
Abstract
The CRISPR/Cas9 system provides an easy way to edit specific site/s in the genome and thus offers tremendous opportunity for human gene therapy for a wide range of diseases. However, one major concern is off-target effects, particularly with long-term expression of Cas9 nuclease when traditional expression methods such as via plasmid/viral vectors are used. To overcome this limitation, we pre-packaged Cas9 protein (Cas9P LV) in lentiviral particles for transient exposure and showed its effectiveness for gene disruption in cells, including primary T cells expressing specific single guide RNAs (sgRNAs). We then constructed an ‘all in one virus’ to express sgRNAs in association with pre-packaged Cas9 protein (sgRNA/Cas9P LV). We successfully edited CCR5 in TZM-bl cells by this approach. Using an sgRNA-targeting HIV long terminal repeat, we also were able to disrupt HIV provirus in the J-LAT model of viral latency. Moreover, we also found that pre-packaging Cas9 protein in LV particle reduced off-target editing of chromosome 4:-29134166 locus by CCR5 sgRNA, compared with continued expression from the vector. These results show that sgRNA/Cas9P LV can be used as a safer approach for human gene therapy applications.
Similar content being viewed by others


Introduction
Within the past few years, several novel technologies have emerged for gene editing: ZFN, TALEN and CRISPR/Cas9 system (reviewed in Gersbach and Perez-Pinera,1 Niu et al.,2 Zhang et al.3 and Manjunath et al.4). Unlike RNA interference, which requires the continued presence of effector moieties to maintain gene silencing, gene-editing technologies allow permanent disruption/deletion of the targeted gene after a single treatment. Moreover, the gene-editing techniques allow insertion of new DNA sequences at the edited sites. All the currently available gene-editing techniques use two components: one for specific DNA recognition and another for nuclease activity to induce double-stranded break at specific sites in the genomic DNA. Following this double-stranded break, the DNA is repaired by either of the two pathways: the error-prone non-homologous end joining pathway attended with small-nucleotide additions or deletions (indels) that results in disruption of the reading frame and gene expression; or homologous recombination (when a DNA template with short homology to the two broken ends is also provided) that results in incorporation of the externally provided DNA at the cleavage site. Thus, these gene-editing systems can be used to knockout/delete genes and also to insert exotic DNA sequences at particular sites.
Although ZFN and TALEN use DNA-binding motifs (of zinc finger proteins or transcription activator-like effector molecules, respectively) fused to the Folk1 endonuclease to mediate sequence-specific DNA cleavage, the most recent CRISPR/Cas9 system uses a short stretch of complementary RNA (that can be easily expressed using a U6 promoter) for DNA recognition and uses Cas9 nuclease for DNA cleavage. Whereas ZFN/TALEN require 10 or 34 amino acids, respectively, to recognize a single nucleotide, CRISPR/Cas9 uses the Watson–Crick complementarity rule via a short single guide RNA (sgRNA) molecule (single nucleotide to single nucleotide) for DNA recognition. Thus, a major advantage is that compared with ZFN/TALEN that usually require labor-intensive design and screening, CRISPR/Cas9 requires much simpler design and a single cloning step. Because of the ease of usage, this system provides tremendous opportunity for human gene therapy applications for a variety of diseases. Within less than 3 years of discovery, this technology has been used to knockout genes, create multiple gene knockout mice and monkeys as well as to knock-in specific DNA sequences in a variety of systems (reviewed in Riordan et al.5 and Sternberg and Doudna6).
However, one major concern for clinical use in humans is the propensity for off-target effects. It has been well documented that off-target effects are a common problem with CRISPR/cas9 system.7, 8, 9, 10, 11 One of the main determinants of off-target effects is the amount and duration of Cas9 expression, implying that a transient exposure to Cas9 would increase its specificity (reviewed in Wu et al.12). Thus, to reduce off-target effects resulting from long-term expression of Cas9, we took advantage of a recently described system to deliver pre-packed proteins within lentiviral particles that has also been used to deliver ZFN protein for safer gene editing in cells.13, 14, 15, 16 Here essentially a vector containing only HIV Gag Pol and mutant integrase was used to express ZFN. Upon transfection of this plasmid along with vesicular virus glycoprotein (VSV-G), the resulting lentiviral particles contained the pre-packaged ZFN protein. In this study, we adopted a similar approach to package Cas9 protein within the lentiviral particles. For ease of application, we also developed a system for simultaneous expression of sgRNA and pre-packaged Cas9 within the lentiviral particles. We show effectiveness of this approach to silence cellular and viral genes involved in HIV replication in cell lines and primary CD4 T cells.
Results and Discussion
Cas9 protein packaged in lentiviral particles is functional in gene editing
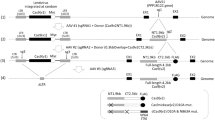
To incorporate Cas9 protein within lentiviral particles (Cas9P LV), FLAG-tagged Cas9 sequence was fused to the N terminus of Gag containing an intervening heterologous phospholipase C-δ1 pleckstrin homology domain. This pleckstrin homology domain is thought to promote the recruitment of Gag and Gag Pol to the membrane.17 To achieve this, we first inserted Cas9 into Gag gene of a packaging construct harboring the integrase D64V mutation (Figure 1a) that renders the viral integrase incapable of mediating vector insertion in the genome. An HIV-1 protease cleavage site (SQNY/PIVQ) was included between Cas9 and the PH domain to allow the release of functional Cas9 protein during particle maturation. To produce viral particles, 293T cells were co-transfected with Cas9-PH-gagpol-D64V, pMDL (wild-type Gag/Pol), and pCMV–VSV-G plasmids (Figure 1b). Concentrated viral particles harvested after 2 days were analyzed for contents by western blot by probing with p24 and Flag tag antibodies. As expected, the particles were positive for gag p24. In addition, the particles were also positive for the expected full-length 160 kDa Cas9/Flag protein, demonstrating that the majority of the virion-associated Cas9 was correctly released from the Gag and GagPol polypeptides during particle maturation (Figures 1c and d). The viral tires were generally 3- to 5-fold lower as compared to standard LV (Supplementary Figure S1A). We also calculated the Cas9 protein packed in the viral particles using Cas9 protein standards by western blot. It appears that ~50 ng of Cas9 protein is packed in ~3.5 × 106 particles (Supplementary Figure S1B). We next tested whether the Cas9 protein packaged within the lentivirus was functional. For this, we transduced TZM-bl cells already expressing a CCR5 sgRNA (by prior transduction with lentiviral vector expressing sgRNA and sorting the GFP+ cells) with Cas9P LV (150 ng p24). CCR5 expression was tested after 72 h by flow cytometry. We observed knockout of CCR5 in sgRNA-expressing TZM-bl cells transduced with VSV-G pseudotyped Cas9 LV (Figure 1e). Thus, the Cas9 protein incorporated within the LV is functional when transduced into cells.

Targeted CCR5 gene disruption by lentiviral delivery of Cas9 protein. (a) Schematic of Cas9 gene expression within the HIV-1 gag/pol vector. After cloning, the gag is composed of the N-terminal Cas9 protein domain, the phospholipase C-δ1 pleckstrin homology domain (PD), matrix, capsid, nucleocapsid, and p6, whereas Pol consists of protease (Pol CP), reverse transcriptase (RT), and integrase harboring the D64V mutation (IN-D64V). (b) LPs harboring Cas9 protein (indicated by red dots inside the virion) was produced by co-transfecting 293T cells with pCMV-VSV-G and pFLAG-Cas9-PH-gag/pol. (c) Analysis of the contents of LPs by western blotting using FLAG- and p24-specific antibodies confirms packaging of Cas9 protein. Con=control (ultracentrifuge pellet from untransfected 293T cells). (d) Analysis of the contents of LPs by western blot using anti-FLAG antibody: Cas9P (LV produced without supplementation with WT gag/pol), Cas9P+WT gag/pol (LV produced with WT gag/pol supplementation), Cas9 en LP (LV encoding Cas9), Cas9 plasmid (Cas9 encoding plasmid transfected TZM-bl cell lysate). Similar sized Cas9 protein was visualized in all cases. (e) CCR5 gene disruption by lentiviurs encapsulated with Cas9 protein. CCR5 sgRNA expressing TZM bl cells were transduced with Cas9 LV and CCR5 expression determined 3 days later by flow cytometry. Dot plots of GFP versus CCR5 staining for one representative of triplicates is shown.
Gene editing of CCR5 in primary CD4 T cells by Cas9p LV
There has been great interest in the HIV field to disrupt the viral co-receptor CCR5 as a therapeutic approach to confer HIV resistance. Thus, we next tested whether Cas9P LV can also be used to edit genes in primary T cells, which are the major targets of HIV. Primary CD4 T cells were activated with CD3/CD28 beads for 48 h and first transduced with a lentiviral vector expressing green fluorescent protein (GFP) as well as a CCR5 sgRNA (Figure 2a). GFP-positive cells were sorted after 72 h, cultured for 3 more days and then transduced with Cas9P LV or a Cas9-encoding LV (2 × 105 cells, 150 ng p24). CCR5 expression in the transduced cells was determined 4 days later by Cel1 assay. We found that 15% of Cas9P LV and 19.5% of Cas9-encoding LV-transduced cells had mutations (indels) in the CCR5 gene (Figure 2b). Thus, Cas9 protein delivery was almost as effective as Cas9 expression for gene editing in primary T cells.

Cas9P LV and Cas9-encoding LV mediate comparable CCR5 gene editing in primary CD4 T cells. (a) Time course of treatment and testing. (b) Disruption of CCR5 gene determined by Cel1 assay. (c) Off-target activity in Cas9p LV and Cas9 encoding LP transduced primary CD4 T cells. Disruption within off-target Chromosome 4:-29134166 locus was determined by Cel1 assay. (d) Persistence of Cas9 protein in cells at indicated times after transduction with Cas9 LV or Cas9 encoding LV was determined by Western blot. Cas9 protein was maximal at 4 h, but declined to be barely detectable at 24 with Cas9 LV, whereas Cas9 was prominent at 48 h with Cas9 en LV.
The main advantage of protein transduction is reduction of off-target effects attended with long-term expression of Cas9. Thus, we expected that Cas9 delivered in LPs would have low off-target relative to on-target activity. One of the off-target sites for this particular sgRNA predicted by in silico analysis is chromosome 4:-29134166. Thus we analyzed this region for off-target gene editing after transduction with Cas9P LV and Cas9 encoding LV in primary T cells. Indeed, we observed ~2.1% off-target cleavage at this locus after transduction with Cas9 encoding LV but did not detect any off-target activity after transduction with Cas9P LV (Figure 2c). On-target activity is shown in Figure 2b and is 25% less with Cas9P LV. We also confirmed that Cas9 expression is longer after transduction with Cas9 en LV compared with Cas9 P LV (Figure 2d). Thus, Cas9 protein delivery is slightly less effective compared with Cas9 expression in inducing on-target gene editing while substantially reducing off-target activity.
Genome editing by ‘all-in-one virus’ (sgRNA/Cas9P LV) vector
In addition to Cas9 protein the CRISPR/Cas9 system needs an sgRNA that targets the gene of interest. As it is also possible to include a vector within the viral particle, we next tested whether we can also express sgRNA along with Cas9 protein transduction using an ‘all-in-one virus’. For this an sgRNA sequence targeting CD4 gene was cloned into the pLB lentiviral vector under the U6 promoter (pLB/sgRNA). To produce sgRNA/Cas9P LV, 293T cells were co-transfected with Cas9-PH-GagPol, pLB/sgRNA, helper pMDL (Gag/Pol), pRSV-Rev and pCMV-VSV-G plasmids (Figure 3a). Presence of p24 and Cas9 expression were tested by western blotting of virus particles harvested from culture supernatants at 48 h as described earlier as well as in in TZM-bl cell lysate obtained 24 h after transduction with sgRNA/Cas9P LV. As shown in Figure 3b, p24 as well as Cas9 protein were detected by western blot both in the viral particles and in transduced cell lysates. We also confirmed the presence of Cas9 protein after transduction with Cas9p LV and Cas9 En LV by fluorescent microscopy (Figure 3c). In addition, we tested CD4 gene modification 48 h after transduction in TZM-bl cells both by flow cytometry and Cel 1 assay. A reduction in CD4 expression was detected by flow cytometry (Figure 3d). Cel1 assay was also positive (Figure 3e). We also confirmed the efficacy of sgRNA/Cas9P LV transduction in Jurkat cells, which is a T cell line in which we were able to show positive CD4 gene editing by Cel1 assay (Figure 3f). To confirm editing within the CD4 gene sequence, we isolated genomic DNA from transduced Jurkat cells (multiplicity of infection 10), amplified the target sequences by PCR, and sequenced the PCR products. We found deletions of various lengths (from −1 to −108 nucleotides) at the expected Cas9 cleavage site in 3/20 clones sequenced (Figure 3g). Collectively these results demonstrate the potential of sgRNA/Cas9P LV for effective gene editing.

Gene-editing by ‘all-in-one virus’. (a) Schematics of ‘all in one virus’ production: red dots inside the virion core indicate Cas9 protein and orange lines indicate lentiviral vector encoding sgRNA. Lentivirus was produced by co-transfecting 293T cells with pCMV-VSV-G, pFLAG-Cas9-PH-gag/pol, pRSV-Rev, pMDL (gag/pol) and pLB vector expressing sgRNA. (b) Analysis of the contents of LPs and LP transduced TZM-bl cell lysates by western blot using FLAG- and p24-specific antibodies. Con=control (ultracentrifuge pellet from untransfected 293T cells). (c) Cellular presence of Cas9 protein after transduction with Cas9 plasmid, Cas 9 en LV or Cas 9P LV in TZM bl cells. Cells were stained with Cas9 antibody and visualized by fluorescence microscopy. (d–f) Targeted CD4 gene disruption by sgRNA/Cas9P LV in TZM-bl and Jurkat cells. (d) Shows flow cytometric analysis of CD4 expression in TZM-bl cells. e and f show Cel1 assay in the sgRNA/Cas9P LV transduced TZM-bl and Jurkat cells, respectively. (g) Shows CD4 gene editing determined by sequencing in Jurkat cells.
sgRNA/Cas9 LV protein can target and delete the HIV-1 provirus in J-Lat cells
Because gene editing provides a way to disable/remove integrated HIV proviral DNA,18, 19 we also tested whether sgRNA/Cas9P LV can be used to inactivate HIV latency. For this, we chose to target viral long terminal repeat (LTR) in a region that is highly conserved in >95% of all HIV clades and strains.20 As the LTR occurs at both ends of the provirus, one sgRNA targeting this region is expected to delete the provirus. The sgRNA targeting LTR was cloned into pLB vector and sgRNA/Cas9P LV produced as described earlier. This lentivirus was then used to transduce J-LAT cells and 48 h post transduction, GFP-positive cells were sorted, cultured for another 48 h and then activated with tumor necrosis factor-α and 5-AZA-dC (Figure 4a). The editing of proviral DNA was determined 48 h after activation by Cel 1 assay and intracellular p24 expression by flow cytometry. Cel1 assay showed a 28% rate of mutation (indels) for LTR (Figure 4b). Similarly, p24 expression in cells transduced with Cas9/sgRNA lentivirus was decreased by ~23% compared with control sgRNA/Cas9-treated cells (Figure 4c). The inactivation of provirus could have been due to mutations in the LTR or excision of the entire provirus by simultaneous targeting of 5′- and 3′-LTR. To distinguish between these possibilities, we determined the DNA gag and pol copy numbers by quantitative reverse transcriptase–PCR. Both the gag and pol copy numbers were significantly reduced, indicating proviral excision (Figure 4d). Thus, Cas9 protein encapsulated in lentivirus can be used to disable proviral DNA. Although clinical application of this approach is currently impractical considering the low numbers of latently infected cells, this strategy can also be potentially used to target other regions in the HIV genome (in addition to CCR5) by judiciously designing the guide RNAs.

Inactivation of proviral DNA by sgRNA/Cas9P LV. (a) Time course of treatment and testing. (b) Cel1 assay of PCR product (LTR) from Cas9P/LTR sgRNA or control sgRNA transduced cells. (c) Flow cytometric analysis of intracellular p24 expression in control or LTR sgRNA transduced cells, 48 after activation of latency. Results from triplicates are shown. (d) Analysis of proviral DNA excision by determining gag and pol copy numbers by quantitative PCR.
In summary, we have developed a system to deliver Cas9 protein in lentiviral particles containing a vector designed to express sgRNA. To ensure adequate amounts of Cas9 protein, we expressed it as a fusion with the Gag protein. Gag-derived polypeptides are abundantly present in lentiviral particles, and each virion has been estimated to contain ~5000 copies of Gag-derived proteins like p24.21 A HIV protease cleavage site between Cas9 and gag would allow the release of functional Cas9 protein during particle maturation. A similar approach has been previously used to express ZFN protein. Although ZFN protein designed to target a particular sequence is all that is needed for gene editing, the CRISPR/Cas9 system will also require sgRNA that confers target specificity. However, our all-in-one lentivirus containing packaged Cas9 protein as well as a vector construct to express sgRNA overcomes this limitation.
The main advantage of our lentiviral particles is the transient exposure to the Cas9 nuclease. Although this reduces the gene editing frequency by ~20%, transient exposure to Cas9 is crucial to avoid the well-recognized off-target effects associated with long-term expression. This approach facilitates a short boost of protein activity (Cas9 protein translocates to the nucleus because of nuclear localization signals inserted into the Cas9 gene) that drives immediate targeted gene disruption without the risks of permanently inserting copies of Cas9 encoding genes into the genome. A lentiviral protein delivery approach also benefits from effective lentiviral transduction, which delivers protein to almost any cell, including hard-to-transfect T cells. Moreover, delivery can be easily further customized by adapting different lentiviral pseudotypes to direct protein transduction to specific cell types of interest (for example, HIV env pseudotyping of the particles for delivery to CD4 T cells).22 Another advantage of our system is the avoidance of the need to express the relatively large Cas9 protein via the lentiviral vector, thereby freeing the vector space to express multiple sgRNAs and/or homology sequence needed for insertion of novel exotic sequences at the desired genomic loci by homologous recombination. We anticipate that lentiviral protein transduction of nuclease proteins will emerge as a versatile alternative to current nuclease delivery techniques and will support continued efforts to promote safe genome editing therapies.
Materials and methods
Cells and culture conditions
293T, TZM-bl, J-Lat and Jurkat cells were cultured as described elsewhere.23 Human peripheral blood mononuclear cells were obtained from Zen-Bio (Research Triangle Park, Durham, NC, USA). CD4 T cells were isolated from peripheral blood mononuclear cells using CD4 T cell enrichment kit (Stem Cell Technologies, Vancouver BC, Canada). CD4 T cells were stimulated with CD3/CD28 (ThermoFisher Scientific, Waltham, MA, USA) before transduction with lentivirus and were cultured at 37 °C in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 U ml−1 of penicillin–streptomycin with recombinant IL-2 (20 U ml−1).
Lentiviral constructs and lentivirus production
The pCas9-PH-gagpol-D64V plasmid was constructed as follows: spCas9 gene was amplified from pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene, Cambridge, MA, USA; #42230) using forward primer 5′-GGTTGGACCGGTACCACCATG-3′ and reverse primer 5′-TTGACACCGGTCTGCACAATCGGATAGTTCTGGCTAAAGTTCTTTTTCTTTTTTGCCTGGCC-3′. PCR product was digested with AgeI and ligated with AgeI/BspEI fragment of pGFP-PH-gagpol-D64V.15 The pLB CAG-Cas9-P2Gm plasmid was constructed as follows: spCas9 gene was cut out from pX330-U6-Chimeric_BB-CBh-hSpCas9 with AgeI/EcoRI and ligated into AgeI/EcoRI fragment of pLB2 CAG P2Gm lentiviral vector (Addgene #19752). pLB-LTR, CD4 and CCR5 sgRNAs (Table 1) were constructed by ligating a synthesized spacer+tracrRNA fragment into HpaI/XhoI fragment of pLB lentiviral vector (Addgene #11619). Lentiviral particles were produced as follows. To produce Cas9P LV, 293T cells were plated to 70–80% confluence in 150-mm dishes 1 day before transfection. The pCas9-PH-gagpol-D64V (80 μg), the env plasmid, pCMV-VSV-G (15 μg each) and pMDL (wild-type Gag/Pol-40 μg), were co-transfected into 293T cells using calcium phosphate precipitation. To produce sgRNA/Cas9P LVs, (with sgRNAs to target LTR, CD4 and CCR5), the lentiviral vector (pLB-sgRNA), pCas9-PH-gagpol-D64V, and the helper pMDLg, Rev and env plasmid, pCMV-VSV-G were co-transfected into 293T cells. To produce Cas9 encoding LV (for Cas9 expression), pLB CAG-Cas9-P2Gm vector, pMDLg/pRRE, Rev and pCMV-VSV-G plasmids were co-transfected into 293T cells. Medium was replaced after 4 h and after another 48 h, the culture supernatants were harvested, centrifuged at 2000 r.p.m. for 10 min, filtered through 0.45 μm filter and ultracentrifuged through a 20% sucrose cushion at 15 000 r.p.m. at 4 °C for 2 h.24 Pellets were re-suspended in phosphate-buffered saline and stored at −80 °C. Concentrations of HIV-1 p24 were measured by ELISA and transduction efficiency was determined by examining for GFP expression by flow cytometry. TZM-bl, Jurkat and primary CD4 T cells were transduced at a multiplicity of infection of 10–50. After transduction, the cells were washed twice with phosphate-buffered saline and cultured in media for 48 or 72 h.
Flow cytometry
Flow cytometry was performed to determine cell surface antigen expression by 30 min incubation on ice with pertinent antibodies. The following monoclonal antibodies were used: human-specific monoclonal antibodies, anti-CCR5 conjugated with allophycocyanin (2D7/CCR5; BD Pharmingen, Franklin Lakes, NJ, USA), anti-human CD4 (allophycocyanin) and corresponding isotype control monoclonal antibodies (BD Pharmingen). Data were acquired on a BD FACS Canto II flow cytometer (Franklin Lakes, NJ, USA) and analyzed on BD FACS Diva software (Franklin Lakes, NJ, USA) v3.0. Overlays were made using FlowJo software (Franklin Lakes, NJ, USA) v3.0 where applicable.
Immunostaining
For immunoflurescence microscopy, TZM-bl cells were seeded on cover slips at a concentration of 2 × 105 cells per well 1 day before transfection or transduction. Cells were then transduced with Cas9 P LV (150 ng p24) or transfected with 2 μg of pX330-U6-Chimeric_BB-CBh-hSpCas9. After 24 h, cells were washed with phosphate-buffered saline and fixed with 4% paraformaldehyde solution at room temperature for 10 min, then permeabilized in phosphate-buffered saline with 3% BSA and 1% Triton-100 for 2 h. The slides were incubated with primary anti-Cas9 antibody (Clone 7A9-3A3, Novus biological, 1:500 dilution at 4 °C overnight) followed by incubation with Alexa Fluor 488 Goat Anti-Mouse IgG secondary antibody (Cat. A-11017, ThermoFisher Scientific, 1:600 dilution at room temperature for 1 h). The cover glasses were mounted with Vectashield Mounting Medium with DAPI (VECTOR, H-1200). Images were obtained using Nikon immunofluorescence microscope (Melville, NY, USA).
J-Lat latency reactivation
The J-Lat cells were transduced with lentiviruses expressing Cas9 protein with LTR-sgRNA or control sgRNA. Successfully transduced (GFP+) cells were sorted by a FACS Aria cell sorter (BD Biosciences, Franklin Lakes, NJ, USA) on day 2 post transduction. After expanding, sorted cells were treated with 10 ng ml−1 of tumor necrosis factor-α (R&D Systems, Minneapolis, MN, USA) and 0.5 μM 5-Aza-dC (Sigma-Aldrich, St Louis, MO, USA) and intracellular staining for the p24 done after 2 more days. For intracellular p24-Ag staining, stimulated cells were washed with Hanks balanced salt solution, fixed and permeabilized using Fix and Perm kit (BD Biosciences), stained with p24-RD1v(Clone KC57/Beckman Coulter, Indianapolis, IN, USA) as described.25
Gag and pol copy numbers
The amount of gag/pol in cellular DNA was quantified by real-time PCR as previously described20 using primers listed in Table 1.
Targeted gene sequencing
DNA was isolated from cells using DNA blood mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. One hundred nanograms of extracted DNA were subjected to PCR and using primers listed in Table 1. To further analyze gene disruption, the above PCR products were cloned into a TA cloning vector (Life Technologies, ThermoFisher Scientific) and cloned products were sequenced using the M13 primer.
Detection of gene disruption by Cel1 assay
The presence of mutations in PCR products was examined using a SURVEYOR Mutation Detection Kit (IDT, Redwood City, CA, USA) according to the manufacturer’s instructions. Briefly, the PCR products were denatured for 10 min in 95 °C and hybridized by gradual cooling using a thermo cycler. Next, 200 ng of hybridized DNA was subjected to digestion 1 μl of SURVEYOR Nuclease in the presence of 1 μl SURVEYOR Enhancer S and 15 mM MgCl2 for 1 h at 42 °C. Then, stop solution was added and samples were resolved on 2% agarose gel together with equal amounts of undigested PCR product controls.15, 18
Western blot analysis of Cas9 protein incorporated into lentiviral particles
Lentiviral particles harvested 72 h post transfection or cell lysates obtained 24 h after transduction/transfection were analyzed by western blotting as previously described.15 Briefly, Lentiviral particles and cells lysates were electrophoresed on a 10% SDS polyacrylamide gel, blotted onto a polyvinylidene fluoride membrane and probed with horseradish peroxidase-conjugated anti-Flag antibody and visualized by enhanced chemiluminescence using horseradish peroxidase substrate. Parallel blot was probed with HIV-1 p24 monoclonal antibody (NIH AIDS Reagent Program: #6457) followed by peroxidase conjugated anti-mouse secondary antibody.15
References
Gersbach CA, Perez-Pinera P . Activating human genes with zinc finger proteins, transcription activator-like effectors and CRISPR/Cas9 for gene therapy and regenerative medicine. Expert Opin Ther Targets 2014; 18: 835–839.
Niu J, Zhang B, Chen H . Applications of TALENs and CRISPR/Cas9 in human cells and their potentials for gene therapy. Mol Biotechnol 2014; 56: 681–688.
Zhang F, Wen Y, Guo X . CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet 2014; 23: R40–R46.
Manjunath N, Yi G, Dang Y, Shankar P . Newer gene editing technologies toward HIV gene therapy. Viruses 2013; 5: 2748–2766.
Riordan SM, Heruth DP, Zhang LQ, Ye SQ . Application of CRISPR/Cas9 for biomedical discoveries. Cell Biosci 2015; 5: 33.
Sternberg SH, Doudna JA . Expanding the Biologist's Toolkit with CRISPR-Cas9. Mol Cell 2015; 58: 568–574.
Mali P, Esvelt KM, Church GM . Cas9 as a versatile tool for engineering biology. Nat Methods 2013; 10: 957–963.
Lin Y, Cradick TJ, Brown MT, Deshmukh H, Ranjan P, Sarode N et al. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res 2014; 42: 7473–7485.
Cradick TJ, Fine EJ, Antico CJ, Bao G . CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res 2013; 41: 9584–9592.
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 2013; 31: 822–826.
Cannon RO 3rd, Leon MB, Watson RM, Rosing DR, Epstein SE . Chest pain and ‘normal’ coronary arteries–role of small coronary arteries. Am J Cardiol 1985; 55: 50B–60B.
Wu X, Kriz AJ, Sharp PA . Target specificity of the CRISPR-Cas9 system. Quant Biol 2014; 2: 59–70.
Skipper KA, Mikkelsen JG . Delivering the goods for genome engineering and editing. Hum Gene Ther 2015; 26: 486–497.
Cai Y, Bak RO, Krogh LB, Staunstrup NH, Moldt B, Corydon TJ et al. DNA transposition by protein transduction of the piggyBac transposase from lentiviral Gag precursors. Nucleic Acids Res 2014; 42: e28.
Cai Y, Bak RO, Mikkelsen JG . Targeted genome editing by lentiviral protein transduction of zinc-finger and TAL-effector nucleases. Elife 2014; 3: e01911.
Cai Y, Mikkelsen JG . Driving DNA transposition by lentiviral protein transduction. Mob Genet elements 2014; 4: e29591.
Urano E, Aoki T, Futahashi Y, Murakami T, Morikawa Y, Yamamoto N et al. Substitution of the myristoylation signal of human immunodeficiency virus type 1 Pr55Gag with the phospholipase C-delta1 pleckstrin homology domain results in infectious pseudovirion production. J Gen Virol 2008; 89: 3144–3149.
Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci USA 2014; 111: 11461–11466.
Ebina H, Misawa N, Kanemura Y, Koyanagi Y . Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep 2013; 3: 2510.
Qu X, Wang P, Ding D, Li L, Wang H, Ma L et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res 2013; 41: 7771–7782.
Swanson CM, Malim MH . SnapShot: HIV-1 proteins. Cell 2008; 133: 742, 742 e1.
Yi G, Choi JG, Bharaj P, Abraham S, Dang Y, Kafri T et al. CCR5 gene editing of resting CD4(+) T cells by transient ZFN expression from HIV envelope pseudotyped nonintegrating lentivirus confers HIV-1 resistance in humanized mice. Mol Ther Nucleic Acids 2014; 3: e198.
Klase Z, Yedavalli VS, Houzet L, Perkins M, Maldarelli F, Brenchley J et al. Activation of HIV-1 from latent infection via synergy of RUNX1 inhibitor Ro5-3335 and SAHA. PLoS Pathog 2014; 10: e1003997.
al Yacoub N, Romanowska M, Haritonova N, Foerster J . Optimized production and concentration of lentiviral vectors containing large inserts. J Gene Med 2007; 9: 579–584.
Lee SK, Dykxhoorn DM, Kumar P, Ranjbar S, Song E, Maliszewski LE et al. Lentiviral delivery of short hairpin RNAs protects CD4 T cells from multiple clades and primary isolates of HIV. Blood 2005; 106: 818–826.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Gene Therapy website
Supplementary information
Rights and permissions
About this article

Cite this article
Choi, J., Dang, Y., Abraham, S. et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther 23, 627–633 (2016). https://doi.org/10.1038/gt.2016.27
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2016.27
- Springer Nature Limited
This article is cited by
-
Viral vectors and extracellular vesicles: innate delivery systems utilized in CRISPR/Cas-mediated cancer therapy
Cancer Gene Therapy (2023)
-
Maybe you can turn me on: CRISPRa-based strategies for therapeutic applications
Cellular and Molecular Life Sciences (2022)
-
Prevalence of CCR5delta32 in Northeastern Iran
BMC Medical Genetics (2019)
-
Novel gRNA design pipeline to develop broad-spectrum CRISPR/Cas9 gRNAs for safe targeting of the HIV-1 quasispecies in patients
Scientific Reports (2019)
-
The therapeutic landscape of HIV-1 via genome editing
AIDS Research and Therapy (2017)