strand break repair through upregulating the expression of Mre1,39 or through phosphorylating MERIT40 and thereby facilitating the assembly of BRCA1-A complex in response to DNA damage.40 Inhibition of AKT by HDAC inhibitors enhances DNA damage induced by chemotherapy drugs, and sensitizes leukemia cells to apoptosis both in vitro and in vivo.
In an attempt to discover the mechanisms by which HDAC inhibitors modulate AKT activity, we find that AKT specifically binds to HDAC3, but not other class I HDACs. The interaction between AKT and HDAC3 is not cell type specific, since we find this phenomenon in different cell lines. Importantly, the expression of HDAC3 is increased following chemotherapy drugs exposure, and is positively correlated with the upregulation of AKT phosphorylation. Besides, the association between AKT and HDAC3 is enhanced by chemotherapy drugs and reduced following HDAC3 inhibition or depletion. Several previous studies have showed the regulation of AKT activity of HDAC3. Gupta et al.41 demonstrated that the knockdown of HDAC3 resulted in increased binding of protein phosphatase PP1 to AKT, thereby dephosphorylating AKT. Another study showed that HDAC3 promoted AKT/mTOR signaling by suppressing the expression of protein phosphatase PHLPP1.42 However, in our study, pretreatment with PP1 inhibitor calyculin A cannot diminish the effect of HDAC inhibitor on AKT phosphorylation (data not shown). Nor did we find any elevation in PHLPP1 expression or increased interaction between AKT and PHLPP1 following HDAC3 inhibition or depletion (data not shown). Therefore, the mechanisms of AKT phosphorylation regulation by HDAC3 may be different in leukemia cells.
Posttranslational modification is a versatile modulator of protein functions and interactions. Among which the reversible acetylation of lysine residues by histone acetyltransferases and HDACs is a posttranslational mechanism that controls the activity of many proteins besides histones, including transcriptional factors and kinases.43, 44 Cross-talk between phosphorylation and acetylation modification has been described for proteins with important functions in both physiology and pathology conditions. For example, Kramer et al.45 found that the signaling of STAT1 was regulated by a phosphorylation-acetylation switch via CBP, HDAC3 and TCP45 complex. The activity of STAT3 is also regulated by cross-talk between phosphorylation and acetylation. Inhibition of HDAC3 triggers hyperacetylation of STAT3 at site Lys685, while downregulates phosphorylation at sites Tyr705 and Ser727.46, 47 Previous study also indicated reversible acetylation as a potential regulatory mechanism that controls the activity of AKT.26 In our study, we find the regulation of AKT phosphorylation by HDAC3 requires its deacetylase activity, suggesting a possible cross-talk signaling between phosphorylation and acetylation of AKT. Indeed, overexpression of HDAC3 reduces the acetylation of AKT and meanwhile promotes its phosphorylation, whereas inhibition or depletion of HDAC3 increases AKT acetylation but decreases its phosphorylation. We further identify the Lys20 at the PH domain of AKT as the posttranslational modification target site of HDAC3. The PH domain contains a bipolar structure in which one side of the domain is a positively charged surface composed of basic amino acids, whereas the other side is populated by acidic residues, which form the ligand-binding pocket in its center.48 Therefore, acetylation at the PH domain might influence the bipolar structure by neutralizing the positive charges, thereby affecting the binding of AKT to PIP3, an important step for AKT activation. In our study, the Lys20 deacetylation mimic mutant of AKT (K20R) shows highly increased phosphorylation and rescues the drug resistance of leukemia cells after HDAC3 depletion or inhibition; whereas the Lys20 acetylation mimic mutant of AKT (K20Q) shows dramatically reduced phosphorylation and has no drug resistance rescue effect.
HDAC3 suppression exhibits in vivo anti-leukemia effect. In consistent with the study by Matthews et al.,49 Hdac3-depleted murine MLL-AF9+ leukemia cells show retarded recurrence of leukemia cells in peripheral blood. Treating MLL-AF9+ leukemia mice with HDAC3-selective inhibitor RGFP966 phenocopies the effects of Hdac3 depletion. Moreover, HDAC3 suppression enhances the response of AML to conventional chemotherapy in vivo, reducing the leukemia burden and further prolonging the survival period of leukemia mice. Since the clinical usage of HDAC inhibitors that target multiple HDAC isoforms has been hampered by dose limiting toxicities, our data suggest the isoform-specific HDAC inhibitor would be a good choice, especially considering that HDAC expression may be upregulated following chemotherapy treatment. Future experimental and clinical studies to investigate the effect of isoform-specific HDAC inhibitors and conventional chemotherapy combination are warranted.
In sum, our study identifies a mechanism by which the HDAC3 regulates AKT activity and mediates DNA damage repair in response to chemotherapy exposure. This indicates disrupting the HDAC3/AKT axis by HDAC inhibitors and combing these inhibitors with cytotoxic agents may constitute a promising strategy to overcome chemotherapy resistance in AML treatment.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files.
References
Dohner H, Weisdorf DJ, Bloomfield CD . Acute myeloid leukemia. N Engl J Med 2015; 373: 1136–1152.
Ferrara F, Schiffer CA . Acute myeloid leukaemia in adults. Lancet 2013; 381: 484–495.
Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M . Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood 2003; 102: 972–980.
Grandage VL, Gale RE, Linch DC, Khwaja A . PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia 2005; 19: 586–594.
Min YH, Eom JI, Cheong JW, Maeng HO, Kim JY, Jeung HK et al. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: its significance as a prognostic variable. Leukemia 2003; 17: 995–997.
West KA, Castillo SS, Dennis PA . Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist Updat 2002; 5: 234–248.
O'Gorman DM, McKenna SL, McGahon AJ, Knox KA, Cotter TG . Sensitisation of HL60 human leukaemic cells to cytotoxic drug-induced apoptosis by inhibition of PI3-kinase survival signals. Leukemia 2000; 14: 602–611.
Brandwein JM, Hedley DW, Chow S, Schimmer AD, Yee KW, Schuh AC et al. A phase I/II study of imatinib plus reinduction therapy for c-kit-positive relapsed/refractory acute myeloid leukemia: inhibition of Akt activation correlates with complete response. Leukemia 2011; 25: 945–952.
Bozulic L, Surucu B, Hynx D, Hemmings BA . PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell 2008; 30: 203–213.
Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FM . Therapeutic opportunities within the DNA damage response. Nat Rev Cancer 2015; 15: 166–180.
Roos WP, Thomas AD, Kaina B . DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer 2016; 16: 20–33.
West AC, Johnstone RW . New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 2014; 124: 30–39.
Liu Y, Cheney MD, Gaudet JJ, Chruszcz M, Lukasik SM, Sugiyama D et al. The tetramer structure of the Nervy homology two domain, NHR2, is critical for AML1/ETO's activity. Cancer Cell 2006; 9: 249–260.
Lahue RS, Frizzell A . Histone deacetylase complexes as caretakers of genome stability. Epigenetics 2012; 7: 806–810.
Eot-Houllier G, Fulcrand G, Magnaghi-Jaulin L, Jaulin C . Histone deacetylase inhibitors and genomic instability. Cancer Lett 2009; 274: 169–176.
Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010; 18: 436–447.
Lane AA, Chabner BA . Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 2009; 27: 5459–5468.
Yang CZ, Luan FJ, Xiong DS, Liu BR, Xu YF, Gu KS . Multidrug resistance in leukemic cell line K562/A02 induced by doxorubicin. Zhongguo Yao Li Xue Bao 1995; 16: 333–337.
Zuber J, Radtke I, Pardee TS, Zhao Z, Rappaport AR, Luo W et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev 2009; 23: 877–889.
Long J, Chang L, Shen Y, Gao WH, Wu YN, Dou HB et al. Valproic acid ameliorates graft-versus-host disease by downregulating Th1 and Th17 cells. J Immunol 2015; 195: 1849–1857.
Toulany M, Kehlbach R, Florczak U, Sak A, Wang S, Chen J et al. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol Cancer Ther 2008; 7: 1772–1781.
Krivtsov AV, Armstrong SA . MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 2007; 7: 823–833.
Milne TA . Mouse models of MLL leukemia: recapitulating the human disease. Blood 2017; 129: 2217–2223.
Scholz C, Weinert BT, Wagner SA, Beli P, Miyake Y, Qi J et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat Biotechnol 2015; 33: 415–423.
Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell 2008; 30: 61–72.
Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V et al. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci Signal 2011; 4: ra46.
Watson PJ, Fairall L, Santos GM, Schwabe JW . Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012; 481: 335–340.
Arrar M, de Oliveira CA, McCammon JA . Inactivating mutation in histone deacetylase 3 stabilizes its active conformation. Protein Sci 2013; 22: 1306–1312.
Adams H, Fritzsche FR, Dirnhofer S, Kristiansen G, Tzankov A . Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin's lymphoma. Expert Opin Ther Targets 2010; 14: 577–584.
Muller BM, Jana L, Kasajima A, Lehmann A, Prinzler J, Budczies J et al. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer—overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013; 13: 215.
Moreno DA, Scrideli CA, Cortez MA, de Paula Queiroz R, Valera ET, da Silva Silveira V et al. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br J Haematol 2010; 150: 665–673.
Wang JC, Kafeel MI, Avezbakiyev B, Chen C, Sun Y, Rathnasabapathy C et al. Histone deacetylase in chronic lymphocytic leukemia. Oncology 2011; 81: 325–329.
Mithraprabhu S, Kalff A, Chow A, Khong T, Spencer A . Dysregulated class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014; 9: 1511–1520.
Quintas-Cardama A, Santos FP, Garcia-Manero G . Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia 2011; 25: 226–235.
Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA non-homologous end-joining. Nat Struct Mol Biol 2010; 17: 1144–1151.
Groselj B, Sharma NL, Hamdy FC, Kerr M, Kiltie AE . Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br J Cancer 2013; 108: 748–754.
Xu N, Lao Y, Zhang Y, Gillespie DA . Akt: a double-edged sword in cell proliferation and genome stability. J Oncol 2012; 2012: 951724.
Toulany M, Lee KJ, Fattah KR, Lin YF, Fehrenbacher B, Schaller M et al. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol Cancer Res 2012; 10: 945–957.
Deng R, Tang J, Ma JG, Chen SP, Xia LP, Zhou WJ et al. PKB/Akt promotes DSB repair in cancer cells through upregulating Mre11 expression following ionizing radiation. Oncogene 2011; 30: 944–955.
Brown KK, Montaser-Kouhsari L, Beck AH, Toker A . MERIT40 is an Akt substrate that promotes resolution of DNA damage induced by chemotherapy. Cell Rep 2015; 11: 1358–1366.
Gupta M, Ansell SM, Novak AJ, Kumar S, Kaufmann SH, Witzig TE . Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood 2009; 114: 2926–2935.
Bradley EW, Carpio LR, Westendorf JJ . Histone deacetylase 3 suppression increases PH domain and leucine-rich repeat phosphatase (Phlpp)1 expression in chondrocytes to suppress Akt signaling and matrix secretion. J Biol Chem 2013; 288: 9572–9582.
Yang XJ, Seto E . Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell 2008; 31: 449–461.
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325: 834–840.
Kramer OH, Knauer SK, Greiner G, Jandt E, Reichardt S, Guhrs KH et al. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev 2009; 23: 223–235.
Gupta M, Han JJ, Stenson M, Wellik L, Witzig TE . Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: implications for therapy. Leukemia 2012; 26: 1356–1364.
Minami J, Suzuki R, Mazitschek R, Gorgun G, Ghosh B, Cirstea D et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia 2014; 28: 680–689.
Rebecchi MJ, Scarlata S . Pleckstrin homology domains: a common fold with diverse functions. Annu Rev Biophys Biomol Struct 1998; 27: 503–528.
Matthews GM, Mehdipour P, Cluse LA, Falkenberg KJ, Wang E, Roth M et al. Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood 2015; 126: 2392–2403.
Acknowledgements
We thank all fellows from Research Center for Experimental Medicine for their technical assistance. In addition, we thank all researchers from Shanghai Institute of Hematology for kind advice and the supply of research reagents. This Study is supported by the Ministry of Science and Technology of China (S2016G9074) and National Natural Science Foundation of China (81270615, 81270621, 81123005).
Author contributions
JL, WYF and LC performed the experiments, analyzed the data and helped to write the manuscript; WHG performed experiments and helped with animal experiments; YS and MYJ helped with animal experiments; YXZ helped to collect primary samples; YW, HBD and WJZ provided advice and revised the paper; JZ provided material and advice, and revised the paper; ABL, JML and JH designed the overall concept, analyzed the data and wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Long, J., Fang, W., Chang, L. et al. Targeting HDAC3, a new partner protein of AKT in the reversal of chemoresistance in acute myeloid leukemia via DNA damage response. Leukemia 31, 2761–2770 (2017). https://doi.org/10.1038/leu.2017.130
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.130
- Springer Nature Limited
This article is cited by
-
Post-translational modification of CDK1–STAT3 signaling by fisetin suppresses pancreatic cancer stem cell properties
Cell & Bioscience (2023)
-
Targeting HDAC3 to overcome the resistance to ATRA or arsenic in acute promyelocytic leukemia through ubiquitination and degradation of PML-RARα
Cell Death & Differentiation (2023)
-
Disruption of mitochondrial oxidative phosphorylation by chidamide eradicates leukemic cells in AML
Clinical and Translational Oncology (2023)
-
HDAC11 mediates the ubiquitin-dependent degradation of p53 and inhibits the anti-leukemia effect of PD0166285
Medical Oncology (2023)
-
The NCOR-HDAC3 co-repressive complex modulates the leukemogenic potential of the transcription factor ERG
Nature Communications (2023)