
Abstract
We studied the mechanisms involved in the human corpora cavernosa (HCC) relaxation induced by a new metal-based nitric oxide (NO) donor, the ruthenium complex cis-[Ru(bpy)2Imn(NO)]+3 (FOR0811). FOR0811 produced relaxation in phenylephrine (PE)-precontracted HCC with a maximal response that achieved 112.9±10.6%. There was no difference between the maximal relaxation induced by FOR0811 when compared with sodium nitroprusside (SNP) (106.8±7.3%), BAY41-2272 (107.6±4.1%) or vardenafil (103.4±3.8%), however, FOR0811 was less potent than SNP and vardenafil. L-NG-nitroarginine methyl ester (L-NAME), a NO synthase inhibitor, had no effect in the concentration–response curve elicited by FOR0811. 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), a heme-site inhibitor of soluble guanylyl cyclase (sGC) was able to either block or reverse the relaxation induced by FOR0811. On the other hand, the relaxation induced by FOR0811 was not affected by glibenclamide, a blocker of ATP-sensitive potassium channels. FOR0811 (10 μM) was able to increase cyclic guanosine monophosphate (cGMP) levels in corpora cavernosa strips. FOR0811 completely relaxes HCC by a sGC-cGMP-dependent mechanism and can be a lead compound in the development of new stable NO donors.
Similar content being viewed by others


Introduction
ED is defined as the inability to achieve or maintain an erection sufficient for satisfactory sexual activity.1 Modern data suggest that 55% of men above 75 years have ED, and the incidence of ED increases from 5.1% at the age of 40 to 15% at the age of 70 years.2 At least 40% of Brazilian men aged 55–59 years suffer from this condition.3
The normal erectile function is achieved when three simultaneous and synergic processes occur: increase of arterial blood influx in penis, relaxation of corpus cavernosum smooth muscle and restriction of venous blood outflow.4
Furchgott and Zawadzki5 first described the endothelium-derived relaxing factor (EDRF), which opened new avenues in the study of the hemodynamics of the erectile mechanism. Later, Moncada et al.6 showed by using a superfusion cascade bioassay that nitric oxide (NO) released from endothelial cells was indistinguishable from EDRF and suggested that EDRF was NO. NO was also demonstrated to be a neurotransmitter released from autonomic nerves. Ignarro et al.7 showed that NO released from non-adrenergic non-cholinergic fibers was the main neurotransmitter involved in penile erection in both rabbit and HCC. After sexual stimulation, NO is released from the cavernous nerve and also from the endothelium to induce relaxation of the corpora cavernosa and helicinal arteries increasing intracavernosal pressure. To evoke such relaxation, NO activates the soluble guanylyl cyclase (sGC), which catalyzes cyclic guanosine monophosphate (cGMP) production from guanosine triphosphate. cGMP acts on intracellular effectors leading to reduced intracellular calcium levels, dissociation of actin and myosin fibers, and smooth muscle relaxation.8
Many different mechanisms have been studied as a target for ED treatment. PDE5 inhibitors (PDE5is), such as sildenafil, vardenafil and tadalafil, were the first effective and safe oral pharmacological treatment for ED.9
NO donors are substances that releases NO in vivo and in vitro, and one of the most studied donors is sodium nitroprusside (SNP). SNP can be used as a powerful vasodilator for the emergency treatment of high blood pressure (hypertensive crisis). This drug has the disadvantage of being very unstable and producing cyanide, which is very toxic to the vascular endothelium.10 Recently, new compounds that are more stable and less toxic have been synthesized. S-nitrosoglutathione and S-nitroso-N-acetylcysteine-ethylester have been shown to promote relaxation of human corpus cavernosum strips showing potential for the treatment of ED refractory to traditional medical treatments.11
During the last decades, many metal-based nitrosyl compounds have been developed as NO donors. Among these compounds, ruthenium-based complexes have dominated the field, where they have showed promising pharmacological activities.12 A new group of substances, which have in common a ruthenium-coordinated complex, has been produced and tested as NO donors. These substances are soluble in water, have thermal stability and do not generate13, 14 cyanide as their byproduct. Testing a new ruthenium compound in rat aortic rings, Bonaventura et al. showed relaxation effects similar to SNP.10 Two other ruthenium compounds, cis-[Ru(bpy)2(SO3)(NO)]PF6 (FOR0810) and trans-[Ru(NH3)4(caffeine)(NO)]C13 (FOR0C13), were evaluated by our group and shown to significantly relax rabbit corpora cavernosa strips.15
The aim of the present study was to test the effect of a new ruthenium complex, cis-[Ru(bpy)2Imn(NO)]+3 (FOR0811), in HCC strips and compare its effects to known corpora cavernosa-relaxing compounds. In addition to this, we tried to confirm the major role of sGC and cGMP pathway in HCC relaxation induced by FOR0811.
Materials and methods
All studies were performed according to a protocol approved by the ethics committee of research with human subjects of the Federal University of Ceara and the National Committee of Ethics of Research of the Brazilian Health Ministry under protocol # 191.528 and the Animal Experimentation Ethics Committee of the Ceara State University under number 08628332-4.
Cis-[Ru(bpy)2(imN)(NO)]+3 (FOR0811; Figure 1) was synthesized by Dr Luiz Gonzaga de França Lopes from the Department of Organic and Inorganic Chemistry, Federal University of Ceara, according to the method described previously, and its NO-releasing properties were demonstrated in vitro.13

Planar structure of cis-[Ru(bpy)2(imN)(NO)]+3 (FOR0811). bpy, bipyridine; imN, imidazole (bound through nitrogen); NO, nitric oxide.
Human corpus cavernosum (HCC) erectile tissues were obtained from donor cadaver (n=16; average 34 years) during surgery for organ transplantation. Tissues from patients who had a previous history of diabetes, hypertension, dyslipidemias, smoking or drug addiction were discarded from the study. A total of 64 strips of human corporal smooth muscle were excised. The experiments were arranged in such way that strips from one donor were used for different experiments and the number of repetitions for a specific protocol was completed with additional donors. Strips that did not present sustained contractions to phenylephrine (PE) or high potassium (80 mM K+) were discarded.
After removal, the tissue was placed immediately in ice-cold transportation buffer (Collin’s solution), maintained at 4 °C and used within 24 h after operation. The corpus cavernosum tissue was dissected following removal of the connective tissues of the tunica albuginea, with each corpora cavernosum providing up to six segments of corpus cavernosum (1 cm × 0.3 cm × 0.2 cm). The samples were mounted in isolated tissue baths containing 5 ml Krebs-Henseleit solution (37 °C; pH 7.4) bubbled with carbogen (95:5 O2/CO2). The samples were mounted in the bath and attached to an isometric force transducer (TRI202P, Panlab, Barcelona, Spain). The strips were mounted under 1 g tension and monitored during 60 min with tension adjustment and nutritive solution change at every 15-min intervals. The data were acquired by using a Powerlab data acquisition system (ADInstruments, Sydney, NSW, Australia).
All the drugs used in the experiments outlined herein were purchased from Sigma Aldrich (Saint Louis, MO, USA) except FOR0811. All the salts were purchased from Vetec (a branch of Sigma Aldrich Brazil, Rio de Janeiro, Brazil).
Following the 60-min resting period, the tissues were precontracted with a isotonic high potassium Krebs–Henseleit solution (80 mM K+) to ‘awake’ the tissues and load internal stores with calcium. The tissues were then washed with Krebs–Henseleit solution until baseline was achieved and were used for the following experimental protocols:
Experiment 1: Initially, the effects of FOR0811 were evaluated in HCC precontracted with 10 μM PE or 80 mM K+. After the plateau phase for the contractile agents was achieved, cumulative concentrations ranging from 10−9 to 10−4 M of FOR0811 were added to the baths. The effect of each concentration was observed during 5 min, and after that, a new concentration was added. Dimethyl sulfoxide was the solvent used to dilute FOR0811 and the maximal concentration used in the baths was 1%. The response of the tissues to dimethyl sulfoxide was tested by incubating this solvent isovolumetrically during the same time interval as the test drugs for the whole experimental period. In another set of experiments, similar concentration–response curves to SNP, a NO donor, BAY 41-2272, an NO-independent guanylyl cyclase stimulator, or vardenafil, a PDE5i, were studied in tissues precontracted with 10 μM PE.
Experiment 2: HCC strips were incubated for 30 min with 30 μM 1H-[1,2,4] oxadiazole [4,3-a]quinoxalin-1-one (ODQ), and thereafter, the tissues were precontracted with 10 μM PE. The concentration–response curve (10−9 to 10−4 M) to FOR0811 was then repeated in the presence of this inhibitor.
Two sets of protocols were carried out in aortic rings of rats to confirm that the heme site of sGC was the major target for FOR0811 and that this effect is species-independent. Male rats (250–275 g) were housed five per cage on 12-h light/dark cycle, and fed a standard chow diet with water ad libitum. Experimental protocols were approved by the animal care and use committee of State University of Ceará.
The descending thoracic aorta was excised and cleaned of perivascular tissue and cut in rings (3 mm). The rings were mounted between two wire hooks under 1 g tension and monitored in organ bath system during 60 min with conditions and data acquisition similar to that described for HCC. In a first set of protocols (n=3), ODQ was added in the plateau of 0.3–1 μM PE-induced contraction, and 25 min later, a single concentration of FOR0811 (10 μM) was added in the organ bath. In other set of experiments (n=3), ODQ was added after a maximal relaxation to 10 μM FOR0811 was achieved to check whether ODQ could reverse this effect. We also added a heme-independent sGC activator, BAY 60–2770, to show that the enzyme was indeed active, but not sensitive to a NO donor.
Experiment 3: To evaluate the potential role of endogenous NO in the relaxation elicited by FOR0811, 100 μM of N-ω-nitro-L-arginine methyl ester (L-NAME) was added to the baths 30 min before precontraction with 10 μM PE. As previously described, concentration–response curves to FOR0811 were constructed in the presence of this inhibitor.
Experiment 4: We also evaluate the role of the KATP channels in the relaxation caused by the test substance. Glibenclamide (30 μM) was added to the baths 30 min before the precontraction with 10 μM PE. Thereafter, the concentration–response curves to FOR0811 were constructed.
Experiment 5: HCC strips were precontracted with 10 μM PE and after the plateau was achieved, the tissues were exposed to 10 μM FOR0811 or dimethyl sulfoxide (0.1% in distilled water). After the maximal relaxant response to this single concentration was achieved, the strips were immediately frozen in liquid nitrogen and stored at −80 °C. The cGMP assay was performed as described by Bonaventura.10 Briefly, the tissues were homogenized with a buffer enriched with 100 μM 3-isobutyl-1-methylxanthine, a nonspecific phosphodiesterase inhibitor. The tissue total protein content was measured by using the PIERCE BCA assay (Thermo Scientific, Rockford, IL, USA). Samples were treated with 10% trichloroacetic acid followed by 2000 g centrifugation for 15 min at 4 °C. Thereafter, the supernatant was washed five times with five volumes water-saturated diethyl ether. The upper ether layer was discarded after each wash. The aqueous extract remaining was dried at 60 °C in a nitrogen atmosphere. The samples were suspended in the cyclic GMP enzyme immunoassay buffer (Cayman Chemical Company, Ann Harbor, MI, USA), and the acetylation procedure was followed. The concentration of cGMP was expressed in fmol per mg protein.
Statistical analysis
The relaxations were normalized to the initial PE or 80 mM K+ contraction and were expressed as a percentage of reversal. The maximal effect (EMAX) was considered as the maximum amplitude observed in the concentration–response curve for each agent. The concentration required to produce half maximal relaxation (EC50) was determined after log transformation of the normalized concentration–response curves and expressed as negative logarithms (pEC50). The statistical analyses were performed with the software GraphPad Prism 5.0 (Graph Pad Software Corporation, San Diego, CA, USA). The data were expressed as mean±standard error (s.e.). The statistical significance was determined using one-way variance analysis (ANOVA), followed by the Bonferroni's Multiple Comparison test or unpaired Student’s t test. The level of statistical significance was set at P<0.05.
Results
Evaluation of the relaxation induced by FOR0811
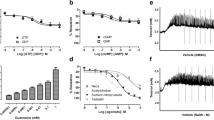
The maximal relaxation (EMAX) produced by FOR0811 was 112.9±10.6% with a pEC50 of 5.0±0.2 in HCC strips precontracted with PE (Figure 2). On the other hand, the maximal relaxation induced by FOR0811 was 87.1±7.3% in strips precontracted with a depolarizing solution (80 mM K+). Neither the maximal amplitude of relaxation nor the potency (pEC50 of 5.1±0.4) of FOR0811 changed when the pharmacomechanical (PE) or electrochemical (80 mM K+) stimulus were compared (Figure 2 and Table 1). In addition, the absolute amplitude of the contractions elicited by those contractile agents were not statistically different (5.7+/−0.9 g –PE vs 6.1+/−1.1 g-K+).

Concentration–response curves for FOR0811 in human corpora cavernosa strips contracted with either phenylephrine or 80 mM K+. Values are expressed as mean±s.e.m.
Comparison of maximal relaxation and potency with SNP, BAY 41-2272 and vardenafil
The maximal relaxant effect and potency of FOR0811 (EMAX=112.9±10.6% and pEC50=5.0±0.2) were compared with the same parameters obtained with SNP, BAY 41-2272 or vardenafil in PE-precontracted HCC. These drugs produced a maximal relaxation of 106.8%±7.3%, 107.6%±4.1% and 103.4±3.8%, respectively. When comparing these drugs, no statistical difference among the maximal relaxations was found (Figure 3 and Table 1). However, FOR0811 was almost 2–3 orders of magnitude less potent than SNP and vardenafil. The pEC50 values for SNP, BAY 41-2272 and vardenafil were 7.8±0.5, 5.6 ±0.2 and 8.2± 0.3, respectively (Table 1).

Concentration–response curves for FOR0811, BAY41-2272, sodium nitroprusside (SNP) or vardenafil in human corpora cavernosa strips contracted with phenylephrine. Values are expressed as mean±s.e.m.
Effect of ODQ or L-NAME on FOR0811-induced relation on HCC strips
Incubation of tissues with 30 μM ODQ, which oxidizes the heme prosthetic group of sGC and turns it unresponsive to NO, completely blunted the response to FOR0811. The EMAX achieved with FOR0811 after 30-min period incubation with ODQ was 37.9%±7.6 (P<0.05) (Figure 4a). There was no difference in the pEC50 values, and no shift in the concentration–response curves was observed (Table 1).

Panel (a) shows the concentration–response curves for FOR0811 in the absence or presence of ODQ, a heme-site soluble guanylyl cyclase inhibitor, or in the presence of NG-nitro-L-arginine methyl ester (L-NAME; a NO synthase inhibitor) in human corpora cavernosa strips contracted with PE. Data are expressed as mean±s.e.m. of n experiments performed on preparations obtained from different donors. *P<0.05, one-way analysis of variance followed by Bonferroni vs FOR0811 alone. Panel (b) depicts the blockade of FOR0811-induced relaxation by 10 μM ODQ (n=3), and panel (c) shows the reversion of the relaxation elicited by 10 μM FOR0811 by the addition of 10 μM ODQ to the bath (n=3). BAY 60-2270 (BAY 60) was used to show that despite being nonresponsive to acetylcholine or to a NO donor, the tissue was still able to respond to a non-NO, non-heme-dependent soluble guanylyl cyclase activator. Ach, acetylcholine; ODQ, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one; PE, phenylephrine; SNP, sodium nitroprusside.
Incubation of tissues with 100 μM L-NAME, a nonspecific NO synthase inhibitor, did not affect the maximal relaxation effect of FOR0811. The EMAX of the drug was 104.8%±16.7% (P>0.05). There was no significant statistical difference in the pEC50 values for FOR0811 in the absence or presence of this drug (Figure 4a and Table 1).
Effect of ODQ on FOR0811-induced relaxation in rat aortic rings
The incubation of rat aortic rings precontracted with PE (0.3–1 μM) with 10 μM ODQ for 25 min before the addition of 10 μM FOR0811 completely blocked the relaxation induced by this single dose challenge (Figure 4b). However, this procedure did not affect the relaxation induced by 3 μM BAY 60-2770 a heme-independent sGC activator. Strikingly, the incubation of 10 μM ODQ was able to fully reverse a maximal relaxation induced by 10 μM FOR0811 (Figure 4c).
Effect of the ATP-dependent potassium channel blockade on relaxation induced by FOR0811
Glibenclamide (30 μM), an ATP-dependent potassium channel blocker, did not blunt the relaxation induced by FOR0811. There is no statistically detected difference between the EMAX or pEC50 of FOR0811 in the presence or absence of this compound (Figure 5 and Table 1).

Concentration–response curves for FOR0811 in the absence or presence of glibenclamide, a KATP channel blocker, in human corpora cavernosa strips contracted with phenylephrine. Data are expressed as mean±s.e.m. of n experiments performed on preparations obtained from different donors.
Measurement of cGMP content
FOR0811 at 10 μM increased the cGMP levels of HCC strips, after 15 min of incubation, from 207.6±27 fmol per mg of protein to 535.6±87 fmol per mg of protein (P<0.005) corresponding to a 2.6-fold increase in this nucleotide levels.
Discussion
In the present study, we demonstrated the relaxant properties of a new metal-based NO donor, FOR0811, in HCC harvested from cadaver donors at the time of organ collection for transplantation. FOR0811 was synthesized along with more than 50 ruthenium- or iron-coordinated complexes and its NO-releasing properties were studied in vitro.13 However, whether this compound would release NO in vivo inducing smooth muscle relaxation was not probed until the present work.
FOR0811 produced a maximal relaxation higher than 110%, on HCC. To the best of our knowledge, this is the first time that substances of the ruthenium-nitrosyl complexes were tested on HCC. Cerqueira et al. tested two substances, cis-[Ru(bpy)2(SO3)(NO)](PF6) (FOR0810) and trans-[Ru(NH3)4(caffeine)(NO)]C13 (FOR0C13), in rabbit corpora cavernosa strips15 and showed that both compounds produced maximal relaxation smaller than SNP. Seidler et al.11 compared the effect of several NO donors in HCC strips, and showed that SNP produced the highest relaxant response. In the present study, FOR0811 produced a HCC maximal relaxant effect, similar to SNP, being, however, less potent. Thereafter, we compared the effect produced by FOR0811 with the relaxant effects of BAY 41-2272, a sGC stimulator that activates purified sGC and strongly synergizes with NO. The concentration–response curves for these substances were quite similar with equal efficacy and potency. Bacarat et al.16 found that the concentration–response curve to BAY 41-2272 is shifted to the right by L-NAME and the authors suggest that BAY41-2272 acts synergistically with endogenous NO to elicit its relaxant effect and induce penile erection in vivo. However, the concentration–response curve to FOR0811 is not affected by L-NAME, and therefore, this metalodrug is probably not synergistic with endogenous NO.
The relaxation induced by FOR0811 is also similar in amplitude to the relaxation induced by vardenafil, a PDE5i, showing that FOR0811 has similar efficacy when compared with some pharmacological groups of known corpora cavernosa-relaxant drugs.
PDE5is do not always produce an erection sufficient for sexual intercourse in patients with endothelial and nitrergic dysfunction, such as diabetic patients or men who have had radical pelvic surgery.17
The concentration–response curve to FOR0811 in PE-contracted strips had no statistical significant differences when compared with values obtained in tissues precontracted with 80 mM K+. As these contractions were of similar amplitude (5.7+/−0.9 g –PE vs 6.1+/−1.1 g-K+), the relaxation induced by FOR0811 is unlikely to depend on a K+-opening mechanism because the relaxation induced by K-channel openers is blunted when the tissues are contracted by a high-potassium solution.
As discussed before, the relaxant response to FOR0811 was strongly blunted by ODQ. Studying another nitrosyl ruthenium complex, [Ru(terpy)(bdq)NO+]3+ (Terpy), in aortic rings of rats, Bonaventura et al.10 showed similar results. Similarly, Wanstall et al. have reported that ODQ reduced the maximal response to SNP rather than induced parallel shifts.18 In addition, the relaxation induced by FOR0811 was either blocked or reversed by ODQ. The inhibitory effect of ODQ is due to changes in the oxidation state of the heme moiety without adverse effects on the catalytic activity of the enzyme, and this compound does not inactivate NO or the activity of NO synthase and adenylyl cyclase.18, 19, 20 This reveals that FOR0811 induces relaxation by the stimulation of the heme site of the sGC because after the oxidation of this site by ODQ, the relaxant effect was blocked or reversed but the effect of a heme site-independent sGC activator, BAY 60-2270, is not affected. The importance of cGMP-dependent signaling pathways to the relaxation induced by NO donors has been shown by many authors.21, 22 For instance, the relaxant response of several NO donors in rat aortic rings, including SNP, were almost abolished by 10 μM ODQ and the response of a NO donor, NCX 4050, was greatly blunted by 1 μM ODQ in HCC and rabbit corpora cavernosa.23, 24
Some studies have shown that some stimulators of sGC have a dual mechanism of action by directly stimulating the native form of sGC and rendering the enzyme more sensitive to endogenous NO and, therefore, such compounds would have their relaxant response blunted by NOS inhibitors.17, 21 Nevertheless, the incubation of HCC for 30 min with 100 μM L-NAME did not change the relaxant effect of FOR0811. Similarly, Filipi et al.24 testing a new class of NO donor drugs, NCX4050, and Seidler et al.11 testing several NO donors in HCC strips observed that L-NAME does not affect the concentration–response curve to those compounds.
Some authors have demonstrated that KATP channels have an important role in HCC smooth muscle relaxation.25 Several substances produce its effects through activation of these specific channels, like pinacidil,26 cromakalim, minoxidil,8 yohimbine27 and phentolamine.28 However, the blockade of such KATP channels by glibenclamide did not change the concentration–response curve to FOR0811. Therefore, the relaxation induced by FOR0811 is not dependent on the activation of ATP-dependent potassium channels. Similar results were reported by29 for the relaxant activity of trans-[Ru(NH3)4(caffeine)(NO)]Cl3 in rabbit corpora cavernosa.
FOR0811 was able to maximally relax HCC and to increase cGMP levels, this relaxation being either blocked or reversed by ODQ. This compound is soluble in water, is stable and does not produce cyanide. This compound could be a lead compound in the search of alternatives to dilate penile blood vessels and relax corpora cavernosa smooth muscle in patients that do not benefit with PDE5is.
References
NIH Consensus Conference. Impotence. NIH Consensus Development Panel on Impotence. JAMA 1993; 7: 83–90.
Johannes CB, Araújo AB, Feldman HA . Incidence of erectile dysfunction in men ages 40-69: longitudinal results from the Massachusetts male aging study. J Urol 2000; 163: 460–469.
Moreira ED Jr, Abdo CH, Torres EB, Lobo CF, Fittipaldi JA . Prevalence and correlates of erectile dysfunction: results of the Brazilian study of sexual behavior. Urology 2001; 58: 583–588.
Bivalacqua TJ, Champion HC, Hellstrom WJ, Kadowitz PJ . Pharmacotherapy for erectile dysfunction. Trends Pharmacol Sci 2000; 21: 484–489.
Furchgott RF, Zawadzki JV . The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980; 288: 373–376.
Palmer RM, Ferrige AG, Moncada S . Nitric oxide accounts for the biological activity of endothelium derived relaxing factor. Nature 1987; 327: 524–526.
Ignarro LJ, Bush PA, Buga GM, Wood KS, Fukuto JM, Rajfer J . Nitric oxide andcyclic GMP formation upon electrical field stimulation cause relaxation of corpuscavernosum smooth muscle. Biochem Biophys Res Commun 1990; 31: 843–850.
Andersson KE . Mechanisms of penile erection and basis for pharmacological treatment of erectile dysfunction. Pharmacol Rev 2011; 63: 811–859.
Corbin JD, Francis SH, Webb DJ . Phosphodiesterase type 5 as a pharmacologic target in erectile dysfunction. Urology 2002; 60 (Suppl 2): 4–11.
Bonaventura D, de Lima RG, Vercesi JA, da Silva RS, Bendhack LM . Comparison of the mechanisms underlying the relaxation induced by two nitric oxide donors: Sodium nitroprusside and a new ruthenium complex. Vascul Pharmacol 2007; 46: 215–222.
Seidler M, Ückert S, Waldkirch E, Stief CG, Oelke M, Tsikas D et al. In vitro effects of a novel class of nitric oxide (NO) donating compounds on isolated human erectile tissue. Eur Urol 2002; 42: 523–528.
Tfouni E, Truzzi DR, Tavares A, Gomes AJ, Figueiredo LE, Franco DW . Biological activity of ruthenium nitrosyl complexes. Nitric Oxide 2012; 26: 38–53.
Silva FON, Araújo SXB, Holanda AKM, Meyer E, Sales FAM, Diógenes ICN et al. Synthesis, characterization, and NO release study of the cis- and trans-[Ru(Bpy)2(SO3)(NO)]+ complexes. Eur J Inorg Chem 2006; 2006: 2020–2026.
Lunardi CN, da Silva RS, Bendhack LM . New nitric oxide donors based on ruthenium complexes. Braz J Med Biol Res 2009; 42: 87–93.
Cerqueira JB, Gonzaga-Silva LF, Lopes LG, Moraes ME, Nascimento NR . Relaxation of rabbit corpus cavernosum smooth muscle and aortic vascular endothelium induced by new nitric oxide donor substances of the nitrosyl-ruthenium complex. Int Braz J Urol 2008; 34: 638–645.
Baracat JS, Teixeira CE, Okuyama CE, Priviero FB, Faro R, Antunes E et al. Relaxing effects induced by the soluble guanylylcyclasestimulator BAY41-2272 in human and rabbit corpus cavernosum. Eur J Pharmacol 2003; 477: 163–169.
Bischoff E, Schramm M, Straub A, Feurer A, Stasch JP . BAY 41–2272: a stimulator of soluble guanylylcyclase induces nitric oxide-dependent penile erection in vivo. Urology 2003; 61: 464–467.
Wanstall JC, Homer KL, Doggrell SA . Evidence for, and importance of, cGMP-independent mechanisms with NO and NO donors on blood vessels and platelets. Curr Vasc Pharmacol 2005; 3: 41–53.
Zhao Y, Brandish PE, Divalentim M, Schelvis JP, Babcok GT, Marletta MA . Inhibition of soluble guanylatecyclaseby ODQ. Biochemistry 2000; 39: 10848–10854.
Feelish M, Kotsonis P, Siebe J . The soluble guanylylcyclase inhibitor 1-H-[1,2,4] oxadiazolo-[4,3-a] quinoxaline 1-one is a non selectivehaem protein inhibitor of nitric oxide synthase and other cytochrome P450 enzymes involved in nitric oxide donor bioactivation. Mol Pharmacol 1999; 56: 243–253.
Badejo AM Jr, Nossaman VE, Pankey EA, Bhartiya M, Kannadka CB, Murthy SN et al. Pulmonary and systemic vasodilator responses to the soluble guanylylcyclase stimulator, BAY 41-8543, are modulated by nitric oxide. Am J Physiol Heart Circ Physiol 2010; 299: H1153–H1159.
Evgenov OV, Ichinose F, Evgenov NV, Gnoth MJ, Falkowski GE, Chang Y et al. Soluble guanylatecyclase activator reverses acute pulmonary hypertension and augments the pulmonary vasodilator response to inhaled nitricoxide in awake lambs. Circulation 2004; 110: 2253–2259.
Hobbs AJ . Soluble guanylatecyclase: the forgotten sibling. Trends Pharmacol Sci 1997; 18: 484–491.
Filippi S, Crescioli C, Vannelli GB, Fazzini A, Natali A, Riffaud JP et al. Effects of NCX 4050, a new NO donor, in rabbit and human corpus cavernosum. Int J Androl 2003; 26: 101–108.
Höppner CK, Stief CG, Jonas U, Mandrek K, Noack T, Golenhofen K . Eletrical and chemical control of smooth muscle activity of rabbit corpus cavernosum in vitro. Urology 1996; 48: 512–518.
Videbaek LM, Aalkjaer C, Mulvany MJ . Pinacidil opens K-selective channels causing hyperpolarization andrelaxation of noradrenaline contractions in rat mesenteric resistance vessels. Br J Pharmacol 1988; 95: 103–108.
Freitas FC, Nascimento NR, Cerqueira JB, Morais ME, Regadas RP, Gonzaga-Silva LF . Yohimbine relaxes the human corpus cavernosum through a non-adrenergic mechanism involving the activation of K+ATP-dependent channels. Int J Impot Res 2009; 21: 356–361.
Gonzaga-Silva LF, Nascimento NR, Fonteles MC, deNucci G, Moraes ME, Vasconcelos PR et al. Phentolamine relaxes human corpus cavernosumby a nonadrenergicmechanism activating ATP-sensitive K channel. Int J Impot Res 2005; 17: 27–32.
Cerqueira JB, Gonzaga-Silva LF, Silva FO, Cerqueira JV, Oliveira RR, Moraes ME et al. Identification of mechanisms involved in the relaxation of rabbit cavernous smooth muscle by a new nitric oxide donor ruthenium compound. Int Braz J Urol 2012; 38: 687–694.
Acknowledgements
This study was funded by the Fundação Cearense de Apoio ao Desenvolvimento Científico e Tecnólogico (FUNCAP). FOR0811 was synthesized by the bioinorganic chemistry lab of the Federal University of Ceará funded by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Conselho Nacional de Desenvolvimento Científico e Tecnológico(CNPq) (LGF Lopes 470054/2011-5) and FUNCAP-MS-CNPq (PPSUS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Leitão Junior, A., Campos, R., Cerqueira, J. et al. Relaxant effect of a metal-based drug in human corpora cavernosa and its mechanism of action. Int J Impot Res 28, 20–24 (2016). https://doi.org/10.1038/ijir.2015.27
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijir.2015.27
- Springer Nature Limited