
Abstract
RGS1 (regulator of G-protein signaling 1) has been associated with multiple autoimmune disorders including type I diabetes. RGS1 desensitizes the chemokine receptors CCR7 and CXCR4 that are critical to the localization of T and B cells in lymphoid organs. To explore how RGS1 variation contributes to autoimmunity, we generated Rgs1 knockdown (KD) mice in the nonobese diabetic (NOD) model for type I diabetes. We found that Rgs1 KD increased the size of germinal centers, but decreased the frequency of T follicular helper (TFH) cells. We show that loss of Rgs1 in T cells had both a T cell-intrinsic effect on migration and TFH cell frequency, and an indirect effect on B-cell migration and germinal center formation. Notably, several recent publications described an increase in circulating TFH cells in patients with type I diabetes, suggesting this cell population is involved in pathogenesis. Though Rgs1 KD was insufficient to alter diabetes frequency in the NOD model, our findings raise the possibility that RGS1 plays a role in autoimmunity owing to its function in TFH cells. This mechanistic link, although speculative at this time, would lend support to the notion that TFH cells are key participants in autoimmunity and could explain the association of RGS1 with several immune-mediated diseases.
Similar content being viewed by others

Introduction
RGS1, a member of the regulator of G-protein signaling (RGS) family,1 has been associated with multiple immune-mediated diseases.2, 3, 4, 5 Data from genome-wide association studies show a significant association of single-nucleotide polymorphisms in the RGS1 region with multiple sclerosis and celiac disease2, 3 (P<10−17), and a suggestive association with type I diabetes.4, 5 RGS proteins are GTPase-activating proteins that modulate chemokine receptor signaling.1 Chemokine receptors depend on heterotrimeric G proteins to activate downstream effectors.6 Upon ligand activation, the G-protein α-subunit (Gα) exchanges guanosine triphosphate for guanosine diphosphate, resulting in dissociation from the Gβγ heterodimer7 and initiating signaling cascades that lead to cytoskeletal rearrangements and cell migration. Hydrolysis of guanosine triphosphate by Gα intrinsic GTPase activity causes signal termination. This enzymatic activity is accelerated by RGS-family proteins.1 RGS1 is highly expressed in lymphoid organs and serves as a negative regulator of chemokine receptor signaling in lymphocytes.1, 8 Ablation of Rgs1 in mice was shown to modify B-cell trafficking.9 In addition, Rgs1 deficiency leads to aberrant architecture of germinal centers.9, 10, 11 Although the phenotype described for Rgs1 knockout (KO) mice was largely attributed to B-cell dysfunction, a subsequent study found that Rgs1 also participates in chemotactic signaling in T cells.12 Rgs1 thus affects the migratory behavior of multiple cell types, and it is as yet unclear how RGS1 gene variation modifies the risk of autoimmunity, and of type I diabetes in particular.
T follicular helper (TFH) cells reside in the follicular areas of secondary lymphoid organs where they promote B-cell expansion and antibody affinity maturation within germinal centers.13 TFH cell maturation is a multistep process that begins in the T-cell zone with the activation of naive CD4+ T lymphocytes and leads to expression of the transcription factor Bcl6. Bcl6 drives the expression of the chemokine receptor CXCR5 that promotes migration from the T-cell zone toward the B-cell follicle.14 This migration also requires downregulation of CCR7 signaling.15 Of interest, Rgs1 expression is markedly upregulated in TFH cells,16 and this likely contributes to desensitizing migrating cells to CCR7 ligands. Notably, several studies have recently implicated TFH cells in type I diabetes.17, 18, 19 The frequency of TFH cells was found to be elevated in patients with type I diabetes. A similar increase in TFH cells was observed in a mouse model for autoimmune diabetes.19
To investigate a possible role for RGS1 in autoimmunity, we developed inducible Rgs1 knockdown (KD) mice within the nonobese diabetic (NOD) mouse model for type I diabetes.20 Rgs1 silencing recapitulated key phenotypes described for Rgs1 KO mice,9 including increased lymphocyte chemotaxis and enlarged germinal centers. Although we found that Rgs1 KD did not alter the risk of diabetes in NOD mice, we observed that loss of Rgs1 reduced the frequency of TFH cells. Furthermore, Rgs1 KD in T cells was sufficient to modify the migration of B cells. These findings suggest that the effects of Rgs1 KO on germinal center formation described previously may be caused in part by changes in TFH cell function. In addition, our data suggest that Rgs1 upregulation is a critical step in the migration of TFH cells that enables cells to downregulate CCR7 signals and to migrate into the follicular area. A link between Rgs1 expression and TFH cell frequency, a T-cell subset implicated in type I diabetes, could explain the association of RGS1 variants with autoimmunity.
Results
Generation of Rgs1 KD NOD mice
To study the role of Rgs1 in autoimmune diabetes, we generated transgenic NOD mice in which Rgs1 gene expression can be silenced by RNA interference in a doxycycline-dependent manner.21 We first validated lentiviral constructs for Rgs1 KD in vitro. Candidate short-hairpin RNA (shRNA) sequences were cloned into the lentiviral vector pUTG21, 22 in which shRNA expression is under the control of a doxycycline-inducible promoter. The vector also contains a constitutively expressed tetracycline repressor and a green fluorescent protein (GFP) reporter. We generated an Rgs1 luciferase reporter where Rgs1 complementary DNA is incorporated into the 3′ untranslated region of the Renilla luciferase gene. We transfected the Rgs1 luciferase reporter into HEK293 cells transduced with lentivirus encoding different shRNA sequences against Rgs1, and quantified Renilla luciferase activity as a measure of gene knockdown. We identified two shRNA sequences that potently inhibited the Rgs1 luciferase reporter (Figure 1a). These shRNA sequences were further validated for their ability to silence expression of a FLAG-tagged Rgs1 construct, as measured by quantitative PCR (Figure 1b) and western blotting (Figures 1c and d). The selected shRNA sequences were then used to generate two distinct Rgs1 KD NOD lines by lentiviral transgenesis (Figure 1e and Supplementary Figure S1). Finally, we confirmed that doxycycline treatment (100 μg ml−1 in the drinking water for 2 weeks) induced Rgs1 KD in vivo (Figures 1f–h and Supplementary Figure S1).

Generation and validation of NOD Rgs1 KD mice. (a–d) HEK293 cells were transduced with lentivirus encoding Rgs1 shRNA sequence #1 or #3 under the control of a doxycycline-inducible promoter. Rgs1 KD or control cells were transfected with the Rgs1 luciferase reporter (a) or with an expression vector containing a FLAG-tagged version of the RGS1 protein (b–d). Rgs1 knockdown efficiency was then analyzed by luminescence measurement (a), quantitative PCR (b) or western blotting (c, d). Results in (a–d) are representative of three experiments. (e) GFP expression in blood samples from Rgs1 KD mice. Representative histograms for Rgs1 KD (white histogram) and WT mice (black histogram) are shown. (f–h) Validation of Rgs1 silencing in vivo. Quantitative PCR (f) or western blotting (g, h) was performed with spleen mRNA or protein, respectively, from individual Rgs1 KD (white bars) and WT mice (black bars). Rgs1 protein levels were compared in mice treated or not with doxycycline. Results in (f–h) are derived from three to five biological replicates and are representative of two experiments. All results show mean values±s.e.m.
Rgs1 silencing sensitizes T cells to CCR7 ligation
Rgs1 modulates CCR7 signaling by promoting the conversion of the guanosine triphosphate-activated Gα subunit to a guanosine diphosphate-quiescent form that results in reassembly of the heterotrimeric G-protein complex,1 thus terminating chemokine receptor signaling. Accordingly, Rgs1 KO was shown to sensitize lymphocytes to CCR7 stimuli.11, 12 We tested the response of Rgs1 KD T lymphocytes to CCR7 chemokine ligands in a transwell assay. Consistent with a role for Rgs1 in diminishing CCR7 signaling, Rgs1 KD increased T-cell migration toward CCL19 (Figure 2a) and CCL21 (Figure 2b). The chemotactic response to the CXCR4 ligand CXCL12 was also affected, though only modestly (Figure 2c). Similar results were obtained with Rgs1 KD B cells (Figure 2d). These data are consistent with a previous report showing that Rgs1 KO sensitized T cells to CCR7 and CXCR4 ligation.12 We concluded that Rgs1 KD modifies lymphocyte chemotaxis in vitro in a manner consistent with observations made with Rgs1 KO cells.

Rgs1 KD sensitizes T cells to CCR7 and CXCR4 stimulation. Migration of T cells from the spleen or lymph nodes (a–c) or splenic B cells (d) from WT (black) and Rgs1 KD (white) in response to CCL19 (a), CCL21 (b, d) or CXCL12 (c, d). Data represents mean±s.e.m. of three biological replicates. *P<0.05, **P<0.01 (unpaired t-test).
Rgs1 KD protects against colitis, but does not change the risk of autoimmune diabetes
A study by Hayday and colleagues12 reported that T cells derived from Rgs1 KO mice were less pathogenic than their wild-type (WT) counterparts in an experimental colitis model based on the transfer of CD4+CD45RBhigh T cells into immunodeficient mice. The authors speculated that the absence of Rgs1 increases the propensity of CD4+ T cells to home to secondary lymphoid organs, reducing their dwell time in the gastrointestinal tract and diminishing colitis severity. To test whether Rgs1 KD would recapitulate this phenotype, we transferred sorted CD4+CD45RBhigh T cells from Rgs1 KD or WT mice into NOD.SCID recipient mice. The results of this experiment suggested that Rgs1 KD T cells were less colitogenic than WT cells. Colons were lighter and longer, and histological scores lower in mice transplanted with Rgs1 KD cells (Supplementary Figure S2). With an indication that Rgs1 KD was able to alter T-cell function in vivo, we proceeded to ask whether Rgs1 silencing would affect the development of autoimmune diabetes in NOD mice. However, we found no difference in the frequency of spontaneous diabetes between WT and Rgs1 KD mice, whether treated or not with doxycycline (Figure 3a). In addition, diabetes onset was tested by two alternative approaches: by administering cyclophosphamide (Figure 3b) that is thought to disproportionally deplete the regulatory T-cell population, leading to a breakdown in immune regulation,23 and by transfer of splenocytes from overtly diabetic Rgs1 KD mice into immunodeficient NOD.SCID mice (Figure 3c). Both approaches indicated again that Rgs1 silencing was insufficient to alter the risk of diabetes in the NOD model.

Rgs1 KD does not change the risk of autoimmune diabetes in the NOD model. (a) Spontaneous diabetes incidence in WT and Rgs1 KD mice treated (bottom panel, P=0.26) or not (top panel, P=0.42) with doxycycline continuously from birth (n=22 mice per group). None of the differences between genotypes or between treated and untreated groups were statistically significant. (b) Cyclophosphamide-accelerated diabetes in WT and Rgs1 KD mice treated with doxycycline for the duration of the experiment (n=7 mice per group, P=0.18). (c) Diabetes in NOD.SCID mice (n=5 per group, P=0.17) treated or not with doxycycline following transfer of splenocytes from an overtly diabetic Rgs1 KD NOD mouse not treated with doxycycline.
Rgs1 silencing affects the differentiation of TFH cells
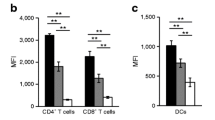
To further dissect the role of Rgs1, we explored its function within secondary lymphoid organs where Rgs1 may fine-tune local migratory signals. Reports from Rgs1 KO mice previously linked loss of Rgs1 to aberrant splenic architecture, characterized by enlarged germinal centers.9, 10 This observation was replicated in Rgs1 KD mice: transgenic mice treated with doxycycline had significantly larger germinal centers after immunization (Figures 4a and b). Accordingly, both the frequency and absolute number of IgDlo B cells were increased by Rgs1 silencing (Figure 4c). Although the proportion of proliferating (Ki-67+) IgDlo cells was not affected by Rgs1 KD, transgenic mice harbored more proliferating B cells overall (Figures 4d and e). When we analyzed different T-cell populations in lymph nodes collected from immunized mice, we found that the frequency of TFH cells, characterized as CD4+PD1highCXCR5high cells, was diminished in Rgs1 KD mice (Figures 5a–c). Further analysis of the TFR population, the regulatory subpopulation of follicular T cells that expresses the transcription factor Foxp3,16 suggested that the frequency of TFR cells may be elevated (Figure 5d), though this difference did not reach statistical significance (P=0.07) and the absolute number of TFR cells was instead decreased by Rgs1 KD (Figure 5e) similarly to the number of TFH cells. Although Rgs1 KD TFH cells were reduced in numbers, they resembled WT TFH cells in their expression of ICOS (Inducible T-cell COStimulator) (Figure 5f) and Bcl6 (Figure 5g). Immunohistochemical staining further confirmed that the numbers of CD4+ T cells within germinal centers were reduced in Rgs1 KD animals (Figure 5h). Collectively, these data show that Rgs1 KD modifies germinal center formation, as shown previously, and indicate that loss of Rgs1 reduces the frequency of follicular T cells.

Rgs1 KD alters germinal center formation. WT and Rgs1 KD mice were treated with doxycycline and immunized subcutaneously with ovalbumin in complete Freund’s adjuvant (CFA). The draining inguinal lymph node (ILN), non-draining axillary lymph nodes (ALNs) and spleen were analyzed 4 days later. (a) Representative images of germinal centers stained with peanut agglutinin (PNA) in the spleen (top) and ILN (middle) of WT and Rgs1 KD mice. The bottom panels show co-staining of PNA with B220 to control for specificity of PNA staining in B-cell areas. Scale bar: 200 μm. (b) Quantification of the area of germinal centers in three (spleen) and five (ILN) mice per group. (c, d) Frequency and absolute number of IgDlo B cells (c) and of Ki-67+ cells within the IgDlo population (d) in immunized mice (n=5 mice per group). (e) ILNs were stained for PNA and Ki-67 to visualize proliferating B cells within germinal centers. Scale bar: 100 μm. *P<0.05, ***P<0.001 (unpaired t-test).

Rgs1 KD decreases the frequency of TFH cells. Representative flow cytometry results (a), frequency (b) and number (c) of CD4+PD-1+CXCR5+ TFH cells in immunized WT and Rgs1 KD mice. Cells shown in (a) are gated on the CD4+ population. (d, e) Representative flow cytometry results (d) and frequency (e) of FoxP3+-expressing cells within the CD4+PD-1+CXCR5+ population gated as shown in (a). (f) Representative flow cytometry results for ICOS expression within the CD4+PD-1+CXCR5+ population as gated in (a). (g) Frequency of CD4+CXCR5+Bcl6+ cells and mean fluorescence intensity (MFI) of Bcl6 staining within this population in immunized WT and Rgs1 KD mice. (h) Spleen sections were stained with PNA and anti-CD4 to quantify CD4+ T cells within germinal centers of immunized WT and Rgs1 KD mice. Representative images and summary data for five WT and three Rgs1 KD mice are shown. Results are representative of two or more experiments. All data show means±s.e.m., *P<0.05, **P<0.01 (unpaired t-test).
Rgs1 KD increases B-cell migration in a B cell-extrinsic manner
Data from the Rgs1 KO mouse model had demonstrated that Rgs1-dependent chemokine signals modify germinal center formation.9, 10, 11 The increase in germinal centers in the absence of Rgs1 had been attributed to a B cell-intrinsic effect. We sought to investigate whether this phenotype is also influenced by Rgs1 function in T lymphocytes. To this end, we transferred B cells from NOD Raspberry mice that express the mRaspberry fluorescent reporter24 into WT or Rgs1 KD mice. Migration of Raspberry+ B cells into the spleen was analyzed by flow cytometry 4 days later. As observed in previous experiments, TFH cell frequency and number was decreased in the spleen of Rgs1 KD mice (Figures 6a and b). Notably, transferred Raspberry+ B cells were more numerous in the spleen of Rgs1 KD recipient animals (Figures 6c and d), suggesting that loss of Rgs1 leads to active recruitment and/or retention of circulating B cells into secondary lymphoid organs owing to B cell-extrinsic effects.

The migratory behavior of WT B cells is modified in Rgs1 KD mice. WT NOD Raspberry B cells were transferred into WT or Rgs1 KD mice treated with doxycycline. Recipient mice were immunized, and spleens were analyzed 4 days later. Representative flow cytometry data (a, c), frequency and absolute number (b, d) of host CD4+PD1highCXCR5high T cells (a, b) and donor WT Raspberry B cells (c, d). Data show mean values±s.e.m. in five WT and six Rgs1 KD mice. *P<0.05, **P<0.01 (unpaired t-test).
Rgs1 deficiency in T cells modifies B-cell migration
In order to investigate whether Rgs1 KD in T cells alone can influence the migratory behavior of B cells, we co-transferred B and T cells from WT and Rgs1 KD mice into NOD.SCID mice in all four possible combinations (Supplementary Figure S3). Mice reconstituted with Rgs1 KD T cells had fewer TFH cells (Figures 7a and b) irrespective of the genotype of co-transferred B cells, indicating that the effect of Rgs1 KD on TFH cell frequency is T-cell intrinsic. The frequency and number of B cells in the spleen of recipient mice was increased in the presence of Rgs1 KD T cells (Figures 7c and d); in contrast, the frequency and number of CD4+ T cells was comparable in all experimental groups (Figure 7e). These results suggest that Rgs1 deficiency in T cells is sufficient to modify B-cell migration. Collectively, the data indicate that Rgs1 KD modifies B-cell migration through both B cell-intrinsic and T cell-mediated effects.

Rgs1 KD in T cells is sufficient to modify B-cell migration. T cells and B cells from WT and Rgs1 KD were co-transferred into NOD.SCID mice in all four combinations. Recipient mice were immunized with ovalbumin in complete Freund’s adjuvant (CFA), and draining lymph nodes and spleen were analyzed 4 days later. Representative flow cytometry data (a, c) used to measure the frequency of CD4+CXCR5+ TFH cells (b), and the frequency and number of CD19+ B cells (d) and total CD4+ T cells (e). Data show mean values±s.e.m. from four to five mice per group. *P<0.05, **P<0.01, ***P<0.001 (unpaired t-test).
Rgs1 KD sensitizes TFH cells to CCR7 ligands
The reduced frequency of TFH cells in Rgs1 KD mice could be because of impaired proliferation after immunization. Alternatively, compromised migration into the follicular area may hinder T-cell differentiation toward a TFH cell phenotype. We found that Rgs1 was upregulated after T-cell activation (Supplementary Figure S4). However, reduced Rgs1 expression in transgenic T cells did not affect their proliferation (Supplementary Figure S4). To test whether Rgs1 KD had a direct effect on TFH cell migration, CD4+ T cells isolated from immunized mice were subjected to a migration assay toward CCL19, CCL21 and CXCL13. TFH cells from WT mice migrated in response to CXCL13 but were irresponsive to CCR7 ligands (Figure 8). In contrast, we found that Rgs1 silencing sensitized transgenic TFH cells to CCL19 and CCL21, without affecting migration toward CXCL13. It was shown previously that the expression of CXCR5 is not sufficient for TFH cell migration into the germinal centers.15 TFH cells must also downregulate CCR7 signaling to skew chemotactic signals in favor of CXCL13. Our results show that silencing Rgs1 in T cells potentiates CCR7 responsiveness, a feature that may compromise the differentiation of TFH cells by positioning them outside of their follicular niche.

Rgs1 KD increases TFH cell sensitivity to CCR7 but not CXCR5 ligation. CD4+ T cells were purified from WT (black bars) and Rgs1 KD mice (white bars) and subjected to a transwell migration assays toward CCL19 and CCL21 (100 ng ml−1, top panel) or CXCL13 (concentration as indicated, bottom panel). After 4 h of incubation, transmigrated cells were labeled for CD4 and CXCR5 and analyzed by fluorescence-activated cell sorting (FACS). Data show the percentage of CD4+CXCR5+ cells within the CD4+ population and represent means±s.e.m. of three biological replicates. *P<0.05, **P <0.01 (unpaired t-test).
Discussion
In this study, we describe a functional link between Rgs1 and TFH cells. Rgs1 had previously been implicated in chemokine receptor signaling in both T and B cells.9, 10, 11, 12 In particular, Rgs1 deletion was shown to modify the migratory behavior of B cells within follicular areas and to increase germinal center formation.9, 10, 11 Even the partial loss of Rgs1 in heterozygous Rgs1 mutant mice was demonstrated to affect chemokine responses,11 and this was confirmed in our experiments with Rgs1 KD mice. Germinal centers are structured into the so-called dark and light zones.13 These zones are functionally distinct, and their organization is strictly dependent on CXCR4 signals that retain centroblasts—dividing B cells undergoing somatic hypermutation—within the dark zone.25 In CXCR4-deficient mice, germinal centers lack distinguishable dark and light zones. The cycling of germinal center B cells from the dark to the light zone and back thus requires sequential up- and downregulation of CXCR4 signals. Expression of CXCR4 itself modifies these signals. In addition, increased Rgs1 expression is required to desensitize B cells to CXCR4 ligation. Consistent with this notion, germinal centers in Rgs1 KO mice feature a more prominent dark zone,9 likely because germinal center B cells stay sensitive to CXCL12 even after decreasing CXCR4 expression. Consequently, the abnormal size and structure of germinal centers in Rgs1-deficient mice had been attributed primarily to a change in B-cell chemotaxis. However, Rgs1 is also expressed in T cells.12 Rgs1 is upregulated following T-cell activation, and is expressed at particularly high levels in TFH cells.16 TFH cells localize to the follicular areas of secondary lymphoid organs, and participate in germinal center reactions. Their correct localization requires not only upregulation of CXCR5 that draws TFH cells towards B-cell areas, but also simultaneous downregulation of CCR7 signals.15 Expression of CXCR5 alone was shown to be insufficient to promote T-cell migration into follicles. Notably, CCR7 expression is reduced but not absent in TFH cells.15 Furthermore, Kehrl and colleagues10 have reported that partial loss of CCR7 is insufficient to significantly diminish CCR7-dependent chemotaxis. Rgs1 expression may thus serve to further desensitize activated T cells to CCR7 ligands that would otherwise retain them in the T-cell zone. We propose that loss of Rgs1 impairs the migration of activated T cells into the B-cell zone and thereby inhibits the full differentiation of TFH cells.26 In a situation analogous to Rgs1 KO B cells being retained in the dark zone by hypersensitivity to CXCL12, Rgs1-deficient T cells are presumably retained in the T-cell zone by hypersensitivity to CCL19 and CCL21. This idea is supported by our observation that Rgs1 KD TFH cells migrated in response to CCR7 ligation, whereas WT TFH cell did not. Of note, CCR7 expression itself was not affected by Rgs1 KD (data not shown). Our data thus indicate that Rgs1 is a key modifier of T-cell localization within lymphoid organs, and that Rgs1 plays a role in the differentiation of TFH cells.
Interestingly, our data suggest that Rgs1 silencing in T cells alone had an effect on B-cell migration. We posit that the germinal center phenotype described for Rgs1 KO mice may be exacerbated by a change in T-cell function, and that this phenotype is not solely caused by B cell-intrinsic effects. Both previously reported data9 and our own experiments support the notion that loss of Rgs1 directly affects B-cell migration. However, T cells appear to contribute to altered B-cell trafficking in Rgs1-deficient mice. Mice devoid of TFH cells fail to form germinal centers. The observations that loss of Rgs1 both reduces TFH cell frequency and increases germinal center formation may seem contradictory. This unexpected correlation could be explained first by the fact that the B cell-intrinsic effects of Rgs1 deficiency are dominant, and second because TFH cells are reduced in number but not entirely absent in Rgs1 KD mice. Rgs1 KD had a moderate impact on TFH cells overall, and it will be of interest to revisit the role of Rgs1 in TFH cells in the context of mice completely deficient in Rgs1.9 Exactly how and to what extent the loss of Rgs1 in T cells might contribute to increased germinal center size remains to be explained. Notwithstanding, our findings that Rgs1 plays a role in TFH cells may provide a possible explanation for the association of RGS1 with several autoimmune disorders including type I diabetes.2, 3, 4, 5 TFH cells have been implicated in multiple sclerosis27, 28 and type I diabetes.17, 18, 19 Conceivably, RGS1 variants that promote TFH cell formation could increase the risk of autoimmunity. In this regard, a disease-associated RGS1 variant has been associated with elevated levels of CXCL13 in the cerebrospinal fluid of multiple sclerosis patients, thus linking a TFH-relevant chemokine to RGS1 gene variation in the context of autoimmune disease.29 Whether an analogous relationship can be found between the type I diabetes-associated variants and TFH cell frequency warrants further investigation.
In sum, we have shown that Rgs1 is a critical modifier of T-cell migration within lymphoid tissue, and that this disease-associated gene participates in the development and function of TFH cells. In light of multiple recent publication reporting an association between elevated TFH cell frequency and type I diabetes,17, 18, 19 our findings raise the intriguing possibility that RGS1 variation affects the risk of autoimmunity owing to its role in TFH cells.
Materials and methods
Mice
NOD mice were maintained under specific pathogen-free conditions at the Joslin Diabetes Center. Transgenic mice were generated as described previously.30 Briefly, lentiviral vectors encoding a shRNA that targets the sequences 5′-CGCAAATAACAGTTGCTATTA-3′ (#1) or 5′-GCATAACAAAGCAGAGAATAT-3′ (#3) in the Rgs1 gene were used in which shRNA expression is under the control of a tetracycline-inducible promoter.21 The same lentiviral construct also encodes the tetracycline repressor and a GFP reporter, both driven by a constitutive promoter.21 Lentiviral particles were microinjected into the perivitelline space of NOD zygotes. Transduced embryos were reimplanted into pseudo-pregnant NOD mice. Transgenic animals were identified by GFP expression, and bred several generations to establish stable lines carrying a hemizygous transgene with Mendelian inheritance. Transgenic mice were treated with 100 μg ml−1 doxycycline in the drinking water to silence Rgs1 in all experiments unless specified. Data for shRNA #1 mice are shown in all experiments, unless otherwise noted. All experiments were approved by the institutional committee for the care and use of animals at the Joslin Diabetes Center (protocol #2014-01).
Luciferase assay
The Rgs1 cDNA (GenBank: BC028634.1) was cloned into the dual-luciferase reporter plasmid psiCheck2 (Promega, Fitchburg, WI, USA). HEK293T cells were transfected using Fugene 6 transfection reagent (Promega) combining 100 ng psiCheck2 plasmid together with 300 ng lentiviral vector pUTG containing a doxycycline-inducible shRNA sequences against Rgs1. Luminescence in cell lysates was measured with a SynergyMx luminometer (Biotek, Winooski, VT, USA) after 48 h to assess reporter silencing.
Western blotting
To validate the knockdown efficiency of selected shRNA sequences in vitro, HEK293T cells were co-transfected with 1 μg of pcDNA3 plasmid containing the Rgs1 gene fused to a N-terminus FLAG-tag peptide and 1 μg of pUTG vector containing a shRNA sequence against Rgs1 transcript. Cell lysates were resolved in a 15% SDS–polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. Proteins were visualized using the following antibodies: HRP-anti-FLAG antibody (Sigma, St Louis, MO, USA) and rabbit anti-actin antibody (Santa Cruz Biotech, Dallas, TX, USA). Endogenous Rgs1 expression in lymphocytes was also analyzed using an anti-rabbit Rgs1 antibody (Thermo Scientific, Waltham, MA, USA).
Quantitative PCR
RNA isolation was performed using TRIzol reagent (Life Technologies, Waltham, MA, USA). Subsequently, cDNA was synthesized using the Superscript III (Life Technologies) according to the manufacturer’s protocol. Quantitative Rgs1 PCR was performed using the following primers: forward 5′-TTTTCTGCTAGCCCAAAGGA-3′ and reverse 5′-TGTTTTCACGTCCATTCCAA-3′. Results were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Lymphocyte migration assays
T and B cells were isolated by magnetic sorting using Pan T cell isolation kit II or CD43 (Ly48) Microbeads respectively (Miltenyi Biotech, Bergisch Gladbach, Germany). Cell purity following magnetic sorting was >95% for all experiments. Cells derived from Rgs1 KD (GFP+) and WT (GFP-) mice were mixed in a 1:1 ratio and placed in the upper chamber of transwell plate. In the lower chamber, CCL19, CCL21, CXCL12 or CXCL13 were added to the media at the indicated concentrations. Cells that transmigrated to the lower chamber over the period of 4 h were analyzed by flow cytometry. Percentage of migration=(number of cells in the lower chamber after migration/input cell number in the upper chamber) × 100.
Diabetes measurements
Diabetes incidence in transgenic mice was compared with non-transgenic littermates. The 4-week-old mice were fed 100 μg ml−1 of doxycycline in drinking water ad libitum and were either left to develop diabetes spontaneously over the course of 30 weeks or administered 200 mg kg−1 cyclophosphamide intraperitoneally to accelerate diabetes onset. In additional experiments, adoptive transfer of diabetes was performed by injecting intravenously 2 × 106 splenocytes from overtly diabetic NOD Rgs1 KD mice into 6-week-old NOD.SCID mice. Cell recipients were then either treated or not with doxycycline. Diabetes was tested by glycosuria measurements using Diastix (Bayer, Leverkusen, Germany). Mice with two consecutive readings of 250 mg dl−1 were considered diabetic.
Histology and immunofluorescence
At 5 days after subcutaneous immunization in the leg with ovalbumin in complete Freund’s adjuvant, mice were killed and spleen, axillary lymph nodes and inguinal lymph nodes were dissected and frozen in optimal cutting temperature medium. Sections mounted on slides were stained with the following antibodies: anti-CD45R (B220) Alexa fluor 488 (eBioscience, Santa Clara, CA, USA), anti-Ki67 (eBioscience), anti-CD4 (Serotec, Raleigh, NC, USA) or biotinylated peanut agglutinin (Sigma), followed by the secondary antibody anti-rat Alexa Fluor 488 or Alexa Fluor-594 streptavidin (Invitrogen, Carlsbad, CA, USA), and analyzed by fluorescent microscopy. Germinal centers size was determined using the measuring tool in the CellSense software (Olympus, Tokyo, Japan).
Flow cytometry
Lymphocytes were purified from the spleen or lymph nodes of WT and Rgs1 KD mice and labeled with the following antibodies: anti-PD1 PE-Cy7, anti-CXCR5 APC, anti-CD4 PE, anti-IgD PE, anti-ICOS PE or anti-CD19 PB (BioLegend, San Diego, CA, USA). Foxp3 intracellular staining kit (eBioscience) was used to label follicular regulatory T cells. Fixation/Permeabilization buffers were used to stain intracellular Bcl6 and Ki-67. Intrinsic GFP and Raspberry fluorophores were also analyzed in specific experiments. Data were acquired on an LSRII flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed using the FlowJo software (TreeStar Inc., Ashland, OR, USA).
B-cell transfer assay
B cells were isolated from NOD Raspberry donors by negative selection using CD43 (Ly-48) magnetic beads (Miltenyi Biotech). Cell purity following magnetic sorting was >95%. 2 × 107 cells were injected intravenously into age- and gender-matched WT or Rgs1 KD NOD mice. After 2 weeks, mice were immunized via intradermal injection in the leg with 160 μg of ovalbumin in complete Freund’s adjuvant. Mice were killed for analysis 4 days later.
Lymphocyte transfer into NOD.SCID mice
T and B lymphocytes were isolated by magnetic sorting from WT or Rgs1 KD mice by negative selection using the Pan T cell isolation kit or CD43 (Ly-48) microbeads (Miltenyi Biotech). WT and Rgs1 KD T and B cells (each 1 × 107) were mixed in a 1:1 ratio in all four combinations. A total of 2 × 107 cells were injected intravenously into age- and gender-matched NOD.SCID recipient mice. After 2 weeks, mice were immunized as described previously. Mice were killed for analysis of lymph nodes and spleens 4 days later.
Statistical analyses
Experimental groups were compared by two-tailed unpaired Student’s t-test using the GraphPad Prism software (Treestar Inc.). A P value of <0.05 was considered significant. Sufficient sample size was estimated without the use of a power calculation. No samples were excluded from the analysis. No randomization was used for animal experiments. Data analysis was not blinded.
References
Moratz C, Harrison K, Kehrl JH . Regulation of chemokine-induced lymphocyte migration by RGS proteins. Methods Enzymol 2004; 389: 15–32.
Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med 2008; 359: 2767–2777.
International Multiple Sclerosis Genetics Wellcome Trust Case Control Consortium 2 Sawcer S, Hellenthal G Pirinen M Spencer CC et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011; 476: 214–219.
Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009; 41: 703–707.
Bradfield JP, Qu H-Q, Wang K, Zhang H, Sleiman PM, Kim CE et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet 2011; 7: e1002293.
Ebert LM, Schaerli P, Moser B . Chemokine-mediated control of T cell traffic in lymphoid and peripheral tissues. Mol Immunol 2005; 42: 799–809.
Oldham WM, Hamm HE . Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol 2008; 9: 60–71.
Reif K, Cyster JG . RGS molecule expression in murine B lymphocytes and ability to down-regulate chemotaxis to lymphoid chemokines. J Immunol 2000; 164: 4720–4729.
Moratz C, Hayman JR, Gu H, Kehrl JH . Abnormal B-cell responses to chemokines, disturbed plasma cell localization, and distorted immune tissue architecture in Rgs1 − / − mice. Mol Cell Biol 2004; 24: 5767–5775.
Han S-B, Moratz C, Huang NN, Kelsall B, Cho H, Shi CS et al. Rgs1 and Gnai2 regulate the entrance of B lymphocytes into lymph nodes and B cell motility within lymph node follicles. Immunity 2005; 22: 343–354.
Hwang IY, Park C, Harrison KA, Huang NN, Kehrl JH . Variations in Gnai2 and Rgs1 expression affect chemokine receptor signaling and the organization of secondary lymphoid organs. Genes Immun 2010; 11: 384–396.
Gibbons DL, Abeler-Dörner L, Raine T, Hwang I-Y, Jandke A, Wencker M et al. Cutting edge: regulator of G protein signaling-1 selectively regulates gut T cell trafficking and colitic potential. J Immunol 2011; 187: 2067–2071.
Victora GD, Nussenzweig MC . Germinal centers. Annu Rev Immunol 2012; 30: 429–457.
Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD et al. Bcl6 mediates the development of T follicular helper cells. Science 2009; 325: 1001–1005.
Haynes NM, Allen CDC, Lesley R, Ansel KM, Killeen N, Cyster JG . Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol 2007; 179: 5099–5108.
Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF et al. Foxp3(+) follicular regulatory T cells control the germinal center response. Nat Med 2011; 17: 975–982.
Xu X, Shi Y, Cai Y, Zhang Q, Yang F, Chen H et al. Inhibition of increased circulating Tfh cell by anti-CD20 monoclonal antibody in patients with type 1 diabetes. PLoS One 2013; 8: e79858.
Ferreira RC, Simons HZ, Thompson WS, Cutler AJ, Dopico XC, Smyth DJ et al. IL-21 production by CD4+ effector T cells and frequency of circulating follicular helper T cells are increased in type 1 diabetes patients. Diabetologia 2015; 58: 781–790.
Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest 2015; 125: 292–303.
Delovitch TL, Singh B . The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity 1997; 7: 727–738.
Zheng P, Kissler S . PTPN22 silencing in the NOD model indicates the type 1 diabetes-associated allele is not a loss-of-function variant. Diabetes 2013; 62: 896–904.
Herold MJ, van den Brandt J, Seibler J, Reichardt HM . Inducible and reversible gene silencing by stable integration of an shRNA-encoding lentivirus in transgenic rats. Proc Natl Acad Sci USA 2008; 105: 18507–18512.
Brode S, Raine T, Zaccone P, Cooke A . Cyclophosphamide-induced Type-1 diabetes in the NOD mouse is associated with a reduction of CD4+ CD25+ Foxp3+ regulatory T cells. J Immunol 2006; 177: 6603–6612.
Dirice E, Kahraman S, Jiang W, El Ouaamari A, De Jesus DF, Teo AKK et al. Soluble factors secreted by T cells promote β-cell proliferation. Diabetes 2014; 63: 188–202.
Allen CDC, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol 2004; 5: 943–952.
Crotty S . T follicular helper cell differentiation, function and roles in disease. Immunity 2014; 41: 529–542.
Romme Christensen J, Bornsen L, Ratzer R, Piehl F, Khademi M, Olsson T et al. Systemic inflammation in progressive multiple sclerosis involves follicular T-helper, Th17- and activated B-cells and correlates with progression. PLoS One 2013; 8: e57820.
Fan X, Jin T, Zhao S, Liu C, Han J, Jiang X et al. Circulating CCR7+ICOS+ memory T follicular helper cells in patients with multiple sclerosis. PLoS One 2015; 10: e0134523.
Lindén M, Khademi M, Lima Bomfim I, Piehl F, Jagodic M, Kockum I et al. Multiple sclerosis risk genotypes correlate with an elevated cerebrospinal fluid level of the suggested prognostic marker CXCL13. Mult Scler 2013; 19: 863–870.
Kissler S . From genome-wide association studies to etiology: probing autoimmunity genes by RNAi. Trends Mol Med 2011; 17: 634–640.
Acknowledgements
We acknowledge support from the Joslin DRC Flow Cytometry Core facility. This work was supported in part by a Career Development Award (2-2010-383) and an Innovative Award (1-INO-2014-169-A-V) from JDRF to SK, and by a DRC award (NIH Award Number P30DK036836) to the Joslin Diabetes Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Genes and Immunity website
Supplementary information
Rights and permissions
About this article

Cite this article
Caballero-Franco, C., Kissler, S. The autoimmunity-associated gene RGS1 affects the frequency of T follicular helper cells. Genes Immun 17, 228–238 (2016). https://doi.org/10.1038/gene.2016.16
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gene.2016.16
- Springer Nature Limited
This article is cited by
-
The NF-κB Pathway: a Focus on Inflammatory Responses in Spinal Cord Injury
Molecular Neurobiology (2023)