
Abstract
Ataxia telangiectasia-mutated (ATM) kinase is a master regulator of the DNA damage response. ATM is frequently inactivated in human B-cell non-Hodgkin lymphomas, including ~50% of mantle cell lymphomas (MCLs) characterized by ectopic expression of CyclinD1. Here we report that early and robust deletion of ATM in precursor/progenitor B cells causes cell autonomous, clonal mature B-cell lymphomas of both pre- and post-germinal center (GC) origins. Unexpectedly, naive B-cell-specific deletion of ATM is not sufficient to induce lymphomas in mice, highlighting the important tumor suppressor function of ATM in immature B cells. Although EμCyclinD1 is not sufficient to induce lymphomas, EμCyclinD1 accelerates the kinetics and increases the incidence of clonal lymphomas in ATM-deficient B-cells and skews the lymphomas toward pre-GC-derived small lymphocytic neoplasms, sharing morphological features of human MCL. This is in part due to CyclinD1-driven expansion of ATM-deficient naive B cells with genomic instability, which promotes the deletions of additional tumor suppressor genes (i.e. Trp53, Mll2, Rb1 and Cdkn2a). Together these findings define a synergistic function of ATM and CyclinD1 in pre-GC B-cell proliferation and lymphomagenesis and provide a prototypic animal model to study the pathogenesis of human MCL.
Similar content being viewed by others


Introduction
Mature B-cell lymphomas represent 85% of non-Hodgkin lymphomas (NHL) and many harbor characteristic chromosomal translocations involving the immunoglobulin (Ig) loci. These translocations result from mis-repair of DNA double-stranded breaks (DSBs) generated during V(D)J recombination or class switch recombination (CSR). V(D)J recombination assembles the productive Ig genes from germline V, D and J gene segments in immature bone marrow B cells. CSR occurs in mature B-cells in specialized structures-germinal centers (GCs) and allows expression of different antibody isotypes (for example, IgG1 or IgE) with different effector functions.1 Naive B cells also undergo somatic hypermutation (SHM) of the Ig variable region in GCs to achieve higher affinities. While V(D)J recombination and CSR are initiated by lymphocyte-specific enzymes, both reactions generate DNA DSB intermediates that are repaired by ubiquitously expressed DNA repair mechanisms. Thus, defects in DNA repair or DNA damage response lead to accumulation of DSB intermediates which, if not repaired appropriately, lead to oncogenic chromosomal translocations in human mature B-cell lymphomas by transposing the strong Ig promoters/enhancers adjacent to cellular oncogenes (for example, c-MYC, BCL2).2
Ataxia telangiectasia-mutated (ATM) kinase encodes a serine/threonine protein kinase, which is activated by DSBs to establish cell cycle checkpoints and promote DNA repair.3 Through this mechanism, ATM promotes efficient and precise DNA repair during both V(D)J recombination and CSR.4, 5, 6, 7 In the absence of ATM, physiological DSBs at Ig and T-cell receptor (TCR) loci accumulate, leading to greatly increased risk of leukemia and lymphoma in Ataxia-telangiectasia patients with germline ATM inactivation.8 ATM is also somatically inactivated in human lymphoid malignancies, most notably in ~50% of mantle cell lymphomas (MCLs), 45% of T-cell pro-lymphocytic leukemia, 10–20% of chronic lymphocytic leukemia, and 5% of diffuse large B-cell lymphomas (DLBCLs).9, 10, 11, 12 ATM-null mice recapitulate many phenotypes of Ataxia-telangiectasia patients and routinely succumb to aggressive T-cell lymphomas at 3–4 months of age with translocations involving the TCR loci, suggesting that ATM suppresses T-cell lymphomas by reducing genomic instability during V(D)J recombination.13 However, the developmental stage and the role of ATM inactivation in B-cell lymphomagenesis is not yet well understood.
Among all B-cell non-Hodgkin lymphomas, MCL has the highest frequency of bi-allelic inactivation of ATM.9 MCL is a mature B-cell lymphoma composed of small- to medium-sized lymphocytes with frequent (~30%) involvement of the gastrointestinal tract. The variable regions of the Ig are unmutated in the majority of MCL cases, consistent with a pre-GC origin. MCL is characterized by deregulated expression of D-type cyclins, especially CyclinD1, via the characteristic t(11;14) chromosomal translocation that joins CCND1 with the active Ig-heavy chain gene (IGH) promoter and enhancer. Yet, ectopic expression of wild-type (WT) CyclinD1 is insufficient to induce MCL in mice,14, 15 implying additional genetic alterations are necessary. Effective treatment for MCL is not available, and animal models recapitulating molecular features of human MCL are yet to be developed.
Here we report that progenitor B-cell-specific inactivation of ATM results in indolent and monoclonal mature B-cell lymphoproliferations that recapitulate the morphological spectrum of ATM-deficient human B-cell non-Hodgkin lymphoma, including both GC and non-GC-derived lymphomas. EμCyclinD1 transgene markedly accelerates the proliferation of genomic instable ATM-deficient naive B cells, leading to early clonal expansion of pre-GC naive B cells and pre-GC lymphomas, and identifies a cooperative effect of these two lesions in immature and naive B cells for lymphomagenesis.
Materials and methods
Mice
ATMC/C,16 EμCyclinD1 transgenic,14 CD21Cre,17 CD19Cre18 and Mb1Cre (ref. 19) mice were previously described. All experimental cohorts carry transgene (CD21Cre, EμCyclinD1) or Cre knock-in (Mb1Cre or CD19Cre) in heterozygosity. All animal experiments were performed in accordance with Columbia University Institutional Animal Care and Use Committee.
Flow cytometry and immunohistochemistry analyses
Flow cytometry analyses were performed on single cell suspensions from the spleen, bone marrow, lymph nodes as previously described.20 Antibodies include CD43(S7), CD5(53–7.3), CD138(281.2), CD21/35(7G6), CD23(B3B4), CD11b/Mac-1(M1/70) and IgK from BD Pharmingen (San Diego, CA, USA); B220(RA3–6B2), CD19(eBio1D3), CD8a(53–6.7), CD4(L3T4), CD3ɛ(145-2C11) and TCRβ(H57-597) from eBioscience (San Diego, CA, USA); Ter119 and Gr1(RB6-8C3) from BioLegend (San Diego, CA, USA); IgM and IgD from Southern Biotech (Birmingham, AL, USA) and peptide nucleic acid from Vector Labs (Burlingame, CA, USA). Immunohistochemistry staining was performed on formalin fixed, paraffin-embedded tissue sections as described previously using the following antibodies: B220, CD3, PAX5, BCL6, IRF4 and CD138.21 Immunofluorescence on paraffin-embedded tissues was performed using antibodies against Ki67 (SP6, Abcam, Cambridge, MA, USA), and B220 (RA-3B62, BD Pharmingen). Images were acquired with a Nikon Eclipse 80i microscope (Nikon, Melville, NY) and processed on NIS-Elements AR 3.10 software (Nikon).
Southern blot and western blot analyses
For Southern blot analyses, genomic DNA from the spleen, tumor and corresponding kidneys were digested with EcoRI (for JH4, c-Myc, constant region of TCRδ probe) KpnI (for 3′ ATM CKO probe) or HindIII (for the IgK probe).13, 22, 23 To avoid cross-reactivity with the EμCyclinD1 transgene that includes a fragment from JH4 to μ enhancer, we generated a new JH4 probe by PCR amplification with the following primers: 5′-TGGTGACAATTTCAGGGTCA-3′ and 5′-TTGAGACCGAGGCTAGATGC-3′. For western blots, stimulated splenic B or T cells were probed with antibodies against ATM (1:2000, MAT3, Sigma, St Louis, MO, USA), pKap1 (1:2000, Bethyl, Montgomery, TX, USA), Kap1/TIF1β (1:1000, Cell Signaling, Danvers, MA, USA), β-Actin (1:10 000, A5316, Sigma) and CyclinD1 (1:1000, Abcam).
CSR and FISH analyses
Splenic B-cells from 8–12-week-old mice were purified with anti-CD43 magnetic-activated cell sorting (MACS) beads and stimulated with LPS/IL4 for 2–4 days as described previously.20 Metaphases were obtained at day 4.5 after stimulation and stained with Cy3-conjugated peptide nucleic acid probe against 3 × telomere sequence as previously described.24 Tumor metaphases were harvested after 6 h incubation in RPMI1640 with15% fetal bovine serum in the presence of colcemid (100 ng/ml, Invitrogen, CA, USA). Chromosome 12 paint was obtained from Applied Spectral Imaging (Carlsbad, CA, USA) and the 5′ c-myc (C10) and 5′ IgH (BAC207) locus-specific FISH (fluorescence in situ hybridization) probes were described previously.22 Images were acquired with Zeiss Axio Imager Z2 equipped with a CoolCube1 camera (Carl Zeiss, Thornwood, NY, USA) and an automatic stage system, and processed with Isis and Metafer4 software packages (MetaSystems, Newton, MA, USA).
Results
Mouse models with B-cell-specific deletion of ATM
To circumvent early lethality due to thymic lymphomas in germline Atm knockout mice (Atm−/−), we generated B-cell-specific deletion of Atm using CD21Cre, CD19Cre or Mb1+/Cre in combination with the ATM conditional allele (ATMC).23 CD21Cre allele17 mediates specific and robust ATM deletion in IgM+-naive B cells and CD19Cre+ATMC/C (ref. 18) results in ATM deletion ranging from 60% in bone marrow pre-B cells to ~100% in naive splenic B cells (Supplementary Figure 1A). Despite efficient deletion of ATM in naive splenic B cells in both CD21Cre+ATMC/C and CD19Cre+ATMC/C mice as evidenced by Southern blot analyses, CSR defects and genomic instability (Supplementary Figures 1A–1C), none of the CD21Cre+ATMC/C (n=23) or CD19Cre+ATMC/C (n=36) mice developed definitive B-cell lymphoproliferations in >28 month follow-up period (Supplementary Figure 1D), by which time the bone marrow samples were virtually devoid of B cells.
On the basis of this observation and the postulated ‘early’ deletion of ATM in human MCL,25 we focused on Mb1Cre,19 which is the earliest B-cell-specific Cre allele available, which leads to specific and robust cre activation in early pro-B/pre-B-cells.26 We generated four cohorts, Mb1+/creATM+/+(C) (hereafter referred to as M) Mb1+/CreATMC/C(−)EμCyclinD1− (MA), Mb1+/cre(+)ATM+/+(C)EμCyclinD1+ (MD/D) and Mb1+/creATMC/C(−) EμCyclinD1+ (MAD). First, we confirmed the efficient and specific deletion of the ATM gene and protein in splenic B cells from MA mice by Southern (Figure 1a) and western blotting (Figure 1b), respectively. In B cells purified from MA mice, irradiation induced phosphorylation of Kap1, an ATM-specific substrate,27 was largely abolished confirming the loss of ATM kinase activity (Figure 1c). Meanwhile, T cells from MA or MAD mice were devoid of the development defects associated with ATM deficiency28—namely reduced surface CD3/TCRβ expression and reduced CD4 or CD8 single-positive T cells in the thymus-consistent with normal ATM function in T cells from MA or MAD mice (Figure 1d). Similarly, myeloid (Gr1+ or CD11b+) and erythroid (Ter119+) lineages were also unaffected in the bone marrow and spleen of MA and MAD mice (Supplementary Figure 2A). Together, these data support the specific and efficient deletion of ATM in developing B-cells. In the Mb1+/Cre mice, the Cre knock-in disrupts the endogenous Mb1/CD79a gene in the targeted allele.19 As Mb1/CD79a is essential for B-cell development and Mb1/CD79a−/− B-cells arrest at the pro/pre- B-cell stage,29, 30 we also confirmed normal B-cell development and spleen cellularity in control MD/D, MA and MAD mice (all carrying heterozygous Mb1+/Cre alleles) and only used Mb1+/Cre for all breeding and final tumor cohorts (Figure 1d, Supplementary Figure 2B). Finally, ectopic expression of CyclinD1 in both B and T cells was also verified in EμCyclinD1+ MD and MAD mice by western blotting (Figure 1b).

B-cell-specific deletion of ATM in Mb1+/CreATMC/− and Mb1+/CreATMC/−EμCylinD1+ mouse models. (a) Southern blot analyses of the Atm locus with genomic DNA harvested from the kidney (Kid), thymus (Thy), bone marrow (BM), total spleen cells (Spl), LPS/IL-4-stimulated splenic B-cells (B) and Con A stimulated splenic T cells (T). (b) Western blot analyses for CyclinD1 and ATM in stimulated splenic B and T cells harvested from MD, MA and MAD mice. (c) Phosphorylation of Kap1 in LPS/IL4 stimulated B cells (day 3) with or without irradiation (IR,10 Gy). Protein lysate is collected 2 h after IR. (d) Representative flow cytometry analyses of bone marrow (BM), spleen and thymocytes from Atm−/− as well as MA, MAD and MD mice.
B-cell-specific deletion of ATM leads to B-cell autonomous lymphomas of both GC and non-GC phenotype
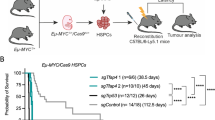
To determine the role of ATM deficiency in B-cell lymphomagenesis, we followed a cohort of MA mice with weekly palpation to identify any abnormal growths. During the 20-month follow-up, 12.5% (3/24) of MA mice developed overt splenomegaly and lymphadenopathy, with frequent intestinal involvement (67%, 2/3) (Figures 2a–c, Table 1). Flow cytometry analyses revealed monoclonal expansion of CD19+ (often B220+) B cells, which was further confirmed by Southern blot analyses that detected clonal IgH rearrangements in all confirmed cases (Figure 2d, Supplementary Figures 3A and B). In the same period, none of the Mb1Cre/+ATM+/+(C/+) mice developed any signs of tumors. At >20 month of age, all tumor cohorts were euthanized for endpoint analyses. One additional MA mouse (2023) was found to have a B-cell lymphoma based on clonal IgH rearrangement and histology (Figure 2c, Supplementary Figures 3A and B), bringing the lifetime risk for monoclonal lymphoproliferative disease to 4/24 (16.7%) in MA mice. Histopathologic and immunophenotypic analyses revealed diffuse large B-cell lymphomas (DLBCL) of splenic or nodal origin in the MA mice, with one manifesting a GC phenotype (BCL6+, IRF4−) and three exhibiting a non-GC phenotype (BCL6±, IRF4+) (Supplementary Figure 3C, Table 1). Given the GC and non-GC immunophenotypes, we analyzed the IgH for SHM and found evidence of SHM in 2/4 MA DLBCL, indicating a post-GC cell of origin. On the basis of these data, we conclude that deletion of ATM in early pre-/pro- B-cells leads to infrequent mature B-cell lymphomas of heterogeneous cell origins with long latency. These results support a role of ATM as a tumor suppressor gene during early B-cell development and also highlight the need for additional genetic hits.

MA and MAD mice develop clonal B-cell lymphoproliferations. (a) Tumor-free survival of MA, MAD and MD/D cohorts up to 20 month of age. Number of mice followed in each cohort is listed in the parentheses. *P<0.05 per Mantel–Cox/log-rank test for statistical significance. (b) Lifetime risk for B-cell lymphoproliferations in MA, MAD and MD/D mice. The gray boxes represent tumors discovered within 20-month follow-up period and the white boxes represent tumors discovered at endpoint analyses (>20-month). (c) Representative images of enlarged spleen and lymph node (from MAD mouse 3468) and small intestine (right, from MA mouse 2023). The black arrow heads point to the lymph nodes. (d) Representative flow cytometry analyses of spleen from tumor-bearing MA and MAD mice. (e) Southern blot analyses of DNA harvested from Kidney (K) and Spleen (S) from MAD mice. Asterisk denotes mouse 3613, in which the tumor primarily resided in bone marrow (B). Mouse 3468 has leukemia with significant infiltration in the kidney, thus the presence of clonal rearrangements. Blue arrow heads point to clonal rearrangements. Probe for the constant region of TCRδ is used as loading control. GL: germ line.
CyclinD1 overexpression accelerates B-cell lymphoproliferations in MA mice and skews lymphomas toward pre-GC origin
Among all human B-cell non-Hodgkin lymphomas, MCL, characterized by ectopic expression of CyclinD1, has the highest frequency of bi-allelic inactivation of ATM.9 Given the early deletion of ATM during MCL development, it was proposed that ATM deficiency might increase IgH-CyclinD1 translocation to promote MCL.9 Alternatively, ectopic CyclinD1 expression might synergize with ATM deficiency to promote cellular proliferation at the price of genomic instability to drive B-cell lymphomagenesis. To test this, we bred MA mice with EμCyclinD1 transgenic mice to generate the MAD and control MD/D mice. At 2 months of age, B-cell number and B-cell development in MAD mice is comparable with MA and MD controls (Figure 1d). By 20-months of age, 3/18 (16.7%) MAD mice had developed overt splenomegaly and lymphadenopathy with occasional intestinal involvement and leukemic presentation (Figures 2a–c, Table 1). The tumor-free survival of MAD mice was significantly shorter than either MA or MD/D control groups (Figure 2a). Moreover, at the endpoint analyses (>20 month), 46.6% (7/15) of MAD mice showed clonal B-cell expansions by flow cytometry analyses and clonal IgH and IgK rearrangements by Southern blotting (Figures 2d and e), bringing the lifetime risk for B-cell lymphoproliferations in MAD mice to 55.6% (10/18) (Table 1). This is in sharp contrast to the control MD/D mice, which did not develop overt splenomegaly within the 20-month follow-up and only 2/12 MD/D mice showed evidence of B-cell lymphoproliferation upon histopathologic analyses at necropsy (Figure 2b, Table 1). By flow cytometry, MA and MAD lymphoproliferations had similar immunophenotypes, namely CD19+B220hi/loIgMhi/lo (Figure 2c). The B-cell identity of B220-negative lymphoproliferations was further validated by PAX5 immunohistochemistry staining (Figure 3, Supplementary Figure 3C). All tumors were CD21/35−CD23−(Supplementary Figure 3D), excluding GC B-cell origin. Two out of 10 (20%) MAD tumors expressed CD5, a marker that is frequently associated with activated B cells and also expressed by human MCL (Supplementary Figure 3D, Table 1). In addition to DLBCL observed in 2/10 (20%) MAD mice, including one exhibiting prominent red pulp involvement, a spectrum of indolent lymphoproliferations was noted in the remainder. All eight lymphoproliferative disorders were characterized by variable degrees of white pulp enlargement, predominantly owing to follicular mantle and marginal zone expansion without discernable GCs. Of note, 5/10(50%) MAD tumors showed significant infiltration of the red pulp by small- to medium-sized lymphocytes, resembling the pattern of splenic involvement by human chronic lymphocytic leukemia and MCL (Table 1, Figure 3). The immunophenotype of the DLBCL indicated a non-GC B-cell origin (BCL6−, IRF4+), as did that of the lymphoproliferative disorders (PAX5+, BCL6−, IRF4± and CD138−). Consistent with these observations, whereas 50% (2/4) of MA lymphomas showed IgH variable region gene mutations, only 2/10 (20%) of the MAD lymphomas showed evidence of SHM. These findings suggest that CyclinD1 overexpression synergizes with ATM deficiency to promote predominantly pre-GC B-cell lymphoproliferations recapitulating some morphological and immune phenotypes of human MCL.

Histopathologic and immunophenotypic analyses of B-cell lymphoproliferations of MAD mice. Representative photomicrographs showing the spectrum of B-cell lymphoproliferations observed. (3468, H&E) DLBCL. The white pulp is markedly expanded and the architecture effaced by an infiltrate of large pleomorphic lymphocytes. The neoplastic cells express B220 (blue). Scattered small disrupted BCL6+ germinal centers are seen and the neoplastic cells are BCL6 negative but they show IRF4 expression, findings compatible with a DLBCL of non-GC phenotype. (4271, H&E) LPD with red pulp lymphocytic infiltrate. The white pulp is disrupted by expansions of the follicular mantle and marginal zones and a diffuse infiltrate of small-sized lymphocytes is present in the red pulp. The neoplastic lymphocytes show B220 (blue), PAX5 and IRF4(variable) expression and they lack BCL6 expression. (4161, H&E) LPD. Clusters of large lymphocytes are seen surrounding the white pulp and also infiltrating the red pulp. The neoplastic lymphocytes are B220 (blue) and BCL6 negative, but they express PAX5 and show IRF4 (variable) expression. (3713, H&E) LPD. The white pulp is variably disrupted by expansions of the follicular mantle and marginal zones with limited red pulp infiltration. The neoplastic cells express B220 (variable), PAX5 and IRF4 (variable) but they lack BCL6 expression.
MAD tumors share a subset of molecular features with human MCL
To further characterize the genetic lesions that underlie lymphomagenesis in the MAD mice, we performed comparative genomic hybridization (CGH) analyses on three MAD, one MA and one MD tumors with high tumor content (>50% by fluorescence-activated cell sorting). CGH identified a large number of gross chromosomal gains and losses in all tumors (Figure 4a and Supplementary Figure 4A). Specifically, two MAD tumors (3468 and 4161) display focal deletion involving the Trp53 tumor suppressor gene, including clear homozygous deletion in tumor 3468 (Figure 4b and Supplementary Figure 4A). Cdkd2a (ink4a/p16) and Rb1 are also deleted in a subset of the MAD and MA lymphomas (Figure 4b). MLL2, a haplo-insufficient tumor suppressor gene in MCL as well as DLBCL, is also partially deleted in all tumors tested (Figure 4b). Meanwhile, Mdm2, Cdk4, and Notch1 are moderately amplified (mostly trisome) in several MAD and MA lymphomas (Figure 4b). These changes are consistent with recurrent genetic alterations that have been characterized for human MCL.25 Meanwhile, the CGH analyses also identified focal deletions in Igκ (chromosome 4) and Igh (chromosome 12) loci indicative of B-cell origin. Fine mapping of the Igh locus showed that the deletions in all three MAD tumors are restricted to the V-D-J portion of the Igh locus and does not involve the switch region located downstream (centrometric of chromosome 12) (Figure 4d), consistent with the pre-GC origin of the MAD tumors. In contrast, MA tumor 2023 shows heterozygous deletion within switch region (Figure 4d). Together with the positive staining for Bcl6 and the presence of SHM, this finding suggests that at least a subset of the MA tumors derived from GC or post-GC cells. CGH is not able to detect reciprocal translocations. To test whether reciprocal translocation involving IgH locus occurred in any of the tumors, we performed chromosome 12 paint together with FISH using a probe that hybridizes to the telomeric end of Igh locus (upstream toward the Vh region, 5′ IgH, 207) in two MAD (3468 and 4161) and one MA (2594) tumors, for which high-quality metaphases were available. The analyses revealed heterozygous deletion, but not translocation involving the 5′IgH in MAD tumor 4161 and MA tumor 2594 (Figure 4c and Supplementary Figure 4C). FISH with c-Myc probes exclude IgH-Myc translocation in the tumors and CGH analyses suggested that c-Myc is not grossly amplified in tested MAD or MA tumors. Finally, targeted Sanger sequencing of 10 MAD and 4 MA tumors did not find mutations in the C-terminal Pro, Glu, Ser, Thr-rich (PEST) domain of Notch1 or the Su(var)3-9 Enhancer-of-Zeste (SET) domain of Multiple Myeloma SET domain (MMSET), two genes that are mutated in ~10% human MCL.9 Together, these findings suggest that MAD tumors share molecular features with human MCL.

Comparative genomic hybridization (CGH) and FISH analyses of MA and MAD tumors. (a) Landscape representation of CGH on MAD tumors-3468 and 4161, with green arrows indicating particular regions of interest. Chromosome (Ch.) numbers indicated at the top. The arrows indicate loci of interest (e.g. Igκ, Igh and Trp53). The y axis (average signal intensity) represents the log ratio of copy number between tumor vs normal at given probe. (b) Heatmap showing focal deletion of Trp53, Cdkn2a, MLL2 and Rb and potential trisomy of Mdm2 and Cdk4. It also shows normal copy number of c-Myc. Different rows represent unique probes used for CGH analyses. (c) Chromosome (Ch.) 12 paint analyses combined with either Telomeric Igh FISH (BAC207 at the Vh region) or c-Myc on MAD tumors-3468 and 4161. The diagram on the right represents the mono-allelic deletion of the Igh in tumor 4161. (d) CGH analysis of the Igh locus of MAD, MA, MD tumors. Demarcations of each region within the Igh locus are shown below. Average signal intensity of −0.6 indicates the value corresponding to heterozygous loss of the allele.
ATM deficiency enhances genomic instability and increases chromosomal fusions in CyclinD1+ B cells
To better understand the mechanism by which ectopic CyclinD1 expression synergizes with ATM deficiency to promote B-cell lymphoproliferations, we analyzed non-neoplastic B cells from 6–12-week-old MAD and MA mice. MCL is thought to originate from pre-GC, mantle zone B cells, which have yet to encounter CSR and SHM.31 As ATM deficiency reduces CSR efficiency,32, 33 we hypothesized that ectopic expression of CyclinD1 and ATM deficiency together might be deleterious to cells undergoing CSR, thus effectively trapping naive B cells at the pre-GC stage of development/maturation. To test this hypothesis, we measured CSR in purified B cells derived from MA, MD, and MAD mice as well as WT or Atm−/− control mice in vitro. Ectopic expression of CyclinD1 by itself did not measurably affect CSR. In vitro CSR to IgG1, measured by flow cytometry analyses, was reduced to ~50% of WT levels in MAD as well as MA and Atm−/− B-cells (Figure 5a), suggesting ATM deficiency compromised CSR in WT and CyclinD1+ B cells at similar levels. Furthermore, irradiation induced G1/S and G2/M checkpoints in LPS/IL4-activated B cells are normal in WT and CyclinD1+ B cells and comparable in Atm−/−, MA and MAD B cells (Supplementary Figures 5A and B), suggesting that ATM deficiency compromised DNA damage induced checkpoints in both WT and CyclinD1+ B cells. Next, we assessed the degree of genomic instability in LPS/IL4-activated B cells from Atm−/−, MA and MAD mice by telomere-FISH analyses. Two kinds of breaks can be quantified by the telomere-FISH assay: chromosomal breaks, which refer to breaks at both sister-chromatids, and chromatid breaks, which refer to breaks in only one of the two sister-chromatids (Figure 5b). The frequencies of chromosomal breaks as well as chromatid breaks were comparable in MA and MAD mice and significantly higher than those observed in MD mice (Figure 5b, Supplementary Table 1). Moreover, the frequency of dicentric chromosomes generated by end–end fusion of chromosomal breaks was significantly higher in activated B cells derived from MAD mice than in those from the MA or MD mice (Figures 5b and c, Supplementary Table 1), suggesting possible increased proclivity of chromosomal translocations. We conclude that ATM deficiency induces genomic instability in CyclinD1+ B cells.

Increased chromosomal fusions in stimulated MAD B cells. (a) Representative flow cytometry plots for surface IgG1 expression in LPS/IL4 stimulated B cells and quantification. Efficiency is calculated as a percentage of IgG1+ cells relative to the control. (b) Representative images of cytogenetic abnormalities observed in T-FISH assay in stimulated splenic B cells and the quantification. (c) Quantification of dicentric chromosomes observed in stimulated B cells derived from MD/D, MA, MAD and Atm−/− mice. P-values were calculated using a two-tailed Student’s t-test assuming unequal variances.
Ectopic CyclinD1 expression increases naive B-cell proliferation and rescues the B-cell lymphocytopenia in older MA mice
Given the majority of the MAD and MA lymphomas are IgM+ and thus derived from cells that have not yet undergone CSR, we compared the naive B-cell population in young (1–3-month-old) vs mid-age (4–6- and 7–12-month-old) MAD and MA mice. Although all mature B-cell population are present in 6- and 12-month-old MAD, MA and MD/D controls, the frequency of naive B cells (CD19+ cells) in spleen declined significantly faster in MA mice, recapitulating the ATM-deficient B-cell lymphocytopenia that worsens over time (Figures 6a and b, Supplementary Figure 6). Ectopic expression of CylinD1 alone has, at most, moderate effects on splenic B-cell frequency in WT mice (MD vs control) but markedly rescued the splenic B-cell lymphocytopenia in MA mice (MAD vs MA, Figure 6b). This is intriguing, as the naive B-cell population is thought to be the progenitor of MCL lymphomas. We then co-stained splenic sections from 6-month-old tumor-free mice with B-cell marker-B220 and proliferation marker-Ki67. Quantification of Ki67+B220+ cells revealed a significant reduction of Ki67+ frequency in B cells from MA mice, which is reverted by ectopic expression of CyclinD1 in the MAD mice (Figures 6c and d). We conclude that ectopic expression of CyclinD1 promotes proliferation of ATM-deficient naive B cells with genomic instability, rescue the B-cell lymphocytopenia, and promote lymphomagenesis in pre-GC B cells that would otherwise diminish owing to ATM deficiency.

CyclinD1 expression rescues progressive B-cell loss in MA mice. (a) Representative flow cytometry plots of splenic B220+CD19+/IgM+ B-cell populations in 6-month-old mice. (b) Quantification of CD19+ B-cell populations from non-tumor-bearing mice analyzed at the indicated time points. (c) Representative immunofluorescence (IF) images, and (d) Quantification of Ki67+ (green, nuclear staining) and B220+ (red, membrane staining) cells in spleens harvested from 7-month-old non-tumor mice. Fields (MD (n=7), MA (n=15) and MAD (n=14)) selected for quantification were taken at × 400 total magnification with equivalent B220+ cellularity. P-values were calculated based on individual two-tailed Student’s t-tests assuming unequal variances.
Discussion
Deletion of ATM in early B-cell development is necessary for mature B-cell malignancies
Given the mature B-cell phenotypes of most ATM-deficient human B-cell lymphomas and the well-characterized role of ATM during CSR, it was speculated that ATM maintains genomic stability in GC B cells to suppress B-cell lymphomas. However, despite complete ATM deletion in naive B cells, our attempt to establish an ATM-deficient mature B-cell lymphoma model with naive B-cell-specific CD21Cre or less robust pre-B-cell-specific CD19Cre was fruitless. Mb1Cre allele is an early and robust B-cell-specific Cre that leads to Cre activation in almost all pre-B-cells (in contrast to ~60% of pre-B-cells via CD19Cre). In this regard, 16.7% of MA and ~55% of MAD mice developed indolent yet clonal ‘mature’ B-cell lymphomas (Figure 2b), recapitulating the disease spectrum of human lymphomas, especially MCL. Even so, most of the MA and MAD tumors are IgM+ and thus have not undergone CSR. Fine mapping of the Igh loci with CGH probe and FISH analyses further revealed V(D)J recombination-related, but not CSR-related chromosomal alterations in MAD tumors. Together these findings highlight the critical tumor suppressor function of ATM in early progenitor or naive B cells, possibly during V(D)J recombination, to prevent mature B-cell lymphomas down the line. In this context, ATM is known to prevent the persistence and propagation of chromosome breaks originating from V(D)J recombination in naive B cells.34 Recent studies reveal that early genetic alterations in hematopoietic stem cells could prime mature B-cell malignancies, including chronic lymphocytic leukemia.35
How ATM promotes lymphomagenesis driven by CyclinD1
Despite strong evidence for a role of CyclinD1 in lymphomagenesis, mice with ectopic expression of WT CyclinD1 (MD/D) rarely developed lymphomas. Genomic analyses of human MCL suggest that ATM deletion coexists with CyclinD1 expression in ~50% of MCLs.9 Here we show that ATM deletion promotes B-cell lymphomagenesis in EμCyclinD1 mice.14, 36 Although ATM deficiency and the resulting genomic instability might simply promote IgH-CyclinD1 translocation in human MCL, our study suggests that loss of ATM also promotes lymphomagenesis after ectopic expression of CyclinD1. In this context, we showed that ATM deficiency increases genomic instability and chromosomal translocations in activated CyclinD1+ B cells (Figure 5). This increased genomic instability in naive B cells likely promotes the loss of additional tumor suppressor genes including Trp53, Cdkn2a, MLL2 or Rb1 that have all been implicated in human MCL to promote transformation.9 In addition, ATM deficiency might potentiate the mitogenic function of CyclinD1 by increasing CyclinD1 protein levels. ATM negatively regulates F-Box proteins (FBX4 and FXBO31) that mediate the degradation of CyclinD1 during DNA damage responses.37, 38 Indeed, germline ATM deficiency accelerates lymphomas in mouse models, expressing a constitutive-nuclear form of CyclinD1.39, 40
How does ectopic expression of CyclinD1 promote B-cell lymphomas in ATM-deficient B cells?
MAD mice developed B-cell lymphomas significantly earlier and more frequently than MA mice, suggesting that mitogenic cues generated by ectopic CyclinD1 expression synergize with ATM deficiency for B-cell transformation. In addition to its checkpoint function, ATM has important roles in DNA repair, as ATM deficiency is detrimental to primary cells owing to ongoing genomic instability. Here we show that B-cell-specific deletion of ATM in MA mice leads to progressive B-cell lymphocytopenia at 6 and 12 months of age (Figure 6), a phenotype that is overlooked in germline ATM-null mice owing to lethal thymic lymphomas. We further showed that EμCyclinD1 promotes the proliferation of naive B-cells and successfully restores B-cell cellularity in older MA mice (Figure 6). Based on this finding, we propose that CyclinD1 promotes B-cell lymphomas by promoting proliferation of ATM-deficient naive B cells with genomic instability, which in turn maintain the cellularity of tumor-prone ATM-deficient B cells. There are three D-type cyclins (D1, D2 and D3) with overlapping functions during embryonic development,41, 42 but GC B cells exclusively depend on CyclinD3 for proliferation and survival,43, 44 which might explain why the pre-GC cells are more susceptible to the mitogenic cues unleashed by ectopic CyclinD1.
In summary, we report the first animal model for ATM-deficient B-cell lymphomas and reveal a synergistic role of CyclinD1 expression and ATM deficiency in naive B cells to promote pre-GC B-cell lymphomas. The MA and MAD mature B-cell lymphomas are largely indolent, similar to the Bcl6-driven mouse lymphomas45 and distinct from Myc-driven aggressive lymphomas. In this regard, MA and MAD lymphomas recapitulate the disease spectrum of human lymphomas, especially MCL,46 and would be an invaluable resource to better understand MCL and the clinical progression of indolent MCL to an aggressive form in the future.
References
Klein U, Dalla-Favera R . Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol 2008; 8: 22–33.
Franco S, Alt FW, Manis JP . Pathways that suppress programmed DNA breaks from progressing to chromosomal breaks and translocations. DNA Repair (Amst) 2006; 5: 1030–1041.
Shiloh Y, Ziv Y . The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14: 197–210.
Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 2006; 442: 466–470.
Pan-Hammarstrom Q, Lahdesmaki A, Zhao Y, Du L, Zhao Z, Wen S et al. Disparate roles of ATR and ATM in immunoglobulin class switch recombination and somatic hypermutation. J Exp Med 2006; 203: 99–110.
Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC . ATM is required for efficient recombination between immunoglobulin switch regions. J Exp Med 2004; 200: 1103–1110.
Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA et al. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J Exp Med 2004; 200: 1111–1121.
Lavin MF . Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 2008; 9: 759–769.
Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA 2013; 110: 18250–18255.
Gronbaek K, Worm J, Ralfkiaer E, Ahrenkiel V, Hokland P, Guldberg P . ATM mutations are associated with inactivation of the ARF-TP53 tumor suppressor pathway in diffuse large B-cell lymphoma. Blood 2002; 100: 1430–1437.
Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 2011; 365: 2497–2506.
Stilgenbauer S, Schaffner C, Litterst A, Liebisch P, Gilad S, Bar-Shira A et al. Biallelic mutations in the ATM gene in T-prolymphocytic leukemia. Nat Med 1997; 3: 1155–1159.
Zha S, Bassing CH, Sanda T, Brush JW, Patel H, Goff PH et al. ATM-deficient thymic lymphoma is associated with aberrant tcrd rearrangement and gene amplification. J Exp Med 2010; 207: 1369–1380.
Bodrug SE, Warner BJ, Bath ML, Lindeman GJ, Harris AW, Adams JM . Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J 1994; 13: 2124–2130.
Lovec H, Grzeschiczek A, Kowalski MB, Moroy T . Cyclin D1/bcl-1 cooperates with myc genes in the generation of B-cell lymphoma in transgenic mice. EMBO J 1994; 13: 3487–3495.
Zha S, Sekiguchi J, Brush JW, Bassing CH, Alt FW . Complementary functions of ATM and H2AX in development and suppression of genomic instability. Proc Natl Acad Sci USA 2008; 105: 9302–9306.
Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K . Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell 2004; 117: 787–800.
Rickert RC, Rajewsky K, Roes J . Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature 1995; 376: 352–355.
Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci USA 2006; 103: 13789–13794.
Yamamoto K, Wang Y, Jiang W, Liu X, Dubois RL, Lin CS et al. Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. J Cell Biol 2012; 198: 305–313.
Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010; 17: 28–40.
Gostissa M, Yan CT, Bianco JM, Cogne M, Pinaud E, Alt FW . Long-range oncogenic activation of Igh-c-myc translocations by the Igh 3' regulatory region. Nature 2009; 462: 803–807.
Zha S, Sekiguchi J, Brush JW, Bassing CH, Alt FW . Complementary functions of ATM and H2AX in development and suppression of genomic instability. Proc Natl Acad Sci USA 2008; 105: 9302–9306.
Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell 2006; 21: 201–214.
Jares P, Colomer D, Campo E . Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer 2007; 7: 750–762.
Kwon K, Hutter C, Sun Q, Bilic I, Cobaleda C, Malin S et al. Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity 2008; 28: 751–762.
Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM and KAP-1 dependent pathway. Nat Cell Biol 2006; 8: 870–U142.
Borghesani PR, Alt FW, Bottaro A, Davidson L, Aksoy S, Rathbun GA et al. Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc Natl Acad Sci USA 2000; 97: 3336–3341.
Pelanda R, Braun U, Hobeika E, Nussenzweig MC, Reth M . B cell progenitors are arrested in maturation but have intact VDJ recombination in the absence of Ig-alpha and Ig-beta. J Immunol 2002; 169: 865–872.
Minegishi Y, Coustan-Smith E, Rapalus L, Ersoy F, Campana D, Conley ME . Mutations in Igalpha (CD79a) result in a complete block in B-cell development. J Clin Invest 1999; 104: 1115–1121.
Walsh SH, Thorselius M, Johnson A, Soderberg O, Jerkeman M, Bjorck E et al. Mutated VH genes and preferential VH3-21 use define new subsets of mantle cell lymphoma. Blood 2003; 101: 4047–4054.
Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA et al. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J Exp Med 2004; 200: 1111–1121.
Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC . ATM is required for efficient recombination between immunoglobulin switch regions. J Exp Med 2004; 200: 1103–1110.
Callen E, Jankovic M, Difilippantonio S, Daniel JA, Chen HT, Celeste A et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell 2007; 130: 63–75.
Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011; 20: 246–259.
Vaites LP, Lian Z, Lee EK, Yin B, DeMicco A, Bassing CH et al. ATM deficiency augments constitutively nuclear cyclin D1-driven genomic instability and lymphomagenesis. Oncogene 2014; 33: 129–133.
Vaites LP, Lee EK, Lian Z, Barbash O, Roy D, Wasik M et al. The Fbx4 tumor suppressor regulates Cyclin D1 accumulation and prevents neoplastic transformation. Mol Cell Biol 2011; 31: 4513–4523.
Santra MK, Wajapeyee N, Green MR . F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 2009; 459: 722–725.
Vaites LP, Lian Z, Lee EK, Yin B, Demicco A, Bassing CH et al. ATM deficiency augments constitutively nuclear cyclin D1-driven genomic instability and lymphomagenesis. Oncogene 2013; 33: 129–133.
Gladden AB, Woolery R, Aggarwal P, Wasik MA, Diehl JA . Expression of constitutively nuclear cyclin D1 in murine lymphocytes induces B-cell lymphoma. Oncogene 2006; 25: 998–1007.
Ciemerych MA, Sicinski P . Cell cycle in mouse development. Oncogene 2005; 24: 2877–2898.
Carthon BC, Neumann CA, Das M, Pawlyk B, Li T, Geng Y et al. Genetic replacement of cyclin D1 function in mouse development by cyclin D2. Mol Cell Biol 2005; 25: 1081–1088.
Peled JU, Yu JJ, Venkatesh J, Bi E, Ding BB, Krupski-Downs M et al. Requirement for cyclin D3 in germinal center formation and function. Cell Res 2010; 20: 631–646.
Cato MH, Chintalapati SK, Yau IW, Omori SA, Rickert RC . Cyclin D3 is selectively required for proliferative expansion of germinal center B cells. Mol Cell Biol 2011; 31: 127–137.
Cattoretti G, Pasqualucci L, Ballon G, Tam W, Nandula SV, Shen Q et al. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell 2005; 7: 445–455.
Zullo K, Amengual JE, O'Connor OA, Scotto L . Murine models in mantle cell lymphoma. Best Pract Res Clin Haematol 2012; 25: 153–163.
Acknowledgements
We wish to thank Dr Frederick W Alt for providing the ATM conditional mouse model, Dr Klaus Rajewsky for providing the CD19Cre and CD21Cre mice and Dr Michael Reth for providing the Mb1Cre mice. We also wish to thank Ms Hongyan Tang and Mr Denis Loredan for their technical assistance. We thank Jennifer L Crowe for critical reading of the manuscript. Research reported in this publication was supported by the NIH/NCI 1RO1CA158073, American Cancer Society (124300-RSG-13-038 DMC) for SZ and NIH/NCI PO1 CA174653 for SZ and GB. SZ was a St Baldrick’s Scholar for Pediatric Cancer and is a Leukemia Lymphomas Society Scholar.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
About this article

Cite this article
Yamamoto, K., Lee, B., Li, C. et al. Early B-cell-specific inactivation of ATM synergizes with ectopic CyclinD1 expression to promote pre-germinal center B-cell lymphomas in mice. Leukemia 29, 1414–1424 (2015). https://doi.org/10.1038/leu.2015.41
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2015.41
- Springer Nature Limited
This article is cited by
-
Chronic lymphocytic leukaemia/small lymphocytic lymphoma and mantle cell lymphoma: from early lesions to transformation
Virchows Archiv (2023)
-
Tia1 dependent regulation of mRNA subcellular location and translation controls p53 expression in B cells
Nature Communications (2017)
-
Role of SOX11 and Genetic Events Cooperating with Cyclin D1 in Mantle Cell Lymphoma
Current Oncology Reports (2017)