
Abstract
The mechanism by which human leukocyte antigen B27 (HLA-B27) contributes to ankylosing spondylitis (AS) remains unclear. Genetic studies demonstrate that association with and interaction between polymorphisms of endoplasmic reticulum aminopeptidase 1 (ERAP1) and HLA-B27 influence the risk of AS. It has been hypothesised that ERAP1-mediated HLA-B27 misfolding increases endoplasmic reticulum (ER) stress, driving an interleukin (IL) 23-dependent, pro-inflammatory immune response. We tested the hypothesis that AS-risk ERAP1 variants increase ER-stress and concomitant pro-inflammatory cytokine production in HLA-B27+ but not HLA-B27− AS patients or controls. Forty-nine AS cases and 22 healthy controls were grouped according to HLA-B27 status and AS-associated ERAP1 rs30187 genotypes: HLA-B27+ERAP1risk, HLA-B27+ERAP1protective, HLA-B27−ERAP1risk and HLA-B27−ERAP1protective. Expression levels of ER-stress markers GRP78 (8 kDa glucose-regulated protein), CHOP (C/EBP-homologous protein) and inflammatory cytokines were determined in peripheral blood mononuclear cell and ileal biopsies. We found no differences in ER-stress gene expression between HLA-B27+ and HLA-B27− cases or healthy controls, or between cases or controls stratified by carriage of ERAP1 risk or protective alleles in the presence or absence of HLA-B27. No differences were observed between expression of IL17A or TNF (tumour necrosis factor) in HLA-B27+ERAP1risk, HLA-B27+ERAP1protective and HLA-B27−ERAP1protective cases. These data demonstrate that aberrant ERAP1 activity and HLA-B27 carriage does not alter ER-stress levels in AS, suggesting that ERAP1 and HLA-B27 may influence disease susceptibility through other mechanisms.
Similar content being viewed by others
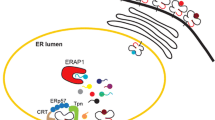


Introduction
Susceptibility to ankylosing spondylitis (AS), the prototypic spondyloarthropathy, is largely genetically determined, with the heritability of disease risk estimated at >90%.1 The endoplasmic reticulum aminopeptidase 1 (ERAP1) gene is strongly associated with AS; only the class I major histocompatibility complex allele, HLA-B27 (human leukocyte antigen B27) shows stronger association.2 ERAP1 (formerly known as ARTS-1) is an aminopeptidase with a ubiquitous tissue distribution, which is expressed in the endoplasmic reticulum (ER). ERAP1 has two reported functions. First, in vitro studies have suggested that ERAP1 may function as a 'sheddase', cleaving cytokine receptors off the cell surface, including interleukin-6 receptor subunit alpha (IL-6R), interleukin-1 receptor type 2 (IL-1R2) and tumour necrosis factor receptor (TNF-R), each of which are encoded by genes associated with AS.3, 4, 5, 6 However, we have demonstrated that spleen cells from ERAP−/− mice did not show altered cleavage of IL-6R and TNF-R in vitro.2 Moreover, in the context of AS, Haroon et al.7 demonstrated that ERAP1 polymorphisms do not alter serum levels of inflammatory cytokines. Second, ERAP1 has been shown to act as a ‘molecular ruler’, trimming peptides that have been partially processed in the proteasome to the optimal length for presentation on major histocompatibility complex class I molecules.8 Polymorphisms in ERAP1 influence the quality and quantity of major histocompatibility complex class I complexes assembled and presented on the cell surface.9, 10
Considering the imputed and genotyped data across ERAP1, association with AS is most strongly localised to a block of single nucleotide polymorphisms (SNPs) lying in a 4.6 kb region between rs27529 (in exon 9) and rs469758 (in intron 12).2, 11 In this region, the only common (minor allele frequency >1%) coding polymorphisms are rs30187 (Arg528Lys) and rs10050860 (Asp575Asn),12 and fine-mapping and haplotype evolutionary studies suggest that rs30187 is directly disease associated, and that a second haplotype, tagged by rs10050860, is also AS associated.2 We previously reported an interaction between ERAP1 and the major HLA class I alleles associated with AS, with association of ERAP1 polymorphisms with AS being restricted to HLA-B27-positive cases.2 In ERAP1, the Arg528Lys substitution encoded by the protective SNP rs30187 causes a significant reduction in aminopeptidase activity towards a synthetic peptide substrate when compared with the wild type, resulting in a two- to threefold decrease in substrate affinity.2 This is consistent with the structural data demonstrating that Lys528Arg disease-protective variants prevent the closed conformation of the protein catalytic site,2 thereby leading to loss of ERAP1 function, though the exact mechanism of how this might contribute to AS pathogenesis remains unclear.
Despite the association between HLA-B27 and AS being known for almost 40 years, how HLA-B27 participates in AS pathogenesis is still unresolved. Leading theories include the arthritogenic peptide theory,13 and two theories involving non-canonical properties of HLA-B27, the unfolded protein response (UPR) and HLA-B27 homodimer formation.5 Compared with other HLA molecules, HLA-B27 is slow to fold and associate with β2-microglobulin.14 The UPR hypothesis postulates that the slow rate of HLA-B27 folding triggers an intracellular signalling cascade in the ER, which, in macrophages, stimulates production and secretion of interleukin 23 (IL23). Evidence in support of the UPR hypothesis is strongest in the HLA-B27-transgenic rat model, 15 but has also been reported in the synovium of AS patients.16
ER stress, buffered by the activation of the UPR, is a homeostatic signalling network that orchestrates the restoration of ER function.17 UPR activation leads to upregulation of expression of pro-inflammatory cytokines, such as TNF, IL17 and IL23, which are also known to be pathogenic in AS. Studies in the HLA-B27-transgenic rat model have implicated the association of HLA-B27 misfolding and ER-stress-dependent activation of UPR in spondyloarthritis. Bone marrow-derived macrophages sampled from colon tissue from rats with inflammatory arthritis were demonstrated to exhibit ER stress in the form of upregulated, UPR-inducing levels of the transcription factor gene X-box-binding protein 1 (XBP-1). Furthermore, the production of IL23 was enhanced in colonic tissue.15 Unlike in human AS though, where patients can have at most two copies of HLA-B27, in the HLA-B27-transgenic rat model, multiple copies of the transgene are required for arthritis to develop.18
To directly assess the role of ERAP1 in AS-attributed ER stress, we examined the gene expression of ER-stress markers 78 kDa glucose-regulated protein (GRP78) and C/EBP-homologous protein (CHOP) using quantitative polymerase chain reaction (qPCR), and correlated their levels with pro-inflammatory cytokine expression levels in AS patients.
Results and discussion
The AS-risk associated rs30187 allele of ERAP1 does not lead to altered levels of ER stress
Accumulation of unfolded proteins in the ER initiates stress responses. The ER-resident chaperone GRP78 (GRP78/BiP) is sequestered which then allows the activation of three main effector molecules: activating transcription factor 6, serine/threonine-protein kinase/endoribonuclease IRE1 and PRKR-like ER kinase (PERK).19 One arm of this pathway involves the activation of CHOP by PRKR-like ER kinase, ultimately resulting in the upregulation of pro-inflammatory cytokines such as IL23.9 To assess whether AS-associated ERAP1 variants alter levels of ER stress, we compared the relative expression of ER-stress genes GRP78 and CHOP in peripheral blood mononuclear cells (PBMCs) from patients carrying either the risk or protective allele of rs30187. Gene expression was normalised to the expression of the constitutive ribosomal gene RPL32. There were no differences in the relative expression levels (ΔCT) of GRP78 or CHOP between AS HLA-B27+rs30187risk (GRP78: ΔCT=0.009±0.001; CHOP: ΔCT=0.024±0.006; mean±s.e.m.), HLA-B27+rs30187protective (GRP78: ΔCT=0.007±0.006; CHOP: ΔCT=0.024±0.002) and HLA-B27−rs30187protective individuals (GRP78: ΔCT=0.009±0.001; CHOP: ΔCT=0.022±0.008; Figures 1a and b). Excluding patients on TNF inhibitors and only studying those that were anti-TNF naive did not alter the findings (data not shown).

GRP78 and CHOP gene expression is not altered by HLA-B27 status or ERAP1 genotype. qRT–PCR was used to determine the level of GRP78 (a and c) and CHOP (b and d) gene expression from active AS patients (n=49). AS patients carrying the rs30187-risk allele of ERAP1 and those with the protective allele of ERAP1 rs30187 showed no significant difference in GRP78 (a) or CHOP (b) gene expression. ERAP1 genotype also did not alter ER-stress response to IFN-γ stimulation as measured by either GRP78 (c) or CHOP (d) gene expression. Furthermore, there was no difference in either GRP78 (e) or CHOP (f) gene expression between HLA-B27+ and HLA-B27- patients irrespective of ERAP1 genotype (P>0.5 in all comparisons). Similar levels of ER-stress gene expression were seen in patient and control samples (g and h).
Interferon gamma (IFN-γ) exposure is an important contributor to ER stress in HLA-B27-expressing PBMC.20 To determine whether ERAP1 variants alter HLA-B27 misfolding and ER stress, we cultured PBMC from HLA-B27+ AS cases in the presence or absence of IFN-γ using conditions described previously.15 After 24 h co-culture with IFN-γ all samples showed elevated expression of GRP78 but not CHOP (Figure 1e). However, no differences were seen in the ER-stress signature between HLA-B27+ cases carrying either rs30187risk or rs30187protective alleles (Figures 1c and d).
ER stress is not different between HLA-B27+ and HLA-B27− AS patients
To assess ER stress in AS patients independent of ERAP1 genotype, we compared GRP78 and CHOP gene expression in HLA-B27+ and HLA-B27− cases without stratification by ERAP1 genotype. GRP78 and CHOP levels are not significantly higher in AS HLA-B27+ patients, with either the rs30187 risk or protective allele of ERAP1, than those who are not carriers of the allele (GRP78: P=0.32; CHOP: P=1.0; Figures 1c and d). Therefore, differences in ER stress are not observed in PBMC samples from AS cases carrying the HLA-B27 allele compared with samples from HLA-B27-negative cases (Figures 1e and f).
To test the possibility that the ER-stress signature observed in AS patients has been saturated by ongoing inflammation, we examined ER-stress gene expression in HLA-B27+ or HLA-B27- healthy controls HLA-B27+rs30187risk (GRP78: ΔCT=0.007±0.001; CHOP: ΔCT=0.023±0.003; mean±s.e.m.), HLA-B27+rs30187protective (GRP78: ΔCT=0.010±0.001; CHOP: ΔCT=0.012±0.003) and HLA-B27−rs30187protective individuals (GRP78: ΔCT=0.012±0.007; CHOP: ΔCT=0.024±0.008). Similar levels of GRP78 and CHOP gene expression were seen in controls (Figures 1g and h) as that was seen in AS patients (Figures 1a and b).
Pro-inflammatory cytokine gene expression is unaffected by ERAP1 variants
Because inflammatory cytokines TNF, IL23 and IL17 are associated with AS pathogenesis, we examined whether carriers of the rs30187-risk allele would display elevated levels of TNF and IL17. Both TNF and IL17 gene expression levels in PBMC were not different between AS patients who were HLA-B27+rs30187risk (TNF: ΔCT=0.002±0.001; IL17A: ΔCT=1.4x10−6±8.3 × 10−7; IL23: ΔCT=0.014±0.004), HLA-B27+rs30187protective (TNF: ΔCT=0.002±0.001; IL17A: ΔCT=7.91x10−7±1.52x10−7; IL23: ΔCT=0.025±0.009) and HLA-B27−rs30187protective (TNF: ΔCT=0.002±0.001; IL17A: ΔCT=2.5x10−6±2.0x10−6; IL23: ΔCT=0.016±0.006; Figure 2).

Inflammatory cytokine gene expression is not altered by ERAP1 genotype. Expression of TNF (a), IL17 (b) and IL23 (c) were determined by qRT–PCR. The relative expression of these genes did not differ between AS patients carrying the ERAP1 rs30187 risk or protective allele in either HLA-B27+ or HLA-B27− individuals (P>0.5 in all comparisons).
Serum levels of pro-inflammatory cytokines were independent of ERAP1 genotype
To confirm that the lack of association of ERAP1 genotype with inflammatory cytokine gene expression corresponded with a similar lack of association at the protein level, we measured serum concentrations of disease-relevant cytokines in HLA-B27+ patients with ERAP1-risk and protective alleles. Serum was analysed only from anti-TNF therapy naive patients, not receiving corticosteroids, at their first presentation at our AS specialist clinic. In total, serum cytokines were measured in 11 HLA-B27+rs30187risk and 20 HLA-B27+rs30187protective patients. ERAP1 genotype did not alter serum expression of either TNF-α (HLA-B27+rs30187risk: 12.46±17.03 pg ml−1, HLA-B27+rs30187protective: 19.34±29.61 pg ml−1) or IL17 (HLA-B27+rs30187risk: 2.45±7.22 pg ml−1, HLA-B27+rs30187protective: 4.23±7.22 pg ml−1; P> 0.05 in all cases; Figure 3).

Serum levels of IL17 and TNF. Serum was collected from 11 HLA-B27+ERAP1Risk and 20 HLA-B27+Protective AS patients not receiving biologic or corticosteroid therapy. IL17 and TNF protein levels were determined in these samples by ELISA. No differences were seen in either IL17 (a) or TNF (b) between patients with risk or protective ERAP1 genotypes (P>0.05 for all comparisons).
Gene expression of markers of ER stress did not correlate with disease severity
Having observed no differences in ER-stress and cytokine gene expression between subjects with risk or protective ERAP1 alleles, we investigated the correlation between measures of ER stress and disease activity in AS patients. For HLA-B27+ individuals with AS, no correlation was observed between GRP78 expression and erythrocyte sedimentation rate (R2=0.05, P=0.14), C-reactive protein (R2=0.0005, P=0.91) or Bath Ankylosing Spondylitis Disease Activity Index scores (R2=0.005, P=0.66; Figures 4a–c). Similarly, no correlation was observed between CHOP expression and erythrocyte sedimentation rate (R2=0.056, P=0.14), C-reactive protein (R2=0.0005, P=0.90) or Bath Ankylosing Spondylitis Disease scores (R2=0.00005, P=0.98). Thus, disease activity appears to operate independently of ER stress. Because ER stress activates the UPR, which is implicated in the production of pro-inflammatory cytokines, correlations between expression of ER-stress markers and that of TNF and IL17 were assessed. GRP78 and CHOP expression showed no linear relationship with expression of either TNF (GRP78: R2=0.043, P=0.19; CHOP: R2=0.084, P=0.06) or IL17 (GRP78: R2=0.016, P=0.44; CHOP: R2=0.004, P=0.88; Figure 4). This indicates that ER stress is unlikely to enhance the production of pro-inflammatory cytokines in HLA-B27+ AS patient PBMC.

No correlation between ER-stress gene expression and inflammation in HLA-B27+ AS patients. Linear regression analyses were performed to compare clinical measures of disease activity with levels of GRP78 (a–c) and CHOP (d–f) gene expression. No relationship was found between ER-stress levels and clinical measures of disease activity.
ER-stress signature in the gut is not altered by ERAP1 genotype
21 HLA-B27+ patients used for analysis of ER stress in the gut were genotyped using TaqMan probes (Applied Biosystems, Mulgrave, VIC, Australia) for a single-coding-region SNP rs30187 in the ERAP1 gene (Supplementary Material); 5 cases were homozygous for the protective ‘G’ allele, 11 cases were heterozygous and 5 cases were homozygous for the risk ‘A’ allele. ER-stress measures were investigated in terminal ileal biopsies from these patients. Gene expression of GRP78 was not altered in the gut by ERAP1 genotype (Figure 5a). Similarly, XBP-1 splicing, as determined by examining the ratio of spliced:unspliced XBP-1, was not affected by ERAP1 genotype in the gut of AS patients (Figure 5b). The protein levels of GRP78 and XBP-1 were also evaluated by immunohistochemistry. No significant differential expression of GRP78 or XBP-1 was observed in AS with risk or protective alleles of ERAP1 (Figures 5c and d).

ER-stress signature is not altered by ERAP1 in the gut of AS patients. Terminal ileal biopsies were taken from AS patients with active disease. ERAP1 genotype (rs30187) was determined using TaqMan genotyping probes. All patients were HLA-B27+. Expression of GRP78 (a) and the ratio of spliced:unspliced XBP-1 (b) were determined by qRT–PCR. Neither the relative expression of GRP78 nor the ratio of spliced:unspliced XBP-1 was altered by carriage of risk or protective alleles of ERAP1 rs30187. (c) Representative immunohistochemical staining for GRP78 and XBP-1 in ileal biopsies from healthy (original magnification x250). The number of GRP78+ (d) and XBP-1+ (e) cells per section was also unaltered by ERAP1 genotype (P>0.05 in all comparisons).
The forces driving the inflammation underlying immune-mediated diseases such as AS remain elusive. Compelling genetic data point to a strong role for antigen presentation in AS but how this altered or aberrant antigen presentation affects disease is still unclear.21 It has been recognised for several years that, relative to other HLA molecules, HLA-B27 is slow to fold and assemble into a functional HLA:peptide complex.14 Studies in the HLA-B27-transgenic rat model of spondyloarthritis have indicated that HLA-B27 misfolding invokes a UPR15 that can drive IL23 production by macrophages.9 The HLA-B27-transgenic rat model also suggests a role for HLA-B27 independent of antigen presentation in this model, since depletion of CD8 T cells had no effect on disease development in this model.22 Polymorphisms in those ERAP1 alleles associated with AS encode protein differences that lead to altered rates of ERAP1 enzyme activity.2 Furthermore, we have demonstrated gene–gene interaction between ERAP1 and HLA-B27 in AS.2 Altered rates of ERAP1 activity may therefore affect the rate at which HLA-B27:peptide complexes fold in the ER which may, in turn, increase the levels of UPR-derived inflammatory cytokines that are secreted by macrophages. To date, this has not been examined in AS patients. In this study, we investigated the effects of the risk allele rs30187 of ERAP1 on ER stress in AS. Our human data show, for the first time, that ER stress is not different in either PBMC or terminal ileal biopsies from AS cases either positive or negative for HLA-B27, or carrying AS risk or protective ERAP1 genotypes. Nor did we observe ERAP1-modified responses to in vitro challenge with UPR-inducing IFN-γ.
In the HLA-B27 transgenic rat model, bone marrow-derived macrophages displaying increased levels of ER stress also secrete elevated levels of IL23, providing a plausible mechanism for induction of IL17-mediated autoinflammatory events in this model.15 However, in AS patients we show there is no difference in the gene expression of ER-stress markers or pro-inflammatory cytokines between individuals carrying the HLA-B27 allele irrespective of rs30187 status. Previous studies examining ER stress and the UPR in HLA-B27+ or HLA-B27− AS patients compared with controls similarly found no difference in ER stress between these groups23, 24 but this study failed to investigate the effects of altered ERAP1 expression on ER-stress signatures.
A lack of support for ER-stress modulation in AS by either HLA-B27 or ERAP1 is suggested in this study. That said, these findings are underpowered for HLA-B27− individuals due to the small number of HLA-B27- samples available to us for this study. In addition, whether PBMC or terminal ileal biopsies isolated from AS patients are the most appropriate target cells for examining ER stress remains uncertain, but these tissues are the only readily accessible disease-relevant tissues in AS patients. Lack of involvement of the UPR in blood samples from AS has been suggested in other studies,23 but our examination of disease-relevant gut biopsies indicates that even in sites of high cellular turnover and likely inflammation in AS patients ER-stress responses are still not upregulated.
Our data show that ERAP1 genotype does not alter expression of ER-stress genes in human PBMCs or terminal ileal biopsies of gut samples. Furthermore, ER-stress gene expression levels are not different between HLA-B27+ and HLA-B27− patients. Together these data suggest that the mechanism of action of HLA-B27 and ERAP1 in AS patients is independent of ER-stress induction. This conclusion is further supported by the lack of correlation seen between clinical measures of disease (erythrocyte sedimentation rate, C-reactive protein and Bath Ankylosing Spondylitis Disease Activity Index) and expression of ER-stress genes. Instead, HLA-B27 may be pathogenic through formation of cell-surface homodimers, which are recognised by auto-reactive T cells, including KIR3DL2+ CD4 T cells.25 Altered ERAP1 function may result in cell-surface presentation of aberrant HLA-B27:self-peptide complexes, or ERAP1-risk alleles in combination with HLA-B27 may result in reduced mucosal immunity and increased bacterial invasion of the gut mucosa, driving IL23 production.26 These theories, along with recent suggestions that autophagy maybe be critical to development of inflammation in AS,27 warrant further investigation.
Subjects and methods
Patient groups
Forty-nine patients diagnosed with AS were included in this study and grouped by their genotype; HLA-B27+rs30187risk (n=14), HLA-B27+ rs30187protective (n=28), HLA-B27−rs30187risk (n=1) and HLA-B27− rs30187protective (n=6). AS was defined according to the modified New York criteria.22 Patient demographics, erythrocyte sedimentation rate, C-reactive protein and Bath Ankylosing Spondylitis Disease Activity Index scores are described in detail in Table 1. Thirteen of the 49 patients have taken TNF inhibitors at the time of blood collection; all other patients had not received TNF inhibitor therapy. No participant received corticosteroids. Peripheral venous blood was collected from AS patients attending the Princess Alexandra Hospital Ankylosing Spondylitis Clinic. Ethical approval was granted by the Princess Alexandra Hospital and University of Queensland Ethics Committees. Written informed consent from all participants was received prior to inclusion in the study.
Terminal ileal biopsies were consecutively sampled from 21 AS patients without clinical symptoms of bowel inflammation at the University of Palermo (Italy) Ankylosing Spondylitis Clinic. Ten out of 21 AS patients have taken non-steroidal anti-inflammatory drugs at the time of ileocolonoscopy, none received DMARDs, corticosteroids or biological agents. As a control group 10 normal subjects (7 male and 3 female patients, age ranging from 35 to 68 years) who have undergone ileocolonscopy for routine evaluation were also evaluated. Paired specimens for histological analysis and qPCR were obtained. All patients and controls provided a signed agreement for this study, and the protocol was approved by the local Ethics Committee of the University of Palermo.
Sample preparation
PBMCs were extracted from heparinised blood using a standard density gradient centrifugation over Ficoll-Paque Plus (GE Healthcare, Uppsala, Sweden). RNA was extracted from isolated PBMC using TRIzol (Life Technologies, Rockville, MA, USA) and cDNA was reverse transcribed (Bioline, Alexandria, NSW, Australia) for qPCR analysis. Serum was collected in serum-separating tubes and centrifuged at 3000 g for 10 min. Ileal biopsies soon after removal were stored in RNAlater solution (Applied Biosystems, Foster City, CA, USA). Each sample was lysed in a tissue homogeniser and RNA was extracted using the commercially available illustra RNAspin mini isolation kit (GE Healthcare, Little Chalfont, UK), according to manufacturer’s instructions. Total RNA was reverse transcribed to cDNA using the high capacity cDNA reverse transcription kit (Applied Biosystems). Samples were stored at –20 °C until use.
Quantitative polymerase chain reaction
Expression of GRP78, CHOP, IL17A and TNF were assayed by SYBR Green (Invitrogen, Carlsbad, CA, USA) qPCR assays using the primers described in Supplementary Material. Gene expression was normalised against expression of 60S ribosomal protein L32 (RPL32). IL23 gene expression was assayed using a TaqMan (Applied Biosystems) pre-designed assay and normalised against expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
IFN-γ stimulation
PBMC from six HLA-B27+rs30187risk and six HLA-B27+rs30187protective individuals were cultured in vitro in the presence or absence of 100 U ml−1 recombinant human IFN-γ (R&D Systems, Minneapolis, MN, USA) for 24 h, as described previously,15 prior to RNA extraction.
Enzyme-linked immunosorbent assay
TNF-α and IL17 enzyme-linked immunosorbent assay (ELISA) were performed on serum samples using ELISA Max Deluxe ELISA kits (BioLegend, San Diego, CA, USA). Assays were performed according to the manufacturer’s instructions using neat serum for IL17 and 1:5 dilution of serum in PBS for TNF-α. ELISA plates were read on FluoStar Optima microplate fluorometer (BMG Labtech, Mornington, VIC, Australia).
TaqMan SNP rs30187 genotyping
SNP rs30187 was genotyped on a ViiA 7 Fast Real-Time PCR System Instrument by using allele-specific TaqMan MGB probes (Applied Biosystems) labelled with fluorescent dyes FAM and VIC (Applied Biosystems, Catalogue no. 4351379), according to the manufacturer's protocols. Allelic discrimination was made with the ViiA 7 Software v1.2.2 (Applied Biosystems). All the cases were run as triplicates to check for genotyping errors. We studied one SNP in the ERAP1 gene, rs30187 (Lys528Arg).
Histology and immunohistochemistry
Specimens from patients with AS were divided into three subgroups as previously described:28 those with normal gut histology, those with acute inflammation and those with chronic inflammation. Briefly, acute inflammation was defined by the presence of neutrophils and/or eosinophils in the crypt and villus epithelium with preservation of normal architecture. Chronic inflammation was defined by alterations of the mucosal architecture, with villous blunting and fusion in the ileal mucosa, an increased mononuclear cell infiltrate and formation of basal lymphoid aggregates in the lamina propria. Immunohistochemistry for XBP-1 and HSPA5 was performed on 5-μm-thick paraffin-embedded sections obtained from intestinal biopsy specimens from patients and controls, and lymph node (used as positive controls) as previously described.27 The primary antibodies, rabbit anti-human GRP78 (1:100 dilution; LSBio, Seattle, WA, USA) and rabbit anti-human XBP-1 (1:100 dilution; Novus Biological, Littleton, CO, USA) were added and incubated for 1 h at room temperature. Ileal and lymph nodal sections incubated with an isotype-matched control antibody were used as negative control. The number of GRP78 and XBP-1-expressing cells was determined by counting the immunoreactive cells on photomicrographs obtained from three random high-power microscopic fields (original magnification x400) under a Leica DM2000 optical microscope using a Leica DFC320 digital camera (Leica Microsystems, North Ryde, NSW, Australia). Results were reported as the median (range).
Statistical analysis
Quantitation data from the qPCR were generated by the Rotor-Gene 1.7.75 software (Qiagen, Doncaster, VIC, Australia). All data displayed in graphs represent the mean±s.e.m. For comparisons between multiple groups differences were compared using the Kruskal–Wallis one-way analysis of variance test with Dunn’s multiple comparison post-hoc test. For comparison of two groups Mann–Whitney U-test was used to determine the statistical significance. Correlation between ER-stress gene expression and clinical measures of disease activity were analysed using linear regression. All statistical analyses were performed in GraphPad Prism 5 software (GraphPad, San Diego, CA, USA). Statistical significance was accepted at a significance level of P<0.05.
References
Brown MA, Kennedy LG, MacGregor AJ, Darke C, Duncan E, Shatford JL et al. Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum 1997; 40: 1823–1828
Evans DM, Spencer CC, Pointon JJ, Su Z, Harvey D, Kochan G et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet 2011; 43: 761–767
Cui X, Hawari F, Alsaaty S, Lawrence M, Combs CA, Geng W et al. Identification of ARTS-1 as a novel TNFR1-binding protein that promotes TNFR1 ectodomain shedding. J Clin Invest 2002; 110: 515–526
Cui X, Rouhani FN, Hawari F, Levine SJ . Shedding of the type II IL-1 decoy receptor requires a multifunctional aminopeptidase, aminopeptidase regulator of TNF receptor type 1 shedding. J Immunol 2003; 171: 6814–6819
Kollnberger S, Bowness P . The role of B27 heavy chain dimer immune receptor interactions in spondyloarthritis. Adv Exp Med Biol 2009; 649: 277–285
Cortes A, Hadler J, Pointon JP, Robinson PC, Karaderi T, Leo P et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet 2013; 45: 730–738.
Haroon N, Tsui FW, Chiu B, Tsui HW, Inman RD . Serum cytokine receptors in ankylosing spondylitis: relationship to inflammatory markers and endoplasmic reticulum aminopeptidase polymorphisms. J Rheumatol 2010; 37: 1907–1910.
Kanaseki T, Blanchard N, Hammer GE, Gonzalez F, Shastri N . ERAAP synergizes with MHC class I molecules to make the final cut in the antigenic peptide precursors in the endoplasmic reticulum. Immunity 2006; 25: 795–806.
Colbert RA, DeLay ML, Klenk EI, Layh-Schmitt G . From HLA-B27 to spondyloarthritis: a journey through the ER. Immunol Rev 2010; 233: 181–202.
Garcia-Medel N, Sanz-Bravo A, Van Nguyen D, Galocha B, Gomez-Molina P, Martin-Esteban A et al. Functional interaction of the ankylosing spondylitis-associated endoplasmic reticulum aminopeptidase 1 polymorphism and HLA-B27 in vivo. Mol Cell Proteomics 2012; 11: 1416–1429.
Reveille JD, Sims AM, Danoy P, Evans DM, Leo P, Pointon JJ et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat Genet 2010; 42: 123–127.
Harvey D, Pointon JJ, Evans DM, Karaderi T, Farrar C, Appleton LH et al. Investigating the genetic association between ERAP1 and ankylosing spondylitis. Hum Mol Genet 2009; 18: 4204–4212.
Benjamin R, Parham P . HLA-B27 and disease: a consequence of inadvertent antigen presentation? Rheum Dis Clin North Am 1992; 18: 11–21.
Mear JP, Schreiber KL, Munz C, Zhu X, Stevanovic S, Rammensee HG et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol 1999; 163: 6665–6670.
Turner MJ, Sowders DP, DeLay ML, Mohapatra R, Bai S, Smith JA et al. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol 2005; 175: 2438–2448.
Dong W, Zhang Y, Yan M, Liu H, Chen Z, Zhu P . Upregulation of 78-kDa glucose-regulated protein in macrophages in peripheral joints of active ankylosing spondylitis. Scand J Rheumatol 2008; 37: 427–434.
Antoniou AN, Lenart I, Guiliano DB . Pathogenicity of misfolded and dimeric HLA-B27 molecules. Int J Rheumatol 2011; 2011: 486856.
Taurog JD, Maika SD, Simmons WA, Breban M, Hammer RE . Susceptibility to inflammatory disease in HLA-B27 transgenic rat lines correlates with the level of B27 expression. J Immunol 1993; 150: 4168–4178.
Oyadomari S, Mori M . Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 2004; 11: 381–389.
Feng Y, Ding J, Fan CM, Zhu P . Interferon-gamma contributes to HLA-B27-associated unfolded protein response in spondyloarthropathies. J Rheumatol 2012; 39: 574–582.
Robinson PC, Brown MA . Genetics of ankylosing spondylitis. Mol Immunol 2014; 57: 2–11.
May E, Dorris ML, Satumtira N, Iqbal I, Rehman MI, Lightfoot E et al. CD8 alpha beta T cells are not essential to the pathogenesis of arthritis or colitis in HLA-B27 transgenic rats. J Immunol 2003; 170: 1099–1105.
Campbell EC, Fettke F, Bhat S, Morley KD, Powis SJ . Expression of MHC class I dimers and ERAP1 in an ankylosing spondylitis patient cohort. Immunology 2011; 133: 379–385.
Neerinckx B, Carter S, Lories RJ . No evidence for a critical role of the unfolded protein response in synovium and blood of patients with ankylosing spondylitis. Ann Rheum Dis 2013; 73: 629–630.
Bowness P, Ridley A, Shaw J, Chan AT, Wong-Baeza I, Fleming M et al. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J Immunol 2011; 186: 2672–2680.
Costello ME, Elewaut D, Kenna TJ, Brown MA . Microbes, the gut and ankylosing spondylitis. Arthritis Res Ther 2013; 15: 214.
Ciccia F, Accardo-Palumbo A, Rizzo A, Guggino G, Raimondo S, Giardina A et al. Evidence that autophagy, but not the unfolded protein response, regulates the expression of IL-23 in the gut of patients with ankylosing spondylitis and subclinical gut inflammation. Ann Rheum Dis 2014; 73: 1566–1574
Ciccia F, Bombardieri M, Principato A, Giardina A, Tripodo C, Porcasi R et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum 2009; 60: 955–965.
Acknowledgements
Written informed consent was obtained from patients and controls for publication of this manuscript and accompanying images. We acknowledge the contribution of the subjects who participated in this study. We thank Murray Hargrave for his assistance in the preparation of this manuscript and Dr Helen Benham for helpful discussions. This study was supported by Australian National Health and Medical Research Council (NHMRC) grant #569830. TJK was supported by an AFA-ARA Heald Fellowship, PCR was supported by NHMRC PhD scholarship and MAB was supported by NHMRC Senior Principal Research Fellowship APP1024879.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Genes and Immunity website
Supplementary information
Rights and permissions
About this article

Cite this article
Kenna, T., Lau, M., Keith, P. et al. Disease-associated polymorphisms in ERAP1 do not alter endoplasmic reticulum stress in patients with ankylosing spondylitis. Genes Immun 16, 35–42 (2015). https://doi.org/10.1038/gene.2014.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gene.2014.62
- Springer Nature Limited
This article is cited by
-
HLA-B27 may modulate the interaction between ERAP1 polymorphisms and smoking in ankylosing spondylitis patients
Molecular Biology Reports (2022)
-
HLA associations in inflammatory arthritis: emerging mechanisms and clinical implications
Nature Reviews Rheumatology (2019)
-
Ankylosing spondylitis: etiology, pathogenesis, and treatments
Bone Research (2019)
-
Stress proteins in the pathogenesis of spondyloarthritis
Rheumatology International (2019)
-
Pathogenesis of ankylosing spondylitis — recent advances and future directions
Nature Reviews Rheumatology (2017)