
Abstract
We studied acute myeloid leukemia (AML) patients with lympho-myeloid clonal hematopoiesis (LM-CH), defined by the presence of DNA methyltransferase 3A (DNMT3A) mutations in both the myeloid and lymphoid T-cell compartment. Diagnostic, complete remission (CR) and relapse samples were sequenced for 34 leukemia-related genes in 171 DNMT3A mutated adult AML patients. AML with LM-CH was found in 40 patients (23%) and was associated with clonal hematopoiesis of indeterminate potential years before AML, older age, secondary AML and more frequent MDS-type co-mutations (TET2, RUNX1 and EZH2). In 82% of AML patients with LM-CH, the preleukemic clone was refractory to chemotherapy and was the founding clone for relapse. Both LM-CH and non-LM-CH MRD-positive AML patients who achieved CR had a high risk of relapse after 10 years (75% and 75%, respectively) compared with patients without clonal hematopoiesis in CR with negative MRD (27% relapse rate). Long-term survival of patients with LM-CH was only seen after allogeneic hematopoietic stem cell transplantation (HSCT). We define AML patients with LM-CH as a distinct high-risk group of AML patients that can be identified at diagnosis through mutation analysis in T cells and should be considered for HSCT.
Similar content being viewed by others

Introduction
The origin of acute myeloid leukemia (AML) is still poorly understood. Our current understanding of AML development is based on the concept that multiple gene mutations and chromosomal aberrations occur in a multistep process leading to epigenetic as well as genetic alterations of genes and their functions. This aspect can be summarized as clonal evolution of AML.1, 2 The majority of AML patients harbor two to six recurrent somatic mutations in the genome of their leukemic cells,1 which is a considerably lower number compared with most solid tumors.3, 4 An important aspect of clonal evolution is that leukemia-related mutations can occur in healthy people resulting in a predisposition for hematologic malignancies.5, 6, 7 This process is now termed as clonal hematopoiesis of indeterminate potential (CHIP).8, 9, 10 Recently, a very important finding was the observation that mutations in DNA methyltransferase 3A (DNMT3A) can be present in T cells of some patients with DNMT3A mutations in their AML blasts.11 Interestingly, in the DNMT3A mutated T cells, NPM1 was not found to be mutated in those patients with NPM1 mutations in AML blasts.11 This phenomenon indicates the clonal expansion of an immature hematopoietic stem/progenitor cell that affects both myeloid and lymphoid lineages before the onset of leukemia, which may be transformed by a leukemia-specific mutation like NPM1.
DNMT3A mutations are among the most frequent mutations in AML and are suggested to have a negative prognostic impact on patients' outcome.12, 13, 14, 15, 16, 17, 18 Very little is known at this point what impact a history of lympho-myeloid clonal hematopoiesis (LM-CH) may have on the clinical picture and course of AML. We also do not know whether the sensitivity to intensive chemotherapy and long-term remissions are different in patients with a history of LM-CH. Thus, the purpose of this study was to analyze the presence of DNMT3A mutations in T cells as a marker for a history of preleukemic LM-CH in a large cohort of DNMT3A mutated AML patients to gain novel information about the clinical and molecular impact of LM-CH in patients with AML.
Patients and methods
Patient samples
A total of 1482 adult AML patients were screened for DNMT3A mutations. One hundred and seventy-one adult AML patients (aged 18–87 years) were found to be DNMT3A mutated and had available diagnostic bone marrow or peripheral blood cell samples. All patients entered multicenter treatment trials AML SHG 0199 (ClinicalTrials Identifier NCT00209833, June 1999 to September 2004, n=44), AML SHG 0295 (February 1995 to May 1999, n=14) or the AMLSG Biology and outcome study (n=113). Patients with PML-RARA or t(15;17) positive AML were excluded from these trials. One hundred and forty patients received intensive therapy,19, 20 21 patients received non-intensive therapy and for 10 patients the therapy is unknown. Written informed consent was obtained according to the Declaration of Helsinki, and the studies were approved by the institutional review board of Hannover Medical School. For 20 out of 71 relapsed patients, a sample at the time of relapse was available. Fifty-five patients had follow-up samples at the time of remission yielding a total of 168 follow-up samples. For 18 patients with non-LM-CH with persistent DNMT3A mutation in remission, AML-matched diagnosis and complete remission (CR) samples were available for mutational analysis with the myeloid panel to detect additional mutations in CR.
From 13 patients (including 2 patients from the LM-CH group), DNA was available from hair follicles serving as a germline control. We had two DNMT3A mutated patients with a blood/bone marrow sample 2–5 years before the diagnosis of AML. At that time both the patients had no malignancy.
Sorting of CD3-positive T cells and hematopoietic progenitor cells
Bone marrow from AML patients were stained with a PE mouse anti-human CD3 (UCHT1) antibody from BD Biosciences (Heidelberg, Germany) and sorted at the cell-sorting facility of Hannover Medical School. Strict purity control measures were administered. Diagnostic AML samples were used for T-cell sorting (CD3+CD11b−CD14−CD33−). The median purity of T cells was 96.8%. A sample was only called DNMT3A mutated in T cells if the mutated allelic burden was higher than the measured percentage of contaminating leukemic cells.
Cytogenetic and molecular analysis
Pretreatment samples from all the patients were studied by conventional banding analysis.21 Only DNMT3A mutated AML patients were selected for our study and DNMT3A mutational status was investigated as previously reported.13 Sequencing was performed on the MiSeq sequencer. Library preparation of samples at the time of diagnosis (n=171) and relapse (n=20) were performed with the TruSight myeloid sequencing panel (Illumina, San Diego, CA, USA). We considered 34 entire genes or hotspot gene regions involved in leukemia for further analysis (Supplementary Table S1). DNMT3A mutational analysis in T cells and follow-up samples was performed by targeted sequencing of DNMT3A with customized NGS primers. For DNMT3A, a primary PCR was performed with customized primers (Supplementary Table S3) and PCR conditions described previously.13
The Illumina Miseq reagent kit v3 (Illumina) was used for library preparation. Sequencing was performed on the MiSeq sequencer aiming for a high coverage per sample (50 000–100 000 average number of aligned reads per sample). This allowed ultra-deep sequencing to gain a high sensitivity. DNMT3A mutations were called in T cells when the variant allele frequency (VAF) was ⩾1%. Using conventional fragment analysis, all the patients were additionally screened for NPM1 and FLT3 mutations. Sanger sequencing of DNMT3A was performed for DNA from hair follicles. The mean coverage of all amplicons was 3262 reads per amplicon (range 101–23 004) in the targeted resequencing of 34 different genes and 82 587 aligned reads per amplicon in the targeted resequencing of DNMT3A (follow-up samples and CD3-positive cells).
Bioinformatic and statistical analysis
The definition of CR, overall survival and relapse-free survival followed recommended criteria.22, 23 Statistical test results are provided for explorative purposes.
In the bioinformatic analysis, short reads from targeted resequencing of 34 genes were aligned to the hg19 reference genome using bwa (mem, paired-end).24 Local realignment around indels was done using local realignment tools from the Genome Analysis Tool Kit.25 Further details are listed in the supplement.
Results
AML with LM-CH occurs in 23% of DNMT3A mutated AML patients and is associated with secondary AML and MDS-related mutations
A total of 171 AML patients with DNMT3A mutation were included in our analysis (143 with R882 and 28 with other DNMT3A mutations, see Supplementary Figure S1, Supplementary Table S2). The somatic origin of DNMT3A mutations was confirmed in the germline DNA from hair follicles in 13 patients, that is, DNMT3A was wild-type in germline DNA (including two with mutation in T cells).
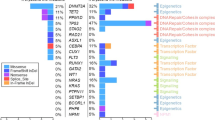
DNMT3A was mutated in both mononuclear and T cells in 40 patients (23%; termed here AML with LM-CH), while it was mutated only in mononuclear cells in 131 patients (77%; termed AML without LM-CH). The mean allele burden was 43.2% in mononuclear cells and 15.7% in T cells of LM-CH patients, while it was 44.6% in mononuclear cells and by definition 0% in T cells in non-LM-CH patients (Figure 1a). In AML patients with LM-CH, the DNMT3A VAF in mononuclear cells did not correlate with VAF in T cells (Pearson correlation R=0.075, Supplementary Figure S2). Comparing the clinical characteristics of AML patients with and without LM-CH revealed that significantly more patients in the LM-CH cohort had secondary AML (P=0.009) and were older at diagnosis (P=0.018, Figures 1b and c, Table 1). There was no difference in the distribution to the three different trials between the patients with or without LM-CH (P=0.22). Other clinical parameters including cytogenetic risk group and blood cell counts did not significantly differ between the two groups (Table 1). The patients with LM-CH were more likely to show a DNMT3A mutation affecting the mutational hotspot R882 of DNMT3A (P=0.007, Table 1, Supplementary Figure S1). Analyzing the mutational profile of 34 genes revealed that the number of mutations per patient between these two groups was similar (mean 3.9 vs 3.5 mutations in patients without vs with LM-CH, P=0.19). The most frequently mutated genes in the cohort were NPM1, FLT3-ITD (internal tandem duplication of the FLT3 gene), TET2 and IDH2 (Supplementary Figure S3 and Supplementary Table S2). Patients with LM-CH were significantly more likely to harbor mutations in TET2 (P=0.008), RUNX1 (P=0.041) and EZH2 (P=0.05), but less likely to have mutated NPM1 (P=0.003) or FLT3-ITD (P=0.037, Figure 1d, Table 1). No significant difference was seen regarding the frequency of the remaining 29 genes (Table 1, Figure 1d). We clustered patients with at least one co-mutation based on their mutation profile by unsupervised hierarchical clustering and found AML patients with LM-CH enriched in a cluster defined by secondary AML-type mutations (ASXL1, RUNX1, TET2) and the two splicing genes SF3B1 and SRSF2, while AML patients without LM-CH clustered with NPM1, FLT3, IDH1, IDH2 and cohesin gene mutations (Figure 1e). Thus, genes related to de novo AML are less common, while MDS-related gene mutations are more common in the LM-CH group. We also compared the VAF of mutated genes between AML patients with and without LM-CH. Confirming our previous result, secondary AML-type mutations had a higher VAF in LM-CH compared with non-LM-CH patients (ASXL1, TP53, EZH2, SF3B1, U2AF1, ZRSR2 and SMC3; Supplementary Figure S4), suggesting that these mutations can initiate AML from a DNMT3A mutated preleukemic clone. Overall, TET2, DNMT3A and NPM1 had the highest VAF (Supplementary Figure S4), confirming that these are driver or preleukemic mutations. In summary, we describe a subgroup of AML that developed on the basis of LM-CH and can be distinguished at diagnosis through mutation analysis in T cells. This patient group constitutes 23% of DNMT3A mutated patients and is associated with secondary AML-type characteristics.

AML with LM-CH is associated with secondary AML and MDS-type mutations. (a) Variant allele frequency of DNMT3A mutations at diagnosis in mononuclear and sorted T cells for AML patients with LM-CH and without LM-CH. (b) Comparison of median age at diagnosis in AML patients with and without LM-CH. (c) Frequency of de novo and secondary AML in patients with and without LM-CH. (d) Mutational landscape of patients with LM-CH or without LM-CH as identified by targeted sequencing analysis.37 (e) Unsupervised hierarchical clustering of all 158 patients based on genes that are mutated in at least 10 patients. LM-CH patients (yellow) cluster on the left side and are enriched for MDS-type mutations.
The preleukemic LM-CH clone is refractory to chemotherapy
We followed the development of the mutation burden of DNMT3A during the course of the disease in 168 remission samples from 55 patients and 20 relapse samples with ultra-deep sequencing. Fifteen (27%) of 55 patients lost the DNMT3A mutation in remission, whereas 40 (73%) patients remained DNMT3A mutated in remission (cut-off ⩾1%). Of the 11 patients with LM-CH who had available follow-up samples, 9 remained mutated during remission (82%) and 2 patients (18%) lost the mutation in remission. These two patients had a VAF of 11% and 27% in T cells at diagnosis, received a hematopoietic stem cell transplantation (HSCT) in first CR, and are alive after long-term follow-up. In the non-LM-CH cohort 13 of 44 patients lost the mutation (30%) and 31 patients with follow-up samples (70%) remained mutated during remission (P=0.45; Figure 2a). Examples of the DNMT3A mutation load during the course of the disease are shown in Supplementary Figure S5. In LM-CH patients, the DNMT3A VAF in T cells largely followed the VAF in MNCs, although at a lower level. Figure 2b shows the summary of the DNMT3A mutational load in AML patients with and without LM-CH from diagnosis to CR (Figure 2b). In LM-CH patients, DNMT3A levels were similar between diagnosis and CR at a mean level of 36.2% (n=7 compared with 46.7% at the time of diagnosis, n=39, P=0.12), while they were much lower at CR compared with diagnosis in non-LM-CH patients (42.8% at diagnosis and 10% at CR, P<0.001, Figure 2b). These data suggest that the preleukemic LM-CH clone is resistant to induction chemotherapy in the majority of patients and persists at high levels in CR, although the morphologically defined leukemic blast clone had been reduced below 5% (CR).

The preleukemic LM-CH clone is refractory to chemotherapy. (a) Schematic overview of DNMT3A mutated AML patients according to their DNMT3A mutation status in T cells at diagnosis and in mononuclear cells at CR. (b and c) Comparison of mean variant allelic frequency of DNMT3A at the time of diagnosis and CR (b) as well as CR and relapse (c).
We sequenced 18 patients with the myeloid panel at the time of CR, who had at least one concomitant mutation at diagnosis, no LM-CH, but a persistent DNMT3A mutation in CR, to monitor the course of the concomitant mutations. In 15 patients NPM1 was lost in CR, in 2 patients each IDH1, TET2, NRAS, TP53, SMC3, RUNX1 and ETV6 were lost, and in 1 patient each RAD21, SMC1A, SF3B1, U2AF1, ZRSR2, WT1 and PTPN11 were lost. As expected, no new mutations were gained in CR. Interestingly, in three patients, one additional gene remained mutated in CR (IDH1, IDH2 or TET2). In each case, the course of the VAF from diagnosis to CR was similar for DNMT3A and the respective additional mutation. These findings suggest that the majority of concomitant mutations are part of the leukemic clone, while some mutations in genes with epigenetic impact may be part of the preleukemic clone.
Next, we compared the DNMT3A allele burden between CR and relapse (Figure 2c). In LM-CH patients, the allele burden remained high between CR and relapse (43% vs 47%, n=3, P=0.02), whereas it considerably increased from CR to relapse in AML patients without LM-CH (from 5% to 29%, n=13, P<0.001; Figure 2c). In LM-CH patients, the allele burden at relapse was comparable to that at diagnosis (P=0.35), whereas patients without LM-CH had a lower allele burden at relapse compared with diagnosis (P=0.07; Supplementary Figure S6).
Of all the patients undergoing HSCT, only patients with full chimerism lost the DNMT3A mutation (patients 3, 9, 12 and 17; Supplementary Figure S5). These data suggest that the preleukemic clone has engaged almost the entire hematopoiesis in LM-CH patients, whereas non-LM-CH patients may have had only a minor or no preleukemic clone before the onset of AML.
AML with LM-CH is preceded by CHIP
Intriguingly, in two AML patients with LM-CH with samples 2 and 5 years before AML diagnosis, we found the identical DNMT3A mutation already in these early samples (Figure 3a). At that time, both the patients had normal blood counts (Figure 3b) and did not have evidence of a hematological malignancy. Thus, the retrospective diagnosis of CHIP can be made for both patients years before AML development. We therefore propose that LM-CH is an indicator of prior CHIP, which can be retrospectively diagnosed at the time of AML diagnosis.

AML with LM-CH is preceded by CHIP. (a) Comparison of mean variant allelic frequency of DNMT3A years before AML diagnosis in healthy condition (CHIP), at the time of diagnosis and during disease course in two patients with LM-CH and available samples before diagnosis. (b) Blood counts of the two patients from a at the time of CHIP and AML diagnosis.
AML patients with LM-CH have a high risk of relapse
We next compared the mutational profile between available diagnosis and matched relapse samples and found a slightly decreased number of mutations covered by our myeloid panel at the time of relapse (3.6 vs 3.2, Supplementary Figure S7). DNMT3A and IDH2 mutations were the most stable markers (95–100% stable) between diagnosis and relapse. Less stable genes were NPM1 (80% stability), FLT3-ITD (75% stability), IDH1 (67% stability) and NRAS (40% stability, Figure 4a).

AML patients with LM-CH have a high risk of relapse. (a) Mutation pattern of paired diagnosis and relapse samples of 20 DNMT3A mutated patients. (b) Cumulative incidence of relapse (CIR) in patients with LM-CH, patients without LM-CH who are DNMT3A mutated in CR (M-CH) and patients without LM-CH who are DNMT3A wild-type in CR.
To better understand the risk of relapse in DNMT3A mutated AML patients, we evaluated the cumulative incidence of relapse in intensively treated AML patients who achieved CR and had follow-up information available. LM-CH patients were included independent of sample availability at CR because clonal hematopoiesis had already been proven at diagnosis. However, non-LM-CH patients were only included if a remission sample was available for DNMT3A mutation analysis (n=64). Twenty-one patients had AML with LM-CH. Of the remaining patients without LM-CH, 30 remained DNMT3A mutated in CR (70%), suggestive of pre-existing myeloid clonal hematopoiesis, M-CH, and 13 lost the DNMT3A mutation in CR (30%). Patients with LM-CH and M-CH had a very high cumulative incidence of relapse at 10 years (75 and 75%, respectively), while non-LM-CH patients who were DNMT3A negative in CR had a lower cumulative incidence of relapse of 27% (P=0.18, Figure 4b). All seven censored LM-CH patients had received allogeneic HSCT in CR1 (100%), 4 of the 10 censored non-LM-CH patients who were DNMT3A mutated in CR had received allogeneic HSCT in CR1 (40%), and 5 of the 10 censored non-LM-CH patients who were DNMT3A wild-type in CR had received allogeneic HSCT in CR1 (50%), suggesting that in LM-CH patients allogeneic HSCT is an essential treatment to achieve long-term relapse-free survival.
Long-term survival of AML patients with LM-CH requires allogeneic stem cell transplantation
We next assessed the clinical outcome of all patients who received intensive induction chemotherapy and had clinical follow-up information (28 LM-CH and 110 non-LM-CH patients). Median follow-up time for these patients was 5.94 years (range, 0.06 to 15.64 years). Complete remission was achieved in 75% of LM-CH and 85% of non-LM-CH patients (P=0.23).
Patients with LM-CH had similar overall survival compared with patients with non-LM-CH (5-year overall survival, 35% and 44%, respectively, P=0.95, Figure 5a). Relapse-free survival was similar in LM-CH and non-LM-CH patients (5-year relapse-free survival, 33% and 36%, P=0.8, Figure 5b). HSCT was performed in 61% of LM-CH and 56% of non-LM-CH patients (P=0.62). All 11 surviving LM-CH patients had received HSCT; of the 47 surviving non-LM-CH patients, 27 had received HSCT and 20 conventional consolidation therapy (Figure 5c). We further evaluated the efficacy of HSCT in CR1 in patients with and without LM-CH. Five-year overall survival was 78% in LM-CH and 57% in non-LM-CH patients receiving HSCT (P=0.17, Figure 5d), suggesting that allogeneic HSCT in CR1 is an effective treatment in LM-CH patients.

Long-term survival of AML patients with LM-CH requires allogeneic stem cell transplantation. (a) Overall survival in intensively treated AML patients with or without LM-CH. (b) Relapse-free survival in intensively treated AML patients with or without LM-CH. (c) Proportion of surviving patients with or without LM-CH who received either chemotherapy alone or an allogeneic transplantation during their course of treatment. Only patients who received intensive induction chemotherapy are considered. (d) Overall survival of AML patients with or without LM-CH who were treated with intensive induction chemotherapy, achieved CR and were transplanted in CR1
Discussion
Clonal hematopoiesis as defined by the presence of preleukemic mutations in DNMT3A or other genes in healthy individuals has been identified as an age-related phenomenon, which is associated with a transformation rate of 0.5–1% per year.5, 6, 7 We have taken advantage of the fact that some AML patients show DNMT3A mutations in their T cells to better understand the role of clonal hematopoiesis as a preleukemic disease.26, 27, 28, 29 In our study, we identified patients with clonal hematopoiesis that preceeded the AML phase already at diagnosis by mutation analysis of their T cells. In two patients, we could prove that DNMT3A mutations were present years before the onset of AML, demonstrating that these patients had CHIP before AML. Most of these patients remained DNMT3A mutated in morphologic CR, often at high VAF, showing that the DNMT3A mutated clone (i) was the founding clone of AML, (ii) existed as an AML-independent clone and (iii) was resistant to induction chemotherapy in most patients. Currently, there are no data on the therapeutic responsiveness of DNMT3A mutated clonal hematopoiesis. However, by determining the presence and extent of clonal hematopoiesis before and after chemotherapy, we might be able to identify drugs that can inhibit this clonally expanded hematopoiesis.
Of our AML patients without LM-CH who achieved morphologic CR, 70% maintained the DNMT3A mutation in mononuclear cells and 30% lost the mutation. Evidence of DNMT3A in CR likely represents myeloid clonal hematopoiesis (M-CH), which distinguishes these patients from LM-CH patients and patients without evidence of clonal hematopoiesis in CR. Our study shows that M-CH patients have a less expanded clonal hematopoiesis than LM-CH patients: LM-CH patients had no significant reduction in the DNMT3A VAF during CR, whereas patients without LM-CH showed a significant reduction in the VAF in CR. As both groups were characterized by a high rate of relapse unless transplanted, and as the DNMT3A mutated clone was very stable at relapse, the preleukemic clone needs to be inhibited to prevent relapse in AML patients, which in our study was only possible by HSCT.
We next evaluated whether AML derived from LM-CH is different than AML without LM-CH. Patients with LM-CH were older and more frequently had secondary AML. In line with this, genes commonly mutated in MDS such as the epigenetic modifiers TET2 and EZH2 as well as the transcription factor RUNX1(ref. 30) were more frequently mutated in AML patients with LM-CH, whereas de novo AML mutations like NPM1 and Flt3-ITD3 were less frequent. This is an important distinction as DNMT3A mutations in general are associated with FLT3-ITD and NPM1 and not associated with TET2.3, 12, 13, 14, 16, 18, 27, 31, 32, 33, 34, 35, 36
From a clinical point of view, our study underscores that DNMT3A is not reliable as MRD marker, although clearance of DNMT3A in CR seems to be associated with a lower risk of relapse. In three patients, we found a TET2, IDH1 or IDH2 mutation in CR samples together with mutated DNMT3A, suggesting that the preleukemic clone may contain several mutations. However, the majority of concomitant mutations disappeared in CR including mutations in IDH1 and TET2. Mutation analysis in T cells at the time of diagnosis may be useful in DNMT3A mutated patients to identify, as early as possible, patients with LM-CH, who may benefit most from allogeneic HSCT (although this should be evaluated prospectively). In addition, mutation analysis in T cells is the only method to identify LM-CH patients for all patients who do not achieve CR.
In summary, our data demonstrate that AML with DNMT3A mutations is derived from LM-CH in approximately a quarter of the patients. CHIP is likely to precede AML with LM-CH. AML patients with LM-CH are more likely to be older and have secondary AML, and the mutational profile is more likely to be characterized by MDS-related mutations (Supplementary Figure S8). Although initial response to therapy is good, the preleukemic CHIP clone is refractory to chemotherapy and long-term survival of AML patients with LM-CH can only be achieved with HSCT. Thus, mutation analysis in sorted T cells already at diagnosis of AML can instruct the search for a suitable stem cell donor.
References
Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012; 150: 264–278.
Chen J, Odenike O, Rowley JD . Leukaemogenesis: more than mutant genes. Nat Rev Cancer 2010; 10: 23–36.
Network TCGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368: 2059–2074.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 8 366: 883–892.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014; 371: 2488–2498.
Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371: 2477–2487.
Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014; 20: 1472–1478.
Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015; 126: 9–16.
Heuser M, Yun H, Berg T, Yung E, Argiropoulos B, Kuchenbauer F et al. Cell of Origin in AML: susceptibility to MN1-induced transformation is regulated by the MEIS1/AbdB-like HOX protein complex. Cancer Cell 2011; 20: 39–52.
Goardon N, Marchi E, Atzberger A, Quek L, Schuh A, Soneji S et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011; 19: 138–152.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014; 506: 328–333.
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010; 363: 2424–2433.
Thol F, Damm F, Lüdeking A, Winschel C, Wagner K, Morgan M et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol 2011; 29: 2889–2896.
Marcucci G, Metzeler KH, Schwind S, Becker H, Maharry K, Mrozek K et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol 2012; 30: 742–750.
Renneville A, Boissel N, Nibourel O, Berthon C, Helevaut N, Gardin C et al. Prognostic significance of DNA methyltransferase 3 A mutations in cytogenetically normal acute myeloid leukemia: a study by the Acute Leukemia French Association. Leukemia 2012; 26: 1247–1254.
Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012; 366: 1079–1089.
Ribeiro AF, Pratcorona M, Erpelinck-Verschueren C, Rockova V, Sanders M, Abbas S et al. Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood 2012; 119: 5824–5831.
Gaidzik VI, Schlenk RF, Paschka P, Stolzle A, Spath D, Kuendgen A et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: results of the AML Study Group (AMLSG). Blood 2013; 121: 4769–4777.
Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008; 358: 1909–1918.
Heil G, Krauter J, Raghavachar A, Bergmann L, Hoelzer D, Fiedler W et al. Risk-adapted induction and consolidation therapy in adults with de novo AML aged </= 60 years: results of a prospective multicenter trial. Ann Hematol 2004; 83: 336–344.
Mitelman F . ISCN (1995): an International System for Human Cytogenetic Nomenclature. SKarger: Basel, Switzerland, 1995.
Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 2003; 21: 4642–4649.
Korn EL . Censoring distributions as a measure of follow-up in survival analysis. Stat Med 1986; 5: 255–260.
Li H, Durbin R . Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010; 26: 589–595.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 2014; 25: 442–454.
Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 2012; 44: 23–31.
Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med 2015; 373: 35–47.
Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015; 518: 552–555.
Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013; 122: 3616–3627, quiz 3699.
Renneville A, Kaltenbach S, Clappier E, Collette S, Micol JB, Nelken B et al. Wilms tumor 1 (WT1) gene mutations in pediatric T-cell malignancies. Leukemia 2010; 24: 476–480.
Scourzic L, Couronne L, Pedersen MT, Della Valle V, Diop M, Mylonas E et al. DNMT3A mutant and Tet2 inactivation cooperate in the deregulation of DNA methylation control to induce lymphoid malignancies in mice. Leukemia 2016.
Meyer SE, Qin T, Muench DE, Masuda K, Venkatasubramanian M, Orr E et al. DNMT3A haploinsufficiency transforms FLT3ITD myeloproliferative disease into a rapid, spontaneous, and fully penetrant acute myeloid leukemia. Cancer Discov 2016; 6: 501–515.
Couronne L, Bastard C, Bernard OA . TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med 2012; 366: 95–96.
Neumann M, Heesch S, Schlee C, Schwartz S, Gokbuget N, Hoelzer D et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood 2013; 121: 4749–4752.
Huether R, Dong L, Chen X, Wu G, Parker M, Wei L et al. The landscape of somatic mutations in epigenetic regulators across 1000 paediatric cancer genomes. Nat Commun 2014; 5: 3630.
Thiede C, Bullinger L, Hernández-Rivas JM, Heuser M, Preudhomme C, Best S et al. Results of the ‘Evaluation of NGS in AML-Diagnostics (ELAN)’ Study—an Inter-Laboratory Comparison Performed in 10 European Laboratories ASH Abstract 2014.
Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood 1998; 92: 2322–2333.
Acknowledgements
We are indebted to all the patients and contributing doctors. We thank the staff of the Central Animal Facility and Matthias Ballmaier from the Cell Sorting Core Facility (supported in part by the Braukmann-Wittenberg-Herz-Stiftung and the Deutsche Forschungsgemeinschaft) of Hannover Medical School, Vishwas Sharma, Michael Morgan, Silke Glowotz, Nicole Ernst and Kerstin Görlich, for their support on this project. This study was supported by the German Federal Ministry of Education and Research grant 01EO0802 (IFB-Tx), grants 110284, 110287, 110292 and 111267 from Deutsche Krebshilfe; grant DJCLS R13/14 from the Deutsche José Carreras Leukämie-Stiftung e.V; DFG grant HE 5240/5-1 and HE 5240/6-1 and BU 1339/8-1; an ERC grant under the European Union’s Horizon 2020 research and innovation program (No. 638035) and by grants from Dieter-Schlag Stiftung.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Rights and permissions
About this article

Cite this article
Thol, F., Klesse, S., Köhler, L. et al. Acute myeloid leukemia derived from lympho-myeloid clonal hematopoiesis. Leukemia 31, 1286–1295 (2017). https://doi.org/10.1038/leu.2016.345
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2016.345
- Springer Nature Limited
This article is cited by
-
Significance of targeting DNMT3A mutations in AML
Annals of Hematology (2024)
-
Hallmarks of T cell aging
Nature Immunology (2021)
-
Distinction of lymphoid and myeloid clonal hematopoiesis
Nature Medicine (2021)
-
Somatic mutations in lymphocytes in patients with immune-mediated aplastic anemia
Leukemia (2021)
-
Engaging chromatin: PRC2 structure meets function
British Journal of Cancer (2020)