
Abstract
The added value of IKZF1 gene deletion (IKZF1del) as a stratifying criterion in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is still debated. We performed a comprehensive analysis of the impact of IKZF1del in a large cohort of children (n=1223) with BCR-ABL1-negative BCP-ALL treated in the EORTC-CLG trial 58951. Patients with IKZF1del had a lower 8-year event-free survival (EFS, 67.7% versus 86.5%; hazard ratio (HR)=2.41; 95% confidence interval (CI)=1.75–3.32; P<0.001). Importantly, despite association with high-risk features such as high minimal residual disease, IKZF1del remained significantly predictive in multivariate analyses. Analysis by genetic subtype showed that IKZF1del increased risk only in the high hyperdiploid ALLs (HR=2.57; 95% CI=1.19–5.55; P=0.013) and in ‘B-other‘ ALLs, that is, lacking classifying genetic lesions (HR=2.22; 95% CI=1.45–3.39; P<0.001), the latter having then a dramatically low 8-year EFS (56.4; 95% CI=44.6-66.7). Among IKZF1del-positive patients randomized for vincristine-steroid pulses during maintenance, those receiving pulses had a significantly higher 8-year EFS (93.3; 95% CI=61.3–99.0 versus 42.1; 95% CI=20.4–62.5). Thus, IKZF1del retains independent prognostic significance in the context of current risk-adapted protocols, and is associated with a dismal outcome in ‘B-other‘ ALL. Addition of vincristine-steroid pulses during maintenance may specifically benefit to IKZF1del patients in preventing relapses.
Similar content being viewed by others


Introduction
Cure rates of children with B-cell precursor acute lymphoblastic leukemia (BCP-ALL) have increased considerably during the last few decades, partly as a result of coupling risk-adapted treatment intensity with an optimized use of traditional antileukemic drugs.1, 2, 3, 4 However, relapses still occur in ~20% of patients, most of them being not considered at high risk initially. This suggests that there is still a need for the improvement of therapeutic stratification using new prognostic markers. Risk stratification in contemporary protocols is based on clinical and biological predictors of relapse, mostly related to genetic lesions defining oncogenic subtypes5, 6 and early response to treatment. High hyperdiploidy and the chromosomal translocation t(12;21)/ETV6-RUNX1 are usually associated with a favorable outcome, whereas t(9;22)/BCR-ABL1, MLL gene rearrangements, low hypodiploidy and intrachromosomal amplification of chromosome 21 (iAMP21) are associated with a high risk of relapse. However, no classifying genetic abnormality can be identified by standard laboratory work-up in about 25% of pediatric BCP-ALL cases, referred to here as ‘B-other‘ ALL.
Besides classifying lesions, a number of cooperating genetic lesions have been identified recently.7 Among these, the deletion of the B-cell transcription factor IKAROS (IKZF1del) emerged as a promising prognostic marker, as initial studies demonstrated a very poor outcome for patients having an IKZF1del, with event-free survival (EFS) rates below 50%.8, 9 However, further studies conducted on larger series of patients treated with risk-directed therapy based on minimal residual disease (MRD) found only a moderately inferior outcome associated with IKZF1del with EFS rates reaching ~70%.10, 11, 12 Consequently, using IKZF1del for treatment stratification could lead to inappropriate over-treatment in a substantial number of patients. Thus, whether outcome can be significantly improved by IKZF1-based risk stratification remains a matter of debate.11 To become a therapeutic stratification criterion, IKZF1del should be an independent prognostic factor and help to identify a subset of patients with a risk of relapse high enough to warrant treatment intensification.
To address these issues we analyzed the prognostic impact of IKZF1del together with several other variables in a large prospective cohort of children with BCP-ALL treated in a single, recent trial.
Subjects and methods
Patients
Between December 1998 and July 2008, 1654 children (⩾1 and <18 years old) diagnosed with BCP-ALL were consecutively enrolled in the Children's Leukemia Group of the European Organisation for Research and Treatment of Cancer (EORTC-CLG) trial 58951 (ClinicalTrials.gov Identifier: NCT00003728).13, 14 The study included 1253 cases for which standard cytogenetic/molecular diagnosis was performed and tumoral DNA was available (Figure 1). This cohort did not differ from the entire cohort with respect to the main features (Supplementary Table 1). Patients with BCR-ABL1-positive ALL (n=30) were excluded from the present study and analyzed separately because from 2005 they were switched to another treatment protocol (EsPhALL) after the induction phase.15, 16 The remaining patients (n=1223) were uniformly treated with a Berlin-Frankfurt-Münster (BFM)-like regimen consisting of four-drug induction, post-induction, late intensification and maintenance, without irradiation (except for transplanted patients who received total body irradiation). Patients were assigned to different risk groups: very low risk (VLR), average risk (AR) and very high risk (VHR). VLR criteria were high hyperdiploidy (⩾51 chromosomes or DNA index >1.16 and <1.5), white blood cell (WBC) counts <10 × 109/l and no central nervous system or gonadal involvement. VHR criteria were the presence of any of the following: 11q23/MLL rearrangement, low hypodiploidy or near haploidy, poor response to prephase (blast counts in peripheral blood ⩾1 × 109/l at completion of the prephase—1 week of corticosteroids and intrathecal injection of methotrexate), lack of complete remission (CR) or MRD ⩾10−2 after induction (day 35). MRD monitoring was based on PCR quantification of T-cell-receptor and immunoglobulin gene rearrangements.17 AR patients were children without VLR or VHR characteristics. The trial included three randomized comparisons: (i) dexamethasone 6 mg/m2/day versus prednisolone 60 mg/m2/day in induction,14 (ii) conventional versus prolonged administration of E. coli asparaginase for non-VHR patients and (iii) the presence versus the absence of vincristine-steroid pulses during maintenance for AR patients only, 6 pulses at intervals of 10 weeks during the first 60 weeks of maintenance therapy.13 The pulses consisted of 7 days of corticosteroids, either prednisolone 60 mg/m2/day or dexamethasone 6 mg/m2/day depending on the first randomization, and vincristine 1.5 mg/m2 on day 1 and day 8. Pulses improved outcome of AR patients, whereas no impact could be demonstrated for the type of corticosteroid.13, 14

CONSORT diagram. Patients eligible for randomization were patients from average risk (AR) group who were still in continuous complete remission at the beginning of maintenance therapy. Randomization was stopped at the end of 2002 when a preliminary analysis of the intergroup trial results suggested that the pulses would fail to provide any benefit.21 BCP-ALL, B-cell precursor acute lymphoblastic leukemia; EORTC-CLG, EORTC Children’s Leukemia Group; IKZF1del, deletion of IKZF1 gene; VLR, very low risk; VHR, vey high risk.
This protocol was accepted by the EORTC Protocol Review Committee and the Ethics Committee of each participating center. Outcome data for patients enrolled in EORTC 58951 were frozen on March 2012; the median follow-up of the study cohort was 6.61 years.
Genomic analyses
Standard karyotype and/or DNA index, fluorescence in situ hybridization and/or reverse-transcriptase PCR and multiplex ligation probe assay (SALSA kit P327 iAMP21, MRC-Holland, Amsterdam, the Netherlands) were used to screen for the most frequent classifying genetic lesions. ERG deletion18 (ERGdel) was detected by breakpoint-specific genomic PCR. Data were centrally reviewed. High hyperdiploidy (⩾51 chromosomes), low hypodiploidy/near haploidy (<40 chromosomes), t(12;21)/ETV6-RUNX1, t(1;19)/TCF3-PBX1, t(9;22)/BCR-ABL1, t(4;11)/MLL-AF4 or other MLL rearrangements, iAMP21 and ERGdel were considered distinct genetic subtypes. BCP-ALLs negative for all of these lesions were pooled and named ‘B-other‘.
IKZF1 deletions were analyzed using both a genomic breakpoint-specific multiplex fluorescent PCR19 and multiplex ligation probe assay method (SALSA P335 ALL-IKZF1-A3 and SALSA P202 IKZF1 kits, MRC-Holland). Cases found positive by either of two methods were considered positive.
Statistical analyses
EFS was calculated from the date of CR to the date of first relapse or death. Patients who failed to reach CR by the end of induction-consolidation were considered as having an EFS at time 0. All patients alive and still in their first CR were censored at their last follow-up. Disease-free survival (DFS) was defined as EFS, but only in patients who reached CR. Overall survival (OS) was calculated from the date of the start of treatment until the date of death; patients still alive were censored at their last follow-up.
Survival distributions were estimated according to the Kaplan–Meier technique and compared using the two-tailed log-rank test. The Cox proportional hazards model was used to obtain the estimate and the 95% confidence interval (CI) of the hazard ratio (HR) of the instantaneous event rate in one group versus another. The possible heterogeneity of the prognostic importance of IKZF1del in the different genetic subgroups was explored by estimation of the HR for each subgroup, together with the 95% CI and a test for interaction. All analyses were based on the intent-to-treat principle.
The relationship between the presence/absence of IKZF1del and categorical variables was tested for significance using the χ2 or Fisher test, and for continuous variables using the Wilcoxon test.
SAS 9.3 statistical software (Cary, NC, USA) was used.
Results
The presence of IKZF1del is associated with high-risk features
Of 1223 BCR-ABL1-negative BCP-ALL cases, 179 (14.6%) had a deletion involving the IKZF1 gene. Clinical and biological features at presentation were analyzed with respect to the presence or absence of IKZF1del (Table 1). Patients with IKZF1del were significantly older (P<0.001), had a higher WBC count at diagnosis (P=0.037) and presented more frequently with Pro-B immunophenotype (10.7% versus 3.9%; P=0.001). There was also a trend toward a more frequent central nervous system involvement at diagnosis in patients with IKZF1del (2.8% versus 1.0%; P=0.056).
IKZF1del was unevenly distributed among genetic subtypes as defined by the main classifying genetic lesions (Table 2). IKZF1del was very frequent in the newly described group of patients having ERGdel (42% of these), as reported recently.18, 20 IKZF1del was relatively frequent in patients having iAMP21, low hypodiploidy/near-haploidy or MLL translocations (37%, 36% and 25%, respectively), which are all known to be associated with poor prognosis, and also in the ‘B-other‘ subgroup (25%), which has an intermediate outcome.18 In contrast, IKZF1del was rarely found in association with the recognized good-prognosis genetic lesions high hyperdiploidy and ETV6-RUNX1 (9.3% and 4.3%, respectively). Altogether, the proportion of genetic lesions of poor and intermediate risk was higher in patients with IKZF1del (10.6% and 51.4%, respectively, versus 3.7% and 29.8% in IKZF1del-negative patients, P<0.001).
Regarding response to treatment, patients with IKZF1del compared with those without IKZF1del more frequently displayed a ‘poor response‘ to prephase (10.6% versus 4.7%; P=0.001), and also had higher levels of MRD at the end of the induction phase (⩾10−2: 8.6% versus 1.9%; ⩾10−3 to <10−2: 12.5% versus 5.6%; P<0.001). Consequently, they received the VHR regimen more frequently (20.1% versus 8.1%; P<0.001).
IKZF1del is an independent predictor of poorer outcome
For the entire group of 1223 BCR-ABL1-negative BCP-ALL patients, the 8-year EFS and OS rates were 83.6% and 91.5%, respectively. As expected, IKZF1del was associated with a lower 8-year EFS rate (Figure 2a), because of a higher rate of relapse (25.7% versus 10.8%; P<0.001). IKZF1del was also associated with a moderately lower 8-year OS rate (Figure 2b).

Kaplan–Meier estimates of event-free survival (a) and overall survival (b) in BCR-ABL1-negative BCP-ALL patients with or without IKZF1del. HR, hazard ratio; CI, confidence interval.
To address the added value of IKZF1del in the context of current risk stratification, we performed multivariate analyses after adjusting for conventional risk criteria (Table 3 and Supplementary Table 2). In a Cox model including National Cancer Institute criteria, response to prephase and genetic risk groups, IKZF1del was significantly related to a lower EFS (HR=1.70; 95% CI=1.22–2.38; P=0.002). It was also related to a lower DFS when including day 35 MRD (⩾ versus <10−3) in the model (HR=1.57; 95% CI=1.10-2.22; P=0.012). Notably, the strong prognostic significance of MRD observed for the entire cohort was also found in patients with IKZF1del (Supplementary Figure 1). Thus, IKZF1del, genetic classification, and MRD have independent prognostic value, definitely confirming that the poor outcome associated with IKZF1del was not merely the result of association with current high-risk features.
Combining IKZF1del and classifying genetic abnormalities refines genetic risk stratification
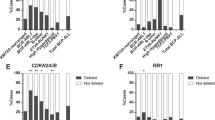
The biology and response to treatment of BCP-ALL cases primarily depends on classifying genetic abnormalities. We hypothesized that the effect of additional lesions would differ according to the oncogenic background. In this regard, we and others have previously observed that IKZF1del does not hamper the good outcome of ERGdel ALL cases.18, 20 To this purpose, we used Forest plots to analyze the prognostic impact of IKZF1del in association with distinct classifying genetic lesions (Figure 3a). Strikingly, IKZF1del was significantly associated with a lower 8-year EFS in only two groups: high hyperdiploidy and ‘B-other‘ (Figures 3b and c). In patients with high hyperdiploidy, the 8-year EFS rate was 76.2% in patients with IKZF1del versus 90.7% in non-IKZF1del patients (HR=2.57; 95% CI=1.19–5.55; P=0.013). In ‘B-other‘ patients, the 8-year EFS rate was 56.4% in patients with IKZF1del versus 79.0% in non-IKZF1del patients (HR=2.22; 95% CI=1.45–3.39; P<0.001). In multivariate analyses focused on each of these two groups, IKZF1del was independently related to a lower EFS and DFS (Supplementary Tables 3 and 4).

Forest plot analysis of IKZF1del according to genetic subgroups (a). Forest plots are based on hazard ratios and interaction tests computed using the Cox model. Kaplan–Meier estimates of event-free survival in BCP-ALL patients with high hyperdiploidy (b) and ‘B-other' ALL (c), according to the presence or absence of IKZF1del. CI, confidence interval; HR, hazard ratio.
Clinical significance of distinct types of IKZF1 deletions
Different types of IKZF1 deletions can be observed, but whether they equally affect prognosis is still an open question. We subdivided IKZF1del cases into three groups: whole-gene deletions resulting in haploinsufficiency, including the loss of chromosome 7p (n=66, 37%), intragenic deletion of exons 4–7, producing dominant negative isoforms (Δ4–7, n=62, 35%) and rare intragenic deletions (n=51, 28%). There was no significant difference (P=0.72) in EFS according to the type of deletion (Supplementary Figure 2). Yet, examination of clinical and biological characteristics revealed that high-risk features were unequally distributed among these three groups (Supplementary Tables 5 and 6). Whole-gene deletions were more frequently associated with poor-prognosis genetic abnormalities (13/66, 20%, as compared with 5/62, 8% for Δ4–7 and 1/51, 2% for rare intragenic deletions). On the other hand, patients with rare intragenic deletions had higher WBC counts (median 20.1 × 109/l) and more frequently presented a poor early response to treatment (poor response to prephase: 17.6%; induction failure: 5.9%; MRD level ⩾10−2: 17.1%). Accordingly, patients with whole-gene deletion and rare intragenic deletions were more often treated with the VHR regimen than patients with Δ4–7 (25.8% and 23.5%, respectively, versus 11.3%; P<0.001), and yet this resulted in similar EFS rates.
Pulses during maintenance prevent relapses in patients with IKZF1del
One of the aims of the EORTC-CLG 58951 trial was to evaluate the benefit of vincristine and corticosteroid pulses during maintenance therapy, as part of a large intergroup I-BFM study.21 The randomization applied to patients from the AR group who were in CR at the end of late intensification. We previously showed that the administration of such pulses improved outcome of these patients.13
When analyzing the prognostic value of IKZF1del in the three distinct risk groups (VLR, AR and VHR), the negative impact was restricted to AR patients (8-year EFS 64.5% versus 87.6% in non-IKZF1del patients; HR=2.90; 95% CI=2.00–4.22; P<0.001; Supplementary Figure 3). Notably, most of the relapses in the IKZF1del-positive AR group of patients occurred rather late, after maintenance therapy. This prompted us to check whether the pulse randomization that was conducted during maintenance in AR patients had an effect on IKZF1del-related relapses. Characteristics of patients eligible for randomization from the studied cohort are shown in Supplementary Table 7. Among them, 220 patients, including 34 (15.5%) having IKZF1del were randomized. Strikingly, in this post-hoc analysis, the outcome of IKZF1del patients who received pulses was identical to that of non-IKZF1del patients (8-year DFS: 93.3% versus 89.5%; P=0.6), whereas the outcome of IKZF1del patients who did not receive pulses was significantly worse than that of non-IKZF1del patients (8-year DFS 42.1% versus 88.8%; HR=6.65; P<0.001; Figure 4). A significant interaction between IKZF1del and treatment (pulses versus no pulses) was also found in multivariate analyses for DFS (Supplementary Table 8). These findings suggest that the intensification of maintenance therapy with vincristine-steroid pulses has contributed to prevent relapses in patients with IKZF1del. Interestingly, in a forest plot analysis including other variables such as age, WBC, MRD and genetic groups (Supplementary Figure 4), IKZF1del was the main factor that influenced the outcome in relation to treatment difference, with HR=0.09 in patients with IKZF1del versus HR=1.02 in patients without IKZF1del (P=0.012).

Kaplan–Meier estimates of disease-free survival (a) and overall survival (b) in the population of patients who were randomized for pulses, according to the presence or absence of IKZF1del.
Discussion
Our results are consistent with previous data showing inferior outcome in patients with IKZF1del mainly in the intermediate risk groups.10, 11, 12, 22 In addition, our large cohort of BCP-ALL children treated in a single MRD-stratified protocol allowed us to definitively confirm the independent prognostic value of IKZF1del, together with MRD and genetic classification. Importantly, the majority of IKZF1del-positive patients who relapsed had no other high-risk features, emphasizing the value of including IKZF1del in risk-stratification algorithms. However, it could be argued that an 8-year EFS rate of nearly 70% is not low enough to warrant the use of IKZF1del for treatment intensification. In addition, the fact that most of these relapses can be rescued by second-line treatment raises the question of the appropriateness of exposing a large number of patients who will not relapse to the toxicity of therapeutic intensification.
We showed here that the significant impact of IKZF1del was restricted to two genetic subgroups, high hyperdiploidy and ‘B-other‘, although in other subtypes the small number of cases and/or low frequency of IKZF1del do not allow definite conclusions. Interestingly, although IKZF1del in those two subgroups was associated with a comparable increased risk in terms of HR, this resulted in strikingly distinct EFS rates. The presence of IKZF1del turned the normally excellent prognosis of patients with high hyperdiploidy into an intermediate prognosis, whereas in the ‘B-other‘ ALLs, the presence of IKZF1del was associated with an EFS of 56%, which is as low as that of patient subgroups with well-recognized very-high-risk features, such as MLL translocations, low hypodiploidy/near-haploidy or MRD≥10−2 (8-year EFS rate of 64%, 55% and 8-year DFS rate of 58%, respectively, in EORTC-CLG 58951). The ‘B-other‘/IKZF1del-positive ALLs represented 7.2% of all patients and accounted for up to 20% of relapses. As genetic classification is already implemented in routine analyses of ALL at diagnosis in most countries, the simple addition of IKZF1del testing provides an easy and relatively cost-effective assay for the identification of a significant fraction of patients at very high risk of relapse. Therapeutic interventions focusing on this subgroup of patients may thus be of particular interest to improve outcome in BCP-ALL while limiting the inappropriate exposure of other patients to intensified treatment.
The genetic basis of ‘B-other‘ ALL is likely to be heterogeneous. Recently, several studies identified a high-risk subtype, termed ‘BCR-ABL1-like‘, having a gene expression profile similar to that of BCR-ABL1-positive ALL, and frequent IKZF1del.8, 23, 24, 25 BCR-ABL1-like ALL harbor a large variety of genomic alterations deregulating signaling pathways26 and their identification is challenging in terms of prospective diagnosis.27 Noteworthy, both IKZF1del and BCR-ABL1-like were independently related to a poor prognosis in a recent study,28 indicating that the poor outcome of ‘B-other‘ patients with IKZF1del is not solely due to ‘BCR-ABL1-like‘ cases.
In addition to the uneven distribution of IKZF1del among genetic subgroups, our data show the preferential association of distinct types of deletions with genetic subgroups. For instance, the ERGdel subtype is frequently associated with IKZF1 Δ4–7. In contrast, ETV6-RUNX1 ALL have virtually no Δ4–7, which is intriguing since this deletion is mediated by V(D)J recombination, a process that is effective in ETV6-RUNX1 ALL.29 In ‘B-other‘ ALL cases, rare intragenic deletions were more often associated with poor response to prephase, lack of CR and high MRD (Supplementary Table 9). Together, these data suggest that the incidence and clinical impact of distinct types of IKZF1del may vary according to the oncogenic environment.
The OS of patients with IKZF1del was much less affected than their EFS, implying that long-term remission could be achieved by second-line treatment, which relied on intensive chemotherapy alone in two-thirds of these patients. The fact that recurring leukemia cells retain chemosensitivity suggests that a more intensive first-line regimen could have prevented relapses. Although based on limited patient numbers, our results support an effective role for the vincristine-steroid pulses during maintenance therapy in preventing relapses in IKZF1del patients. The administration of pulses in several ongoing pediatric ALL protocols should therefore improve the outcome of IKZF1del patients. In addition, the fact that the benefit of pulses seems to be restricted to IKZF1del patients could allow pulses to be avoided in non-IKZF1del patients, restricting needless toxicity. It would have been interesting to confirm these findings in the patient cohorts of the I-BFM intergroup study, a meta-analysis evaluating the value of vincristine-dexamethasone pulses in intermediate risk patients.21 Unfortunately, IKZF1del was not studied in these patients. Moreover, by contrast with the EORTC study, no benefit of pulses could be observed in the intergroup study,13, 21 which could be explained by noticeable differences in risk group definition, and heterogeneity between participating groups. For instance, the EORTC randomized cohort although including a larger proportion of patients, excluded those with high MRD levels.
Considering the timing of IKZF1del-associated relapses in AR patients and the chemosensitivity to second-line therapy, it is plausible that other modifications intensifying maintenance treatment will yield a similar effect. In conclusion, IKZF1 status is a valuable criterion for risk-adapted stratification in the treatment of children with BCP-ALL. The addition of vincristine-steroid pulses during maintenance in patients with IKZF1del seems an effective and reasonable strategy for preventing relapses. It would thus be worthwhile to confirm our data in other randomized trials.
References
Pui CH, Robison LL, Look AT . Acute lymphoblastic leukaemia. Lancet 2008; 371: 1030–1043.
Gaynon PS, Angiolillo AL, Carroll WL, Nachman JB, Trigg ME, Sather HN et al. Long-term results of the children's cancer group studies for childhood acute lymphoblastic leukemia 1983–2002: a Children's Oncology Group Report. Leukemia 2010; 24: 285–297.
Pui CH, Evans WE . A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin Hematol 2013; 50: 185–196.
Roberts KG, Pei D, Campana D, Payne-Turner D, Li Y, Cheng C et al. Outcomes of children with BCR-ABL1-like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol 2014; 32: 3012–3020.
Moorman AV . The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev 2012; 26: 123–135.
Teachey DT, Hunger SP . Predicting relapse risk in childhood acute lymphoblastic leukaemia. Br J Haematol 2013; 162: 606–620.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007; 446: 758–764.
Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009; 360: 470–480.
Kuiper RP, Waanders E, van der Velden VH, van Reijmersdal SV, Venkatachalam R, Scheijen B et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia 2010; 24: 1258–1264.
Dorge P, Meissner B, Zimmermann M, Moricke A, Schrauder A, Bouquin JP et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica 2013; 98: 428–432.
Palmi C, Lana T, Silvestri D, Savino A, Kronnie GT, Conter V et al. Impact of IKZF1 deletions on IKZF1 expression and outcome in Philadelphia chromosome negative childhood BCP-ALL. Reply to "incidence and biological significance of IKZF1/Ikaros gene deletions in pediatric Philadelphia chromosome negative and Philadelphia chromosome positive B-cell precursor acute lymphoblastic leukemia". Haematologica 2013; 98: e164–e165.
Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 2012; 119: 3512–3522.
De Moerloose B, Suciu S, Bertrand Y, Mazingue F, Robert A, Uyttebroeck A et al. Improved outcome with pulses of vincristine and corticosteroids in continuation therapy of children with average risk acute lymphoblastic leukemia (ALL) and lymphoblastic non-Hodgkin lymphoma (NHL): report of the EORTC randomized phase 3 trial 58951. Blood 2010; 116: 36–44.
Domenech C, Suciu S, De Moerloose B, Mazingue F, Plat G, Ferster A et al. Dexamethasone (6 mg/m2/day) and prednisolone (60 mg/m2/day) were equally effective as induction therapy for childhood acute lymphoblastic leukemia in the EORTC CLG 58951 randomized trial. Haematologica 2014; 99: 1220–1227.
Biondi A, Schrappe M, De Lorenzo P, Castor A, Lucchini G, Gandemer V et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 2012; 13: 936–945.
van der Veer A, Zaliova M, Mottadelli F, De Lorenzo P, Te Kronnie G, Harrison CJ et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 2014; 123: 1691–1698.
Guidal C, Vilmer E, Grandchamp B, Cave H . A competitive PCR-based method using TCRD, TCRG and IGH rearrangements for rapid detection of patients with high levels of minimal residual disease in acute lymphoblastic leukemia. Leukemia 2002; 16: 762–764.
Clappier E, Auclerc MF, Rapion J, Bakkus M, Caye A, Khemiri A et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 2014; 28: 70–77.
Caye A, Beldjord K, Mass-Malo K, Drunat S, Soulier J, Gandemer V et al. Breakpoint-specific multiplex polymerase chain reaction allows the detection of IKZF1 intragenic deletions and minimal residual disease monitoring in B-cell precursor acute lymphoblastic leukemia. Haematologica 2013; 98: 597–601.
Zaliova M, Zimmermannova O, Dorge P, Eckert C, Moricke A, Zimmermann M et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 2014; 28: 182–185.
Conter V, Valsecchi MG, Silvestri D, Campbell M, Dibar E, Magyarosy E et al. Pulses of vincristine and dexamethasone in addition to intensive chemotherapy for children with intermediate-risk acute lymphoblastic leukaemia: a multicentre randomised trial. Lancet 2007; 369: 123–131.
Waanders E, van der Velden VH, van der Schoot CE, van Leeuwen FN, van Reijmersdal SV, de Haas V et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia 2011; 25: 254–258.
Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009; 10: 125–134.
Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 2010; 116: 4874–4884.
Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 2014; 371: 1005–1015.
Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 2012; 22: 153–166.
Izraeli S . Beyond Philadelphia: 'Ph-like' B cell precursor acute lymphoblastic leukemias - diagnostic challenges and therapeutic promises. Curr Opin Hematol 2014; 21: 289–296.
van der Veer A, Waanders E, Pieters R, Willemse ME, Van Reijmersdal SV, Russell LJ et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 2013; 122: 2622–2629.
Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet 2014; 46: 116–125.
Acknowledgements
The authors thank all the EORTC-CLG study group members. We thank the many clinicians and biologists for their participation in the study. The authors also thank the EORTC HQ Data Management Department members (Séraphine Rossi, Lies Meirlaen, Liv Meert, Aurélie Dubois, Christine Waterkeyn, Alessandra Busato, Isabel VandeVelde and Gabriel Solbu) for their support of this trial. We thank S Rasika for English editing of the manuscript. This work was supported by a donation from the La Ligue Nationale Contre le Cancer from France through the EORTC Charitable Trust and has received founding from the European Union’s Seventh Framework Program (PF7/2007-2013) under the ERA-net TRANSCAN project TRANSCALL.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
About this article

Cite this article
Clappier, E., Grardel, N., Bakkus, M. et al. IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children's Leukemia Group study 58951. Leukemia 29, 2154–2161 (2015). https://doi.org/10.1038/leu.2015.134
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2015.134
- Springer Nature Limited
This article is cited by
-
Prevalence and prognostic significance of IKZF1 deletion in paediatric acute lymphoblastic leukemia: A systematic review and meta-analysis
Annals of Hematology (2023)
-
Cytogenetics or MRD in B-cell ALL. Do both reign supreme?
Leukemia (2022)
-
Maintenance therapy for acute lymphoblastic leukemia: basic science and clinical translations
Leukemia (2022)
-
Hsp90/C terminal Hsc70-interacting protein regulates the stability of Ikaros in acute myeloid leukemia cells
Science China Life Sciences (2021)
-
Ikaros family zinc-finger 1 mutation is an independent factor for the poor prognosis of adult B-cell acute lymphoblastic leukemia, and allogeneic hematopoietic stem cell transplantation can improve clinical outcomes
Bone Marrow Transplantation (2019)