
Abstract
Background/Objectives:
The current world-wide obesity epidemic partially results from a vicious circle whereby maternal obesity during pregnancy predisposes the offspring for accelerated weight gain and development of metabolic syndrome. Here we investigate whether low-grade inflammation, characteristic of the obese state, provides a causal role for this disastrous fetal programming in mice.
Methods:
We exposed pregnant and lactating C57BL/6JBom female mice to either high-fat diet (HFD), or continuous infusion of lipopolysaccharide (LPS), a potent trigger of innate immunity, and studied offspring phenotypes.
Results:
Both maternal LPS or HFD treatments rendered the offspring hyperphagic and inept of coping with a HFD challenge during adulthood, increasing their adiposity and weight gain. The metabolic effects were more pronounced in female offspring, while exposed male offspring mounted a larger inflammatory response to HFD at adulthood.
Conclusions:
This supports our hypothesis and highlights the programming potential of inflammation in obese pregnancies.
Similar content being viewed by others

Introduction
Maternal obesity significantly increases complication risk during pregnancy and at delivery, and exerts detrimental long-term consequences for the health of the offspring.1 Offspring born to obese mothers are at higher risk of becoming obese and developing metabolic syndrome already in childhood, with dire consequences for their future health.2, 3, 4 These effects are particularly important in the context of the staggering global obesity epidemic that affects an increasing proportion of young women.5 By 2012 one-third of women of childbearing age in the USA were obese6 implicating a substantial risk of exposure to maternal obesity during prenatal development for a large part of the future population.
Animal studies clearly demonstrate that obesity and consumption of diets rich in saturated fat during pregnancy or lactation increase the risk for obesity and metabolic syndrome in the offspring, even when they consume normal diets throughout adulthood.7 These effects are further exacerbated when the offspring themselves consume a high-fat diet (HFD).8, 9, 10 Consequently, this may propagate development of a vicious circle, where the prevalence of obesity and resulting diseases are reinforced in the following generations. While the association between maternal and offspring obesity is rather well described by epidemiological and animal studies, the underlying mechanisms remain elusive.
Obesity is strongly associated with a state of low-grade, chronic inflammation that has been put forward as the culprit in development of insulin resistance, atherosclerosis and cardiovascular disease.11 The exact source of inflammation in obesity is yet to be identified, but gut derived endotoxemia—increased level of lipopolysaccharide (LPS) in the circulation—may be an important contributor. LPS is naturally present in the intestinal lumen as a component of the cell wall of Gram-negative bacteria and is a potent trigger of the innate immune system.12, 13 Following ingestion of a meal, LPS can be translocated across the intestinal wall into the circulation along with uptake of nutrients, thereby initiating a transient postprandial endotoxemia.14, 15 Frequent consumption of HFD chronically elevates LPS in the circulation and thus contributes to propagation of the low-grade inflammation characteristic to obesity.13, 14, 16, 17
Low grade, chronic inflammation characteristic to obesity18 has emerged as a compelling underlying factor capable of programming offspring obesity and metabolic syndrome by maternal obesity.19 This is backed by evidence that elevated levels of inflammatory markers or an induced inflammatory event during pregnancy can alter metabolism and future health of the offspring.20, 21, 22 Due to the proposed role of LPS in obesity-related inflammation, it is applicable to use chronic infusion of LPS to experimentally induce enduring, low-grade inflammation in animal models. This allows for investigation of the role of obesity-related inflammation in the etiopathophysiology of associated comorbidities in absence of other co-variates.14, 23 Chronic intraperitoneal administration of LPS mimics the origin of endotoxemia from the gut without affecting glucose tolerance in mice and rats.24, 25, 26 Hence chronic intraperitoneal infusion of LPS during pregnancy and/or lactation can be used to evaluate the mediating role of low level, chronic inflammation in programming of offspring phenotype by maternal obesity or high-fat feeding in absence of other confounding factors.
We hypothesized that exposing female mice to chronic low doses of LPS during pregnancy and lactation will have similar programming effects on offspring phenotype as maternal consumption of HFD. We exposed female mice to either control diet and LPS (chronically delivered for a period of 60 days via intraperitoneal slow release pellet), or ad libitum access to HFD during pregnancy and lactation. We kept the offspring on a control diet from weaning until 19 weeks of age, and then switched them onto a diet rich in saturated fat (HFD). While consuming control diet, female offspring from LPS and HFD groups were only slightly heavier than controls, but the consequent HFD challenge uncovered the phenotypic differences induced by maternal treatments. The challenge rendered LPS and HFD offspring hyperphagic, obese and altered their inflammatory phenotype in comparison to control offspring, emphasizing the programmed impaired capacity to cope with a post-natal HFD exposure. The resulting similarities between offspring phenotypes induced by maternal LPS or HFD provide experimental evidence that inflammation can be a key programming factor in pregnancies affected by such multifactorial conditions as obesity and HFD.
Materials and methods
The experiments were approved by The Danish Animal Experiments Inspectorate, permit # 2012-15-2934-00042.
Maternal treatment
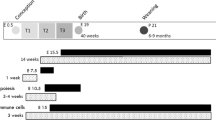
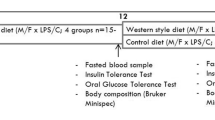
Six-week old (w.o.) female (n=28), virgin C57BL/6JBomTac mice (Taconic, Lille Skensved, Denmark) were group-housed at 24 °C, 12:12 h light:dark with lights on at 0630 hours, at 60% humidity and fed laboratory chow. Food and water were available ad libitum at all times, except when food was removed for 6 h before oral glucose tolerance test (OGTT) and 4 h before euthanasia. After 2 weeks of acclimation, females were weighed and divided into 3 groups with similar body mass (mean 19.8±0.3 g, n=9–10). Custom-made slow release pellets (Innovative Research of America, Sarasota, FL, USA) lasting 60 days to cover pregnancy and lactation, and designed to deliver a constant dose of LPS (6.45 μg day−1, Escherichia coli 055:B5, Sigma-Aldrich, Brøndby, Denmark) or only the biodegradable pellet matrix, were implanted intraperitoneally. Nine mice received LPS pellet, while only pellet matrix was implanted in control (n=9) and HFD (n=10) mice. The LPS dose was chosen to resemble the level of endotoxemia induced by high-fat feeding in mice.14 Immediately after surgery, control and LPS mice were switched onto control diet (#D12450B, 10% kcal from fat), whereas the remaining 10 mice were switched onto a lard-based HFD (#D12492, 60% kcal from fat, both diets from Research Diets Inc., New Brunswick, NJ, USA). Mice were housed individually until mating and were kept on the corresponding diets during post-surgical recovery, mating, pregnancy and lactation.
One week after pellet implantation each female was placed in a cage with an age matched, 9 w.o. C57BL/6JBomTac male. Until breeding all males consumed control diet, but during mating males were feeding on the same diet as the corresponding females. Females were weighed twice a week, and the male was removed from the cage during the last week of pregnancy. Females were allowed to give birth and the day of birth was recorded. The offspring were weaned at the age of 24 days, and the mothers were subjected to OGTT. They were euthanized 4 days later.
Offspring
Offspring were counted and weighed 3 days after birth. Litters with fewer than 3 pups were culled on the 3rd day after birth (n=1 in HFD group). All other offspring remained with their mothers during lactation, and were weighed twice per week. At the age of 24 days, when nursing is finished in mice,27 a maximum of 2 male and 2 female offspring per litter were randomly chosen and weaned onto control diet, and the rest of the litter was euthanized. Female offspring were group-housed throughout the experiment (3–6 females per cage), while all male offspring were group-housed until 13 w.o. and most were then switched to single housing to avoid aggressive behavior.
Until 19 w.o. all offspring were fed a control diet (#D12450B). From weaning until 13 w.o. all offspring were weighed weekly but repeated individual body mass development was not tracked, therefore data was analyzed separately for each time point. After 13 w.o. caloric intake and individual body mass were measured weekly until 19 w.o. when all offspring were switched to HFD (#D12492), whereupon food intake and individual body mass were measured twice a week.
OGTT was performed at 15 and 23 w.o. and body composition was measured at 18 and 21 w.o. using echo magnetic resonance (MR) scanner for rodents. All offspring were euthanized and tissue samples collected at 24 w.o.
Detailed description of the other methods used is provided in Supplementary Material.
Data analysis
Data is presented as means±s.e.m. and ‘n’ refers to number mice included in the analysis. Data from male and female offspring was always analyzed separately and ‘Maternal ID’ was always included in the initial analysis as a co-factor to control for litter effect, however it never had a significant effect, and was then removed from further analysis.
Data was compared using one-way analysis of variance (ANOVA) to test for the effect of maternal treatment and Tukey’s range test was used for post hoc analysis. Area under the curve (AUC) was calculated for blood glucose levels during OGTT and compared using ANOVA. Continuous changes in body mass and food intake, the effect of post-natal diet on body composition and same time point insulin levels before and after HFD challenge, were compared using Repeated measures (RM) ANOVA. Fasting and 15 min insulin within the same OGTT were compared using RM ANOVA. Body mass, along with maternal treatment, was included as a co-variate (ANCOVA) when analyzing plasma leptin and cholesterol levels. Analyses were performed using JMP 13.0 (SAS Institute Inc., Cary, NC, USA).
Results
Maternal phenotype and pregnancy outcomes
Post-operative body mass loss occurred in all experimental groups, but was largest among LPS mice (Figure 1a), indicating successful LPS delivery and resultant systemic immune response and sickness behavior.28 Throughout pregnancy and lactation HFD females gained more weight than control or LPS females (Figure 1a, RM ANOVA: P=0.0002, Tukey’s test: control vs HFD P<0.01, Control vs LPS non-significant (n.s.), HFD vs LPS P<0.04).

Maternal phenotype. (a) Maternal body mass throughout the experiment. (b) Maternal oral glucose tolerance test (OGTT) at the time of weaning (24 days post-partum), calculated area under the curve (AUC) for plasma glucose, and plasma insulin before and 15 min after oral glucose gavage. (c) Relative mRNA expression in liver and visceral adipose tissue of mothers 4 days after weaning the offspring. Data presented as means±s.e.m. Body mass and plasma insulin were compared using RM ANOVA; AUC and relative gene expression were compared using one-way ANOVA followed by Tukey’s test. e Indicates statistically significant difference (P<0.05) between LPS and Control, f between HFD and control, and g between LPS and HFD. Bars not connected by the same letter, are statistically different (Tukey’s test, P<0.05).
HFD dams had impaired glucose tolerance (Figure 1b), and increased visceral fat mass (Supplementary Table 3) compared with controls and LPS, as well as increased plasma insulin levels during fasting and 15 min after glucose gavage, compared with LPS females that had the lowest insulin levels (Figure 1b).
We compared the level of inflammation induced by HFD or LPS by measuring plasma levels of several pro-inflammatory markers using Luminex assay, however did not find a statistically significant effect of maternal treatment. Only plasma leptin was significantly elevated in HFD females (Supplementary Table 4). Even though the mean levels of inflammatory markers (assessed by measuring relative mRNA expression) were almost two-fold higher in livers of LPS mothers compared with controls, there was no statistically significant effect of maternal treatment on relative mRNA expression of TNF-α, interleukin (IL)-1β or CD14 in neither liver, nor adipose tissue (Figure 1c).
Mating success (23 litters — nControl=7, nLPS=7, nHFD=9 — from 28 initial mating pairs), time from introducing the mating pair to delivery (21.4 days) and litter size (6 pups) were not affected by maternal treatment.
Offspring body mass and body composition on control diet
Maternal treatment did not affect offspring body mass 3 days after birth, however LPS and HFD female offspring, and HFD male offspring gained significantly more body weight during lactation compared with controls (RM ANOVA: Female and male offspring P=0.03; Tukey’s test, Females: LPS and HFD vs control P<0.03 between 11 and 15 days old, n.s. rest of time, LPS vs HFD n.s. throughout lactation; Males: HFD vs control P<0.05 between 11 and 19 days old, LPS vs control and LPS vs HFD n.s.; Figure 2). From weaning until 9 w.o. LPS and HFD female offspring retained the higher body mass (3-13 w.o.: ANOVA: P<0.03 throughout the period, Tukey’s test, 3-9 w.o.: control vs LPS P<0.05; control vs HFD P<0.01; LPS vs HFD ns; 10–13 w.o.: control vs LPS n.s, control vs HFD P<0.04, LPS vs HFD n.s.). However differences in male offspring body mass disappeared already after 1.5 weeks (3–4.5 w.o., ANOVA: P<0.001, thereafter n.s.). After 13.5 w.o. only LPS and HFD female offspring retained elevated body mass (RM ANOVA, Females: 13–18 w.o.: P=0.007, Tukey’s test: control vs HFD P<0.04, control vs LPS P in range of 0.01–0.2, LPS vs HFD n.s.).

Offspring body mass. Circles represent female offspring and squares male offspring. Data presented as means±s.e.m. f Indicates statistical effect of maternal treatment among female and m among male offspring (RM ANOVA: P<0.05). Data from lactation shown on light gray background, data acquired during a HFD challenge shown on dark gray background. OGTT, oral glucose tolerance test; body comp., body composition.
MR showed a statistically significant elevation of lean body mass among female LPS and HFD offspring at 18 w.o. (Figure 3, ANOVA, P=0.01, Tukey’s test: control vs LPS and HFD P<0.03, LPS vs HFD n.s.), but no significant changes in fat mass or fat percent (17% fat for LPS and HFD female offspring, 15% fat for controls). Male offspring lean body mass was not affected by maternal treatment at the age of 18 weeks, and fat percent was similar among all the groups (12%).

Offspring body composition. Offspring body composition determined using EchoMRI at the age of 18 and 21 weeks (before and during the HFD challenge), and visceral fat mass measured after euthanasia at the age of 24–25 weeks. Data acquired during or after a HFD challenge shown on dark gray background. Data presented as means±s.e.m. and compared using RM ANOVA. Bars not connected by the same letter within the same sex are statistically different (Tukey’s test P<0.05).
Offspring body mass and body composition on HFD
Post-natal HFD challenge increased offspring body mass in all groups (Figure 2), but exposure to maternal LPS or HFD magnified weight gain among female offspring (RM ANOVA: P=0.0006, Tukey’s test: LPS and HFD vs Control P<0.02, LPS vs HFD n.s.). The rise in body mass stemmed from increased deposition of fat (Figure 3, RM ANOVA, 18 vs 21 w.o. P<0.001) that was larger among LPS and HFD females (Figure 3, Tukey’s test, 21 w.o., control vs LPS and HFD P<0.05, HFD vs LPS n.s.) and a small rise in lean body mass (Figure 3, RM ANOVA, 18 vs 21 w.o. P<0.0001, Tukey’s test, 21 w.o., control vs LPS and HFD P<0.001, LPS vs HFD n.s.). This lead to a near-significant difference in fat percent (control=23.7±1.9%, LPS=28.8±2.0%, HFD=29.4±1.8%; ANOVA, P=0.08) and an elevation of visceral fat mass at 24 w.o. (Figure 3, ANOVA, P=0.01, Tukey’s test, control vs LPS and HFD P<0.03, LPS vs HFD n.s.). Male offspring showed a similar pattern but did not reach significant differences between maternal treatments.
Offspring energy intake
Maternal LPS or HFD increased daily caloric intake among female offspring already on control diet (Figure 4, RM ANOVA: P<0.0001, Tukey’s test: LPS vs control P<0.003 (14–19 w.o.), HFD vs control P<0.001 (14–16 and 17–19 w.o.), LPS vs HFD P<0.001 (13–15 w.o.), n.s. after 16 w.o.), but the HFD challenge further magnified the differences. The switch to HFD increased caloric intake among all females for the first 3 days, but LPS and HFD female offspring retained the higher daily caloric intake throughout the experiment (RM ANOVA: P<0.0001, Tukey’s test, LPS and HFD vs Control P<0.0002 at all time points; HFD vs LPS n.s., Figure 4).

Offspring daily caloric intake. Offspring daily caloric intake measured once or twice per week, as the weight of feed consumed per animal, per day and multiplied by energetic content of the diet. Data acquired during a HFD challenge shown on dark gray background. Data presented as means±s.e.m. for female (top) and male (bottom) offspring. f Indicates statistical effect of maternal treatment among female and m among male offspring (RM ANOVA: P<0.05).
Maternal treatment did not affect daily caloric intake of males on control diet (Figure 4), but HFD challenge elicited a similar but lesser response as among female offspring. Caloric intake spiked during the first 3 days among all groups, but in spite of a significant effect of the overall maternal treatment (RM ANOVA: P=0.01) the statistical differences between treatments varied throughout the period (Tukey’s test, n.s.).
Offspring OGTT
Offspring glucose tolerance and insulin secretion were not affected by maternal treatment at the age of 15 weeks (Figure 5). After 4 weeks of HFD, glucose tolerance decreased among all offspring, as indicated by increased AUC values compared with the first OGTT (RM ANOVA, HFD exposure P<0.0001 for males and females), however no differences appeared between the treatment groups. Nevertheless, after the HFD challenge LPS and HFD female offspring had higher fasting plasma insulin levels than controls (ANOVA, P=0.02, Tukey’s test, LPS and HFD vs control P<0.04, LPS vs HFD ns., Figure 5b).

Offspring oral glucose tolerance tests (OGTT). (a) Plasma glucose values and the calculated area under the curve (AUC) during OGTT. (b) Plasma insulin values during OGTT. Results from OGTT performed on control diet (15 weeks old) are depicted on white background, and on high-fat diet (HFD; 23 weeks old) on dark gray background. Data presented as means±s.e.m. d Indicates significant effect of post-natal diet (control diet vs HFD) on insulin at the specific time point, and t indicates significant effect of time of measurement (fasting vs 15 min) within the same diet (RM ANOVA: P<0.05 within the same diet, or time point, respectively). Bars not connected by the same letter display a statistically significant effect of maternal diet on insulin level at that time point (ANOVA, followed by Tukey’s test).
Offspring inflammatory markers
Maternal LPS significantly increased blood leukocyte count in males (ANOVA: P=0.01, Tukey’s test: LPS vs control P=0.001, HFD vs control n.s., LPS vs HFD n.s.) and had a similar trend in female LPS and HFD offspring (ANOVA, P=0.07; Figure 6). Despite this, no differences were detected in plasma inflammatory cytokines assessed by Luminex (Supplementary Table 4) with the exception of IL-12 (p40) and IL-13 that were significantly higher in LPS female offspring compared with controls.

Offspring blood leukocyte count. Offspring blood leukocyte count was performed on the day of euthanasia, at the age of 24-25 weeks. Data presented as means±s.e.m. and compared using one-way ANOVA, followed by Tukey’s test. Bars not connected by the same letter are statistically different (P<0.05).
We also used a Luminex assay to measure levels of inflammatory markers in offspring hypothalamus. Here, LPS male offspring had significantly higher levels of most measured inflammatory markers, with HFD males being generally between LPS and Controls, and Controls presenting with the lowest levels of inflammatory markers (Supplementary Table 5). Hypothalamic inflammatory markers were not affected by maternal treatment among female offspring.
Despite the pronounced inflammation in the hypothalamus we did not observe effects of maternal treatment on relative mRNA expression of a selected set of inflammatory genes in offspring liver or adipose tissue for male or female offspring (Supplementary Figure 1).
Offspring leptin levels and tissue lipid content
At the age of 24 weeks LPS and HFD female offspring had increased plasma leptin levels compared with controls (ANOVA, P=0.005, Tukey’s test, LPS and HFD vs control P<0.01, LPS vs HFD n.s., Supplementary Table 6). However inclusion of body mass in the analysis as a co-variate revealed that plasma leptin was associated with body mass rather than maternal treatment in both female (ANCOVA, body mass: P<0.0001; maternal treatment: n.s.) and male offspring (ANCOVA, body mass: P<0.0001; maternal treatment: n.s.). Offspring muscle and liver lipid content was not significantly affected by maternal treatment (Supplementary Table 7).
DNA methylation in female offspring
To link the phenotypic changes induced by maternal treatment to epigenetic alterations we examined overall DNA methylation among female offspring, as they had a more pronounced metabolic phenotype than male offspring. We measured Line-1 methylation and methylation patterns of candidate genes (insulin-like growth factor 2 (Igf-2) –differential methylated region 2 (DMR2) and peroxisome proliferator-activated γ coactivator-1α (PGC-1α), a transcriptional coactivator involved in mitochondrial biosynthesis and oxidative metabolism) in muscle tissue, however we did not observe effects of maternal treatment on any of the measured parameters (Supplementary Figure 2).
Discussion
Here we show that chronic maternal inflammation during early development changes offspring phenotype at adulthood, and worsens their ability to cope with a HFD challenge. Importantly, offspring exposed solely to maternal inflammation resemble offspring born to obese mothers. As their phenotypes are programmed in the absence of many confounding factors characteristic to maternal obesity (hyperleptinemia, hyperglycemia, hyperinsulinemia or increased fat deposition), our study demonstrates the potent programming role of chronic inflammation in early development.
Although maternal treatment did not affect birth weight, LPS and HFD offspring became heavier during lactation (Figure 2). This outcome could be partially explained by exposure to maternal HFD during late lactation among HFD offspring, as mice pups start consuming hard chow from ca. 17 days of age.27 As we weaned the offspring onto control diet only at the age of 24 days to allow for nursing to completely cease27 the pups were exposed to some amounts of HFD for up to 7 days during late lactation, and it could have affected their early body mass. LPS offspring, however, were also heavier than controls during lactation, and as they only had access to control diet, in addition to milk, these effects must stem from maternal inflammation.
Maternal obesity or high-fat feeding does not universally induce alterations in offspring body mass at adulthood when feeding a normal post-weaning diet9, 29 meaning that sometimes the detrimental metabolic phenotype induced by maternal programming may remain concealed. However, a HFD challenge, imposed at adulthood, can unveil the programmed alterations.9 Similarly to previous studies on male offspring exposed to maternal HFD and then challenged with a HFD at adulthood9 we observed that HFD female offspring gained more weight than controls (Figure 2) and developed insulin resistance (Figure 5) following exposure to HFD after the age of 19 weeks. Interestingly, LPS female offspring had a very similar response to the HFD challenge—increased weight gain and appetite, and development of insulin resistance (Figures 2 and 5). This shows that maternal LPS or HFD induced similar changes in offspring phenotype, rendering them less capable to deal with high-fat challenge at adulthood. Response among male HFD and LPS offspring was very similar to that among female offspring, however it did not reach statistical significance.
Notably, the elevation of body mass among LPS and HFD female offspring was accompanied by a large increase in body fat, and particularly elevated visceral fat deposition (Figure 3). Again, the pattern was similar, but not statistically significant among affected male offspring (Figure 3). The increased visceral fat deposition points to programming of a particularly ‘dangerous’ phenotype among LPS and HFD offspring, as visceral obesity is tightly linked with insulin resistance and development of metabolic syndrome.30, 31 In line with this, the HFD challenge increased fasting levels of insulin among LPS and HFD female offspring indicating development of insulin resistance (Figure 5).32 In addition to increase in body weight, and body fat, we also observed a consistent increase of lean body mass of all female offspring, that was larger among LPS and HFD female offspring (Figure 3). This is curious, as other studies have reported the opposite—reduced offspring lean body mass in response to post-natal HFD challenge.33 The underlying cause however remains to be investigated.
The increase in offspring body mass and adiposity during a HFD challenge was likely caused by altered appetite regulation or increased preference for high-fat food. Both female and male LPS and HFD offspring majorly increased their caloric intake when exposed to HFD at adulthood (Figure 4). Maternal high-fat or ‘junk food’ feeding during pregnancy and lactation increases offspring appetite even on control diet7 and their preference for high-fat food during adulthood.34 Here we show that maternal inflammation has a similar effect (Figure 4) and both factors are likely contributors. The observed elevation of such appetite regulating hormones as ghrelin, insulin and leptin in hypothalamus of LPS and HFD male offspring (Supplementary Table 5) support altered appetite regulation by maternal inflammation. Even though these were assessed in the whole hypothalamus, and not differentiated by hypothalamic nuclei, the general effects of ghrelin in appetite stimulation and long-term anorexic effects of insulin and leptin are universal.35 However as we observed differences in calorie intake in both male and female offspring, but only observed alteration in appetite hormones in male offspring, and hormones with opposite action were upregulated, it is difficult to pinpoint the exact interactions. It is therefore necessary to conduct further studies including food preference tests, and calorie-adjusted pair feeding, and more detailed analysis of central appetite regulation.
Male offspring showed chronic immune activation, with the largest effects observed in LPS, and intermediate in HFD male offspring evident from elevated leukocyte counts (Figure 6) and increased levels of a number of pro-inflammatory cytokines in the hypothalamus (Supplementary Table 5). A similar effect has been previously induced by maternal obesity and high-fat feeding, where male offspring developed a pro-inflammatory phenotype in the adipose tissue even on control diet.29 Hypothalamic inflammation can precede development of metabolic syndrome17, 36 and LPS and HFD male offspring had not only hypothalamic and systemic inflammation, but also increased caloric intake, and elevated levels of ghrelin, leptin and insulin during the HFD challenge (Figures 3 and Figures 6; Supplementary Table 5). This shows that maternal LPS or HFD imposes programming effects on male offspring phenotype at adulthood and affects their response to a HFD challenge. We did not observe any effects of maternal treatment on inflammatory measures among female offspring.
Sexual dimorphism is common in studies of maternal programming, however the general direction of effects is still not clear. Exposure to maternal inflammation during pregnancy has induced obesity only in female offspring21 or in both sexes,20 while maternal obesity or high-fat feeding has produced obese phenotypes in both sexes,7, 34, 37 or only male offspring38 and even resulted in some temporal variation.39 Here we observed a more pronounced metabolic phenotype among female offspring, and immune activation among male offspring. The underlying mechanisms governing sexual dimorphism are not elucidated and various actors have been suggested at play, for example, different developmental vulnerabilities,38 altered neuroendocrine regulation20 or placental and epigenetic effects.40 It is therefore important to always investigate effects in both offspring sexes, and conduct thorough meta-analyses that can aid in understanding the crucial factors.
Consumption of HFD or LPS injections in rodents exclusively during pregnancy increase offspring adiposity and affect metabolic phenotype21, 38, 41 while the lactation period is crucial for programming of offspring appetite.42 Here we observed elevated caloric intake among LPS and HFD offspring, especially during the HFD challenge (Figure 4) indicating possible programming of appetite regulation by maternal treatments. Maternal obesity during pregnancy and lactation impairs appetite regulation through offspring leptin resistance and the consequent hyperphagia that persists into adulthood.37, 39 Offspring from LPS and HFD groups presented with elevated central and peripheral leptin levels indicating development of leptin resistance (Supplementary Tables 5 and 6) resembling previous studies.39 The similarities between LPS and HFD offspring phenotypes indicate involvement of a convergent programming mechanism. Increased levels of pro-inflammatory cytokines and immune cells in the milk are a possible candidate because maternal obesity can increase milk’s inflammatory properties in mice and humans.43, 44, 45 It is therefore possible that both maternal HFD and LPS altered inflammatory properties of the milk in the present study, thus imposing programming effects during lactation, but this needs to be further elucidated.
We attempted to link the marked metabolic changes among female offspring to epigenetic alterations, but did not observe changes in any of the investigated methylation patterns in muscle tissue. This is unexpected, as PGC-1α, a key regulator of energy metabolism46 is hyper-methylated in muscle tissue of female mice exposed to maternal HFD during early development.47 In the present animal model, maternal effects of LPS and HFD were likely conveyed via different pathway.
Here we show that maternal low-grade, chronic inflammation induced by LPS during pregnancy and lactation leads to long-term programming of offspring phenotype rendering them more susceptible to development of obesity and metabolic syndrome, and less suited to deal with a HFD challenge even as adults. This phenotype is highly similar to that induced by maternal HFD. The similarities between offspring exposed to maternal LPS or HFD strongly indicate that inflammation is a key mechanism underlying the observed programming effects. Pinpointing a single programming effector in an otherwise multifactorial setting of obesity allows for future development of targeted anti-inflammatory interventions aimed at obese pregnancies that may protect future generations from development of obesity and metabolic syndrome. It is important to further identify the exact programming window (pregnancy or lactation) as well as the mechanism underlying programming by inflammation. It is also important to study the effects of mild anti-inflammatory interventions such as mild exercise48 and maternal food supplements.49, 50
References
Poston L, Harthoorn LF, van der Beek EM, Workshop CIE. Obesity in pregnancy: implications for the mother and lifelong health of the child. A consensus statement. Pediatr Res 2011; 69: 175–180.
Must A, Strauss RS . Risks and consequences of childhood and adolescent obesity. Int J Obesity 1999; 23: S2–S11.
O'Reilly JR, Reynolds RM . The risk of maternal obesity to the long-term health of the offspring. Clin Endocrinol 2013; 78: 9–16.
Bider-Canfield Z, Martinez M, Wang X, Yu W, Bautista M, Brookey J et al. Maternal obesity, gestational diabetes, breastfeeding and childhood overweight at age 2 years. Pediatr Obes 2016; 12: 171–178.
Flegal KM, Carroll MD, Ogden CL, Curtin LR . Prevalence and trends in obesity among US Adults, 1999-2008. JAMA 2010; 303: 235–241.
Catalano P, deMouzon SH . Maternal obesity and metabolic risk to the offspring: why lifestyle interventions may have not achieved the desired outcomes. Int J Obes 2015; 39: 642–649.
Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EHJM et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance - A novel murine model of developmental programming. Hypertension 2008; 51: 383–392.
White CL, Purpera MN, Morrison CD . Maternal obesity is necessary for programming effect of high-fat diet on offspring. Am J Physiol 2009; 296: R1464–R1472.
Kruse M, Seki Y, Vuguin PM, Du XQ, Fiallo A, Glenn AS et al. High-fat intake during pregnancy and lactation exacerbates high-fat diet-induced complications in male offspring in mice. Endocrinology 2013; 154: 3565–3576.
Fante T, Simino LA, Reginato A, Payolla TB, Vitoreli DC, Souza M et al. Diet-induced maternal obesity alters insulin signalling in male mice offspring rechallenged with a high-fat diet in adulthood. PLoS ONE 2016; 11: e0160184.
Hotamisligil GS . Inflammation and metabolic disorders. Nature 2006; 444: 860–867.
Alexander C, Rietschel ET . Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res 2001; 7: 167–202.
Laugerette F, Vors C, Peretti N, Michalski MC . Complex links between dietary lipids, endogenous endotoxins and metabolic inflammation. Biochimie 2011; 93: 39–45.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007; 56: 1761–1772.
Laugerette F, Vors C, Geloen A, Chauvin MA, Soulage C, Lambert-Porcheron S et al. Emulsified lipids increase endotoxemia: possible role in early postprandial low-grade inflammation. J Nutr Biochem 2011; 22: 53–59.
Moreira AP, Texeira TF, Ferreira AB, Peluzio Mdo C, Alfenas Rde C . Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr 2012; 108: 801–809.
Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 2012; 122: 153–162.
Gregor MF, Hotamisligil GS . Inflammatory mechanisms in obesity. Annu Rev Immunol 2011; 29: 415–445.
Segovia SA, Vickers MH, Gray C, Reynolds CM . Maternal obesity, inflammation, and developmental programming. Biomed Res Int 2014; 2014: 418975.
Dahlgren J, Nilsson C, Jennische E, Ho HP, Eriksson E, Niklasson A et al. Prenatal cytokine exposure results in obesity and gender-specific programming. Am J Physiol Endocrinol Metab 2001; 281: E326–E334.
Lambin S, van Bree R, Vergote I, Verhaeghe J . Chronic tumor necrosis factor-alpha infusion in gravid C57BL6/J mice accelerates adipose tissue development in female offspring. J Soc Gynecol Invest 2006; 13: 558–565.
Gaillard R, Rifas-Shiman SL, Perng W, Oken E, Gillman MW . Maternal inflammation during pregnancy and childhood adiposity. Obesity 2016; 24: 1320–1327.
Nøhr MK, Dudele A, Poulsen MM, Ebbesen LH, Radko Y, Christensen LP et al. LPS-enhanced glucose-stimulated insulin secretion is normalized by resveratrol. PLoS ONE 2016; 11: e0146840.
Dudele A, Fischer CW, Elfving B, Wegener G, Wang T, Lund S . Chronic exposure to low doses of lipopolysaccharide and high-fat feeding increases body mass without affecting glucose tolerance in female rats. Physiol Rep 2015; 3.
Fischer CW, Liebenberg N, Madsen AM, Müller HK, Lund S, Wegener G . Chronic lipopolysaccharide infusion fails to induce depressive-like behaviour in adult male rats. Acta Neuropsychiatr 2015; 27: 189–194.
Nguyen AT, Mandard S, Dray C, Deckert V, Valet P, Besnard P et al. Lipopolysaccharides-mediated increase in glucose-stimulated insulin secretion: involvement of the GLP-1 pathway. Diabetes 2014; 63: 471–482.
Weber EM, Olsson IAS . Maternal behaviour in Mus musculus sp.: an ethological review. Appl Anim Behav Sci 2008; 114: 1–22.
Gruys E, Toussaint MJ, Niewold TA, Koopmans SJ . Acute phase reaction and acute phase proteins. J Zhejiang Univ 2005; 6: 1045–1056.
Alfaradhi MZ, Kusinski LC, Fernandez-Twinn DS, Pantaleao LC, Carr SK, Ferland-McCollough D et al. Maternal obesity in pregnancy developmentally programs adipose tissue inflammation in young, lean male mice offspring. Endocrinology 2016; 157: 4246–4256.
Despres JP . Abdominal obesity as important component of insulin-resistance syndrome. Nutrition 1993; 9: 452–459.
Kihara S, Matsuzawa Y . Fat distribution and cardiovascular disease risk. Curr Cardiovasc Risk Rep 2015; 9: 1–6.
Laakso M . How good a marker is Insulin level for insulin-resistance. Am J Epidemiol 1993; 137: 959–965.
Desai M, Jellyman JK, Han G, Beall M, Lane RH, Ross MG . Maternal obesity and high-fat diet program offspring metabolic syndrome. Am J Obstet Gynecol 2014; 211: 237.e1–237.e13.
Bayol SA, Farrington SJ, Stickland NC . A maternal 'junk food' diet in pregnancy and lactation promotes an exacerbated taste for 'junk food' and a greater propensity for obesity in rat offspring. Br J Nutr 2007; 98: 843–851.
Suzuki K, Simpson KA, Minnion JS, Shillito JC, Bloom SR . The role of gut hormones and the hypothalamus in appetite regulation. Endocr J 2010; 57: 359–372.
Cai DS, Liu TW . Hypothalamic inflammation: a double-edged sword to nutritional diseases. Ann N Y Acad Sci 2011; 1243: E1–E39.
Kirk SL, Samuelsson AM, Argenton M, Dhonye H, Kalamatianos T, Poston L et al. Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS ONE 2009; 4: e5870.
Dahlhoff M, Pfister S, Blutke A, Rozman J, Klingenspor M, Deutsch MJ et al. Peri-conceptional obesogenic exposure induces sex-specific programming of disease susceptibilities in adult mouse offspring. Biochim Biophys Acta 2014; 1842: 304–317.
Sun B, Purcell RH, Terrillion CE, Yan JQ, Moran TH, Tamashiro KLK . Maternal high-fat diet during gestation or suckling differentially affects offspring leptin sensitivity and obesity. Diabetes 2012; 61: 2833–2841.
Tarrade A, Panchenko P, Junien C, Gabory A . Placental contribution to nutritional programming of health and diseases: epigenetics and sexual dimorphism. J Exp Biol 2015; 218: 50–58.
Liu XJ, Wang BW, Zhao M, Zhang C, Chen YH, Hu CQ et al. Effects of maternal LPS exposure during pregnancy on metabolic phenotypes in female offspring. PLoS ONE 2014; 9: e114780.
Bouret SG, Simerly RB . Developmental programming of hypothalamic feeding circuits. Clin Genet 2006; 70: 295–301.
Du Y, Yang MR, Lee S, Behrendt CL, Hooper LV, Saghatelian A et al. Maternal western diet causes inflammatory milk and TLR2/4-dependent neonatal toxicity. Gene Dev 2012; 26: 1306–1311.
Collado MC, Laitinen K, Salminen S, Isolauri E . Maternal weight and excessive weight gain during pregnancy modify the immunomodulatory potential of breast milk. Pediatr Res 2012; 72: 77–85.
Ballard O, Morrow AL . Human milk composition nutrients and bioactive factors. Pediatr Clin N Am 2013; 60: 49–74.
Liang H, Ward WF . PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ 2006; 30: 145–151.
Laker RC, Lillard TS, Okutsu M, Zhang M, Hoehn KL, Connelly JJ et al. Exercise prevents maternal high-fat diet-induced hypermethylation of the Pgc-1a gene and age-dependent metabolic dysfunction in the offspring. Diabetes 2014; 63: 1605–1611.
Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA . The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. Nat Rev Immunol 2011; 11: 607–615.
Wall R, Ross RP, Fitzgerald GF, Stanton C . Fatty acids from fish: the anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr Rev 2010; 68: 280–289.
Krishnan AV, Feldman D . Mechanisms of the anti-cancer and anti-inflammatory actions of vitamin D. Annu Rev Pharmacol Toxicol 2011; 51: 311–336.
Acknowledgements
We thank Niels Jessen, Gitte Marie Rasmussen and Christian Buhl for help during experimental setup and design and Heidi M Jensen and Rasmus Buchanan for help with animal care. The study was funded by The Danish Council for Independent Research, Medical Sciences, and by the Toyota Foundation. AD was supported by the Graduate School of Science and Technology, Aarhus University.
Author contributions
AD, TW and SL conceived and designed the work; AD, KSH, MK, MH, GWint, BE, ALN and MKN acquired the data and participated in data analysis; AD, KSH, MK, BE, GWeg, ALN, AL, SBP, TW and SL interpreted the results. AD wrote the manuscript draft and all authors participated in manuscript revision and have read and approved the final draft.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on International Journal of Obesity website
Supplementary information
Rights and permissions
About this article

Cite this article
Dudele, A., Hougaard, K., Kjølby, M. et al. Chronic maternal inflammation or high-fat-feeding programs offspring obesity in a sex-dependent manner. Int J Obes 41, 1420–1426 (2017). https://doi.org/10.1038/ijo.2017.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2017.136
- Springer Nature Limited
This article is cited by
-
Metabolic and feeding adjustments during pregnancy
Nature Reviews Endocrinology (2023)
-
A maternal high-fat/low-fiber diet impairs glucose tolerance and induces the formation of glycolytic muscle fibers in neonatal offspring
European Journal of Nutrition (2021)