
Abstract
The objective of this study was to investigate the regulatory effects of TGF-β1 on CCL3/4 expression and inflammation-related pain during intervertebral disc degeneration (IVDD). TGF-β1 and CCL3/4 expression patterns in different degenerative human nucleus pulposus (NP) tissues were measured by qPCR and immunohistochemistry (IHC), and the effects of TGF-β1 on CCL3/4 expression were measured by qPCR, ELISA and immunofluorescence. The roles of NF-κB and MAPK in TGF-β1-mediated CCL3/4 promoter activity were studied using siRNAs, western blotting and qPCR. After establishing an IVDD rat model in vivo, we administered intradiscal injections of TGF-β1. The effects of TGF-β1 on IVDD were determined by MRI and histological analyses, and the effects of TGF-β1 on dorsal root ganglion (DRG) inflammation and pain development were determined by IHC staining and pain-behavior testing, respectively. TGF-β1 and CCL3/4 expression was elevated in degenerative NP tissue. CCL4 expression was significantly inhibited by TGF-β1 treatment. Pharmacological inhibition or siRNA knockdown of the ERK1/2 signaling attenuated TGF-β1-mediated suppression of CCL4 expression. In vivo, TGF-β1 injection inhibited the development of degenerative features in the IVDD model. Moreover, TGF-β1 prevented the inflammatory response and pain development. The results of this study show that TGF-β1 downregulates CCL4 expression through ERK1/2 signaling activation in NP cells. Furthermore, TGF-β1 can prevent degenerative processes, inhibit inflammatory responses in the DRG and prevent pain development in the IVDD rat model. The results of this study indicate that TGF-β1 may represent a therapeutic target for the control of inflammation-related pain associated with IVDD.
Similar content being viewed by others

Introduction
Lower back pain (LBP), a worldwide health problem that perplexes spine surgeons, has attracted considerable attention.1, 2 Approximately 80% of the general population has experienced or will experience LBP during their lives,1 and increasing amounts of evidence show that intervertebral disc degeneration (IVDD) plays an important role in LBP.3 Biological therapies have been proposed as novel strategies for treating IVDD and LBP.4, 5 Therefore, understanding the mechanisms underlying IVDD is helpful for preventing and treating IVDD-related diseases and LBP.6
Several growth factors, such as transforming growth factor-β (TGF-β), insulin-like growth factor-1 (IGF-1), epidermal growth factor (EGF), and fibroblast growth factor (FGF), have the potential to inhibit IVDD.7 A strong immune suppressor, TGF-β1, has been reported to inhibit the inflammatory response in multiple cell types,8 and recent studies have demonstrated that TGF-β1 has protective effects on IVDD. For example, TGF-β1 promotes nucleus pulposus (NP) cell proliferation, stimulates ECM synthesis, and inhibits MMP and ADAMTS expression.9, 10, 11, 12, 13 Moreover, researchers have determined that TGF-β1 has anti-inflammatory effects in NP cells.14 However, the mechanisms underlying the effects of TGF-β1 on IVDD and on pain development are largely unknown.
Chemokines are tiny chemotactic cytokines that play vital roles in the inflammatory response,15, 16 and inflammatory factors, such as TNF-α and IL-1β, can stimulate chemokine expression in the ‘inflammatory soup’.16 In our previous study, we demonstrated that TNF-α- and IL-1β-dependent induction of CCL3 expression in NP cells promotes macrophage migration, which plays a critical role in IVDD.17 More recently, we showed that resistin regulates CCL4 through Toll-like receptor-4, p38-MAPK, and activation of the NF-κB signaling pathway in IVDD.18 Interestingly, recent studies have demonstrated that CCL3 and CCL4 participate in the induction of neuropathic pain.19, 20, 21 However, the influence of TGF-β1 on CCL3/4 expression in IVDD and pain development remains unknown.
Thus, the objective of this study was to investigate the regulatory effects of TGF-β1 on CCL3/4 expression during IVDD and pain development. We showed that CCL4 expression was significantly down-regulated by TGF-β1 in an ERK1/2-dependent fashion and that TGF-β1 prevented IVDD and pain development in an IVDD model in rats.
Materials and methods
All experimental protocols were approved by the Ethics Committee of the First Affiliated Hospital of Sun Yat-sen University, and all experiments were performed in accordance with the approved guidelines for the care and use of laboratory animals of the above institution.
Human-tissue collection and grading
From February 2014 to April 2016, we collected a total of 19 disc samples from 19 patients (male:female, 9:10), whose average age was 40.9 years (range, 15–77 years). Detailed information regarding these samples is provided in Table 1. Each patient provided informed consent for sample collection before participating in the study. Patient disease states were evaluated using the Pfirrmann grading scheme, which entails the use of T2-weighted magnetic resonance imaging (MRI) and independent image analyses performed by three observers. Additional data on the NP tissue specimens used for cell isolation and the donors of the specimens can be made available by the corresponding author upon request.
Isolation and culture of primary NP cells
NP cells were isolated from adult Wistar rats (350 g) using a method reported by Risbud et al.17 NP cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA, USA) and 10% fetal bovine serum (FBS, Invitrogen) supplemented with antibiotics (Invitrogen) at 37 °C in a humidified atmosphere containing 5% CO2.
Real-time quantitative reverse transcription PCR (RT-qPCR)
Total RNA was extracted from NP cells using Trizol reagent (DingGuo, China). This purified, DNA-free RNA was converted to cDNA using a ReverTraAce qPCR RT Kit (TOYOBO, Japan). Reactions were set up in triplicate in 96-well plates using SYBR Green PCR Master Mix (TIANGEN, China). RT-qPCR analyses were performed to detect target mRNA expression using a real-time IQ5 system (Bio-Rad, West Berkeley, CA, USA), and β-actin was used as an internal control. The total volume of the reaction mixture used for each PCR was 20 μl, which included 10 μl of SYBR Green mixture, 0.6 μl of each primer (10 μmol l−l), 2 μl of cDNA template and 6.8 μl of ddH2O. The following PCR conditions were used for the experiments: 95 °C for 15 min, 95 °C for 10 s, 60 °C for 20 s and 72 °C for 30 s for 40 cycles. Relative gene-expression levels were normalized to β-actin expression levels, and the data were presented as fold changes using the formula 2−ΔΔCT, as recommended by the manufacturer. The sequences of the primers used herein are listed in Table 2.
Immunohistochemical (IHC) staining for the levels and distributions of TGF-β1 and CCL3/4 in the NP
iImmunohistochemical (IHC) staining of tissue sections collected from the discs of adolescent patients with idiopathic scoliosis was performed to determine the levels and distributions of TGF-β1 and CCL3/4 in the NP. After retrieval of antigens, quenching of endogenous peroxidase and blocking of nonspecific binding, the sections were incubated with the following antibodies: a mouse polyclonal antibody against TGF-β1 (Novus, Littleton, CO, USA; 1:200 dilution), rabbit polyclonal antibodies against human CCL3 (Abcam, Cambridge, UK, 1:4000 dilution), CCL4 (Abcam, 1:2000 dilution), and an HRP-conjugated secondary antibody. The immunopositive cells in the NP were counted in five high-power fields (× 400) by independent researchers blinded to information pertaining to the study, and the percentage of immunopositive cells was calculated by dividing the number of immunopositive cells by the total number of NP cells and then multiplying the resulting number by 100.
Enzyme-linked immunosorbent assay (ELISA)
NP cells were trypsinized, dispersed into single-cell suspensions and seeded in six-well plates. Each well contained 5 × 104 cells. Recombinant human TGF-β1 (R&D, Minneapolis, MN, USA) was the transforming growth factor used in all the groups. The concentration of CCL3/4 in the cell supernatant was detected using an MIP-1α/β ELISA Kit (Elabscience, Wuhan, China), according to the manufacturer’s instructions.
SiRNA and transfection
The rat ERK siRNA used herein was constructed by GenePharma (Suzhou, China). Its sequences were as follows: sense, 5'-CUGGCUAAGUGCCAUCAAATT-3'; and antisense, 5'-UUUGAUGGCACUUAGCCAGTT-3'. Rat NP cells were transferred to a 6-cm dish (5 × 106 cells) one day before transfection. Lipofectamine 2000 (Invitrogen) served as the transfection reagent. Fifteen microliters of siRNA (10 nM) and 20 μl of Lipofectamine were separately diluted in 500 μl of opti-MEM. Five minutes later, they were mixed together and incubated for 20 min. Then, the mixture was transferred to the above dish with 5 ml of opti-MEM. After 6 h, the medium was replaced with fresh complete medium. The cells were collected for RNA extraction 3 days after transfection.
Western blot analysis
NP cells were placed on ice immediately after treatment and washed twice with ice-cold phosphate-buffered saline. The cells were lysed in RIPA buffer, and total protein was extracted and quantified via BCA assay using a BCA Protein Assay Kit (Pierce, Billings, MT, USA). Total cellular proteins (20 μg per well) were resolved via 10% SDS-PAGE and then transferred to PVDF membranes (Millipore, Boston, MA, USA) via electroblotting. The membranes were blocked in 5% non-fat dry milk with TBST and incubated with the following primary antibodies overnight at 4 °C: an anti-ERK1/2 antibody (CST, Danvers, MA, USA; 1:1000), an anti-phospho-ERK1/2 antibody (CST; 1:1000), and an anti-GAPDH antibody (CST). After being washed with TBST, the membranes were incubated with anti-rabbit or anti-rat HRP-conjugated secondary antibodies, which were subsequently detected with ECL Plus Reagent (Millipore). The results of this experiment were quantified using a multi-gauge densitometry system (Fujifilm, Tokyo, Japan).
Immunofluorescence confocal microscopy
NP cells were transferred to flat-bottom 96-well plates (5 × 103 per well). After treatment and incubation, the NP cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton-X 100 in phosphate-buffered saline for 10 min, blocked with phosphate-buffered saline containing 1% BSA, and incubated with rabbit polyclonal antibodies against CCL3 (Abcam, 5 μg ml−l) and CCL4 (Abcam, 5 μg ml−1). Then, the cells were incubated overnight at 4 °C. After being washed, the NP cells were incubated with an Alexa Fluor-488 conjugated anti-rabbit secondary antibody (Invitrogen), diluted 1:1000, for 1 h at room temperature. The cells were then imaged with a laser scanning confocal microscope using a 20 × /0.4 LCPlanFl objective (Olympus, Tokyo, Japan).
IVDD model and surgical technique
The animals used in this study were male Sprague-Dawley rats, weighing 200–250 g (equivalent to 8–9 weeks old). All surgical procedures were similar to the protocol used in the posterior disc-puncture group in our previous study.22 IVDD induction proceeded as follows:
The rat was placed prone, and a midline longitudinal incision was made in the back. The left facet joint between the fourth and fifth lumbar vertebra was removed. The L4 DRG, L5 nerve root, and the L4/5 IVD were visualized. A 21-gauge needle was inserted parallel to the endplates, 3.0 mm into the disc, where it was held for 30 s. A clamp was used as a stopper to control the depth of the needle. The muscles were closed with 3-0 silk suture, and the skin margins were closed with 4-0 nylon suture.
One week post-surgery, 8 μl (1.6 ng) of TGF-β1 was injected into the IVD through an anterior approach using a microliter syringe (10 μl, Gaoge, Shanghai, China). The same procedure was repeated 2 weeks post-surgery.
Lumbar MRI examinations were performed 0, 2, 4, 6 and 8 weeks after surgery.
Histological examination
Histological examinations were performed on samples from the two groups at the indicated time points. The rats were killed, and the L4/5 IVDs were excised along with the adjacent vertebrae. After being fixed with 10% neutral buffered formalin, the specimens were decalcified in 10% ethylene-diamino-tetraacetic acid solution and embedded in paraffin wax. Then, the samples were sectioned longitudinally in the sagittal plane at 5-mm intervals and stained with hematoxylin and eosin (H&E) so that the morphological changes in the discs could be assessed.
IHC staining for TNF-α and IL-1β in the DRG
IHC staining was performed to determine TNF-α and IL-1β expression levels in the DRG in the two groups. Immunohistochemical examinations were performed 0, 2 and 4 weeks after surgery. The protocol of dissecting L5 DRG was similar to that used in our previous study.22 Then, the sections were incubated with a rabbit anti-rat polyclonal TNF-a antibody (Cell Signaling, Danvers, MA, USA; 1:200 dilution) and rabbit polyclonal IL-1β antibody (Cell Signaling; 1:200 dilution), followed by biotinylated goat antirabbit IgG antiserum (Vector Laboratories, Burlingame, CA, USA; 1:200 dilution). The sections were subsequently rinsed and mounted on gelatin-coated slides. Images of the stained sections were captured with a fluorescence microscope attached to a CCD spot camera (Leica, Germany) and processed with Leica IM50 software (Germany).
Behavioral study
Researchers blinded to the surgical protocol conducted the behavioral tests. Reflex reactions to both mechanical and thermal harmful stimuli applied to both hind paws were measured in eight rats from each group within the week of surgery and during each of the 6 weeks following.
Mechanical hyperalgesia and thermal hyperalgesia
The detailed testing protocol used herein was similar to that used in our previous study.22
Mechanical hyperalgesia
Sensitivity to non-noxious mechanical stimuli was tested by determining hind-paw withdrawal response to von Frey hair (Stoelting, Wood Dale, IL, USA) stimulation of the plantar surface. The rats were placed in a clear plastic cage with a wire mesh bottom, allowing full access to the hind paw. Both the left and right hind paws were assessed, and the average of the results from the left and right sides was calculated.22
Thermal hyperalgesia
To examine thermal withdrawal latency, noxious thermal stimulation was applied to the paw using a radiant heat source (Plantar Test, Ugo Basile Biological Research Apparatus, Comerio, Italy). Each rat was placed unrestrained under a clear glass floor and allowed to habituate before testing. Radiant heat was focused on the plantar surface of a hind paw when it was in contact with the glass. The time until the rat withdrew its paw was recorded to the nearest 0.1 s. The mean withdrawal latency was determined from the last four measurements. The average of the results from the left and right sides was calculated for further analysis.
Statistical analysis
Statistical analyses were performed using SPSS 18 software (SPSS, Inc., Chicago, IL, USA). One-way variance (ANOVA) and Student’s t-test were used to analyze the difference between groups. Typically, the results are presented as the mean±s.e.m. A P<0.05 was considered statistically significant.
Results
TGF-β1, CCL3 and CCL4 expression in different degenerative human IVD tissues
Representative MRI images of different degenerative discs are shown in Figure 1a (from left, Grades I, II, III, IV and V). We examined CCL3/4 and TGF-β1 expression in different degenerative human IVD tissues using real-time RT-PCR analysis. We found that CCL3/4 and TGF-β1 mRNA expression levels were significantly higher in NP tissue than in AF tissue (Figures 1b–d). CCL3/4 and TGF-β1 mRNA expression levels (Grade IV (n=4), V (n=5)) were 2.96-fold (P=0.001) (Figure 1e), 5.26-fold (P<0.001) (Figure 1f), and 3.43-fold higher (P=0.002) (Figure 1g) in severely degenerative NP tissue than in mildly degenerative NP tissue (Grade I (n=1), II (n=2), III (n=3)). We also evaluated CCL3/4 and TGF-β1 expression in normal NP tissues via immunohistochemical staining analysis. CCL3 (54.60±6.35% vs 22.67±3.76%, P=0.013), CCL4 (40.67±4.49% vs 20.33±3.18%, P=0.021) and TGF-β1 immunoreactivity levels (31.00±4.36% vs 14.67±2.60%, P=0.032) were significantly higher in NP tissue than in AF tissue (Figure 1h).

TGF-β1, CCL3 and CCL4 expression in different degenerative human intervertebral disc (IVD) tissues. (a) Representative MRI images of different degenerative discs (from left, Grades I, II, III, IV, and V). (b–d) CCL3/4 and TGF-β1 mRNA expression levels were significantly higher in NP tissue than in AF tissue. (e–g) CCL3/4 and TGF-β1 mRNA expression levels were significantly higher in severely degenerative NP tissue (Grade IV (n=4), V (n=5)) than in mildly degenerative NP tissue (Grade I (n=1), II (n=2), III (n=3)). (h) CCL3 and CCL4 and TGF-β1 immunoreactivity was significantly higher in NP tissue than in AF tissue (*P<0.05 compared with the control group). The tissue samples in each group were obtained from three different donors. All experiments were repeated three times. TGF, transforming growth factor.
TGF-β1 inhibits CCL3 and CCL4 expression in NP cells
To investigate the effects of TGF-β1 on CCL3 and CCL4 mRNA expression and protein secretion, we treated NP cells with different doses of TGF-β1 at different time points, as indicated previously. Our RT-PCR results showed that TGF-β1 decreased CCL3 mRNA expression to levels 0.87-fold (P=0.018), 0.80-fold (P=0.002), and 0.81-fold (P=0.004) lower than their previous levels at 4, 8 and 24 h after TGF-β1 treatment, respectively, as shown in (Figure 2a). TGF-β1 decreased CCL3 mRNA expression levels at multiple doses, as shown in (Figure 2b). Our RT-PCR results showed that TGF-β1 decreased CCL4 mRNA expression levels to 0.41-fold (P<0.001), 0.31-fold (P<0.001), and 0.21-fold (P<0.001) lower than their previous levels at 4, 8 and 24 h after TGF-β1 treatment, respectively, as shown in (Figure 2c). Moreover, our results showed that the effects of TGF-β1 on CCL4 mRNA expression were time- and dose-dependent (Figure 2d). Our ELISA results showed that CCL3 protein levels in the conditioned medium were also decreased after TGF-β1 stimulation (Figures 2e and f). However, the difference in CCL3 levels between the 0 ng ml−l and 1 ng ml−l TGF-β1-stimulated groups was not statistically significant. Moreover, TGF-β1 significantly inhibited CCL4 protein expression in a time-dependent manner (Figure 2g). The effects of TGF-β1 on CCL4 protein levels peaked in cells treated with 1 ng ml−l TGF-β1 and remained significantly elevated compared to those in cells treated with 0.1 and 10 ng ml−l TGF-β1 (Figure 2h). CCL3 and CCL4 protein levels in TGF-β1-treated (10 ng ml−l for 24 h) NP cells were confirmed using immunofluorescence microscopy (Figures 2i and j).

TGF-β1 inhibits CCL3 and CCL4 expression in NP cells. (a) RT-PCR showed that TGF-β1 decreased CCL3 mRNA levels at 4, 8 and 24 h after TGF-β1 treatment. (b) TGF-β1 decreased CCL3 mRNA levels at various doses. (c) RT-PCR showed that TGF-β1 decreased CCL3 mRNA levels at 4, 8 and 24 h after TGF-β1 treatment. (d) Moreover, the effects of TGF-β1 on CCL4 mRNA expression were time- and dose-dependent. (e, f) The ELISA results showed that CCL3 protein levels were decreased in the conditioned medium after stimulation. (g) The difference between the groups treated with 0 ng ml−1 TGF-β1 and 1 ng ml−1 TGF-β1 was not statistically significant. TGF-β1 significantly inhibited CCL4 protein expression in a time-dependent manner in NP cells. (h) CCL3 and CCL4 protein expression in TGF-β1-treated NP cells was confirmed using immunofluorescence microscopy. (i, j) (*P<0.05 compared with the control group) All experiments were repeated three times. TGF, transforming growth factor.
ERK1/2 signaling-pathway inhibition blocks TGF-β1-mediated CCL4 suppression
To ascertain whether TGF-β1 (10 ng ml−l)-mediated inhibition of CCL3/4 expression requires MAPK and NF-κB signaling, we pretreated NP cells with the indicated NF-kB inhibitor (SM7368, 20 μM), ERK1/2 inhibitor (PD98059, 20 μM, 1:1000 dilution), p38 inhibitor (SB203580, 20 μM, 1:1000 dilution) or c-Jun N-terminal kinase (JNK) inhibitor (SP600125, 20 μM, 1:1000 dilution). Pretreatment with the above agents significantly inhibited TGF-β1-mediated suppression of CCL3 mRNA and protein expression (Figures 3a and c).

ERK1/2 signaling pathway inhibition blocks TGF-β1-mediated CCL4 suppression. (a, c) Pretreatment significantly attenuated TGF-β1-mediated suppression of CCL3 mRNA and protein expression. (b, d, e) ERK1/2 inhibition significantly attenuated TGF-β1-mediated suppression of CCL4 mRNA and protein expression (*P<0.05 compared with the control group). All experiments were repeated three times. TGF, transforming growth factor.
Moreover, TGF-β1-mediated inhibition of CCL4 mRNA and protein expression was significantly attenuated by ERK1/2 inhibition (Figures 3b, d and e).
TGF-β1 activates the ERK1/2 signaling pathway in NP cells, and ERK siRNA infection blocks TGF-β1-mediated CCL4 suppression
To confirm that the ERK1/2 signaling pathway is required for TGF-β1-mediated inhibition of CCL4 expression in NP cells, we used western blotting to evaluate ERK1/2 signaling pathway activity after the cells were treated with TGF-β1. We noted a rapid increase in ERK1/2 phosphorylation post-TGF-β1 treatment. We also noted that ERK1/2 activity levels peaked 5–15 min post-treatment and declined thereafter but remained elevated until 6 h post-treatment (Figure 4a). To ascertain whether TGF-β1 activates ERK1/2 signaling, we constructed ERK-specific small interfering RNA (siRNA). Our RT-PCR results showed that ERK1/2 mRNA expression levels decreased significantly after siRNA pretreatment (Figure 4b). Furthermore, TGF-β1-mediated downregulation of CCL4 mRNA and protein expression was significantly inhibited by ERK siRNA (Figures 4c and d).

TGF-β1 activates the ERK1/2 signaling pathways in NP cells, and ERK siRNA infection blocks TGF-β1-mediated CCL4 suppression. (a) Following TGF-β1 treatment, ERK1/2 phosphorylation rapidly increased. ERK1/2 activity peaked 5–15 min post-treatment and declined thereafter but remained elevated until 6 h post-treatment. (b) RT-PCR showed that ERK1/2 mRNA decreased significantly after siRNA pretreatment. (c, d) ERK siRNA significantly inhibited TGF-β1-mediated suppression of CCL4 mRNA and protein expression. (*P<0.05 compared with the control group). All experiments were repeated three times. TGF, transforming growth factor.
TGF-β1 inhibits IVDD and the expression of CCL4 in vivo
To determine the role of TGF-β1 in IVDD in vivo, we established a rat disc-degeneration model, which we had used successfully in our previous study.22 MR imaging and H&E staining data were recorded after disc puncture and TGF-β1 intradiscal injection. MR imaging showed significantly increased disc degeneration (decreased signal intensity) at 4–8 weeks after disc puncture (Figure 5a). However, the intensity of the T2 signal was significantly higher in the group treated with intradiscal injections of TGF-β1 1 and 2 weeks following puncture than in the group treated with puncture only, indicating that TGF-β1 had a positive effect on the IVDs (Figure 5a). The results of our MRI-guided quantitative evaluation, which was performed in accordance with the Pfirrmann grading system, confirmed that TGF-β1 has protective effects on IVDD (P=0.011 and P=0.03 for 6 and 8 weeks, Figure 5b). Regarding the results of our histological assessment, which were demonstrated by HE staining, disc height decreased significantly, the NP shrunk and, in some cases, even disappeared, the number of NP cells decreased, the collagen fibers became disorganized, and the NP was partially or fully replaced by fibrous tissue between 2 and 8 weeks after AF puncture (Figure 5c). In contrast, in the TGF-β1-treated group, the collagen fibers showed slight disorganization, the collagen structure was fuzzy, and the NP displayed proteoglycan and water loss (Figure 5c). Moreover, the TGF-β1-treated group displayed significantly preserved disc heights and NP content compared with the puncture group between 2 and 8 weeks after puncture (Figure 5c).

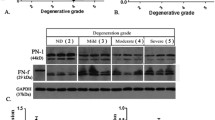
TGF-β1 inhibits intervertebral disc degeneration (IVDD) and the expression of CCL4 in vivo. (a) The intensity of the T2 signal was significantly higher in the group treated with intradiscal injections of TGF-β1 1 and 2 weeks following puncture than in the group treated with puncture only (N=8 for each group). (b) The MRI-guided quantitative evaluation performed according to the Pfirrmann grading system confirmed that TGF-β1 exerts protective effects on IVDD (N=8 for each group). (c) H&E staining showed severe degenerative changes between 2 and 8 weeks after AF puncture. Notably, the TGF-β1 group displayed significantly preserved disc heights and NP content compared with the puncture group between 2 and 8 weeks after puncture. (d, e) IHC staining showed strong CCL4 immunoreactivity in NP cells at 4 and 8 weeks after puncture (N=3 for each group at different time point). However, after TGF-β1 injection, IHC staining showed significantly decreased immunoreactivity in these cells. Notably, the number of NP cells was significantly decreased at 8 weeks after surgery (*P<0.05 compared with the control group). TGF, transforming growth factor.
IHC staining showed strong CCL4 immunoreactivity (67.00±5.20%, 50.67±4.09%) in NP cells at 4 and 8 weeks after puncture (Figures 5d and e). However, after TGF-β1 injection, IHC staining showed significantly decreased CCL4 immunoreactivity in these cells (42.00±4.04%, 34.33±2.03%; P=0.019 and 0.023 for 4 and 8 weeks; Figures 5d and e). Notably, the number of NP cells was significantly decreased at 8 weeks after surgery. These results indicated that the positive effects of TGF-β1 on the IVD may be associated with CCL4 suppression.
TGF-β1 injection attenuates the inflammatory response in the DRG and improves pain behavior in vivo
To determine the effects of TGF-β1 on DRG inflammation and pain development, we performed an IHC staining analysis of the DRG and conducted pain-behavior testing in the animal model. IHC staining revealed strong TNF-α (64.67±5.46%, 41.00±4.93%) and IL-1β immunoreactivity (50.33±4.33%, 30.67±3.76%) in the DRG at 2 and 4 weeks after puncture (Figures 6a–c). In contrast, in the TGF-β1 injection group, IHC staining revealed significantly decreased TNF-α (45.67±4.81%, 27.00±2.89%) and IL-1β immunoreactivity (33.33±4.10%, 19.33±3.28%; P<0.05; Figures 6a–c) in the DRG. Regarding mechanical sensitivity, the puncture group displayed greater mechanical hyperalgesia at 1–6 weeks post-puncture than did the TGF-β1 group (P<0.05; Figure 6d). Interestingly, the TGF-β1 group displayed mechanical hypersensitivity at 5–6 weeks after puncture was normal compared with before puncture (Figure 6d). Evidence of thermal hyperalgesia was noted in both groups at 1 and 2 weeks after puncture compared with before puncture (P<0.05) (Figure 6e). However, no significant difference in thermal hyperalgesia severity was noted between the puncture and TGF-β1 groups at any time point (Figure 6e). These results indicate that TGF-β1 application is associated with reductions in DRG inflammation and improvements in pain behavior in vivo.

TGF-β1 injection attenuates the inflammatory response in the dorsal root ganglion (DRG) and improves pain behavior in vivo. (a–c) Immunohistochemical (IHC) staining showed strong TNF-α and IL-1β immunoreactivity in the DRG at 2 and 4 weeks after puncture (N=3 for each group at different time point). In contrast, in the TGF-β injection group, IHC staining showed significantly decreased TNF-α and IL-1β immunoreactivity. (d) The puncture group displayed greater mechanical hyperalgesia at 1–6 weeks post-puncture than the TGF-β1 group (N=9 for each group). The TGF-β1 group displayed normal mechanical hypersensitivity at 5–6 weeks after puncture compared with before puncture. (e) No significant difference in thermal hyperalgesia was noted between the puncture and TGF-β1 groups at any time points (N=9 for each group; *P<0.05 compared with the control group, #P<0.05 compared with preoperative data). (f) A proposed model of the relationship between TGF-β1 and CCL4 in intervertebral disc degeneration (IVDD) and the development of inflammation-related pain. TGF, transforming growth factor.
Discussion
In this study, we showed that TGF-β1 and CCL3/4 expression levels are positively correlated with IVD degeneration grades. Moreover, we also demonstrated the regulatory mechanism underlying the effects of TGF-β1 on CCL3 and CCL4 expression in NP cells for the first time. Specifically, we demonstrated that TGF-β1 inhibits CCL3 and CCL4 expression, especially CCL4 expression, through ERK signaling-pathway activation in NP cells. TGF-β1 regulates CCL4 expression in a time-dependent manner but does not regulate CCL3 expression in a specific manner. We found that intradiscal injections of TGF-β1 could inhibit disc degeneration and CCL4 expression in NP cells, as well as prevent inflammation in the DRG in vivo. Moreover, we found that the pain behavior of the rats in the TGF-β1 group was significantly improved compared with that of the rats in the control group.
Previous studies have shown that TGF-β1 is involved in IVDD. It is well documented that TGF-β1 is overexpressed in degenerative discs.23, 24, 25 Furthermore, TGF-β1 can suppress IL-1β and TNF-α expression and thus inhibit inflammatory responses in degenerative discs.26 IVDD is also an inflammatory disease, and NP cells produce high levels of inflammatory cytokines and chemokines, which accelerate the progression of IVDD.27 However, little is known about the role of TGF-β1 in regulating chemokine expression in NP cells. According to our results, TGF-β1 and CCL3/4 expression levels were positively correlated with IVDD grades, indicating that TGF-β1 and CCL3/4 may play roles in in IVDD.
Chemokines play a vital role in the inflammatory response associated with IVDD.28, 29 In our previous study, we demonstrated that TNF-α- and IL-1β-dependent induction of CCL3 expression in NP cells promotes macrophage migration, which plays a critical role in IVDD.17 More recently, we showed that resistin regulates CCL4 through Toll-like receptor-4, p38-MAPK and NF-κB signaling pathway activation in IVDD.18 Additionally, it has also been reported that TGF-β1 can inhibit IL-1β- and TNF-α-induced inflammatory responses in degenerative discs.14, 26 However, the regulatory mechanisms underlying the effects of TGF-β1 on chemokines, including CCL3/4, in IVDD are unknown. Thus, in this study, we demonstrated for first the time that TGF-β1 downregulates CCL3 and CCL4 expression in NP cells.
The MAPKs are a class of serine/threonine kinases that respond to extracellular stimuli, such as growth factors, neurotransmitters, hormones, cell stress and cell adhesion, in mammalian cells. The families involved in the MAPK signaling pathway in mammalian cells include the p38 MAPK, ERK and JNK families.30 It has been reported that MAPK and NF-κB play important roles in the inflammatory response associated with disc degeneration.31 Our previous study showed that the MAPK and NF-κB pathways regulate CCL3 promoter activity in NP cells,17 findings consistent with the results of this study. We also demonstrated that the regulatory mechanism underlying the effects of resistin on CCL4 in NP cells involves the p38-MAPK and NF-κB signaling pathways.18 However, in this research, we found that the ERK pathway is a main regulator of the effects of TGF-β1 on CCL4 in NP cells. The ERK1/2 signaling pathway has been shown to play a crucial role in IVDD. However, the effects of ERK1/2 on inflammatory and catabolic responses during IVDD are complicated and remain unclear. The ERK1/2 signaling pathway activation tightly regulates MMP and ADAMTS expression, thereby causing proteoglycan depletion.32, 33 However, Yang H et al.34 demonstrated that ERK1/2 inhibitors abolished the antagonistic effect of TGF-β1 on TNF-α-mediated catabolic responses in IVD, indicating a role of ERK1/2 pathway in suppressing the inflammation-induced catabolic responses. Furthermore, a study by Xiao YQ suggested that TGF-β inhibits inflammatory cytokine production through cross-talk between MAPKs, specifically ERK-dependent inhibition of p38 MAPK caused by up-regulation of MAPK phosphatase-1.35 Moreover, cross-talk between the ERK1/2 and Smad signaling pathways may also enhance TGF-β-dependent anti-inflammatory and anabolic responses.36, 37, 38, 39 Similarly, our silencing studies using ERK siRNA transfection showed that TGF-β1-dependent CCL4 expression was increased, a finding that highlighted the importance of the ERK1/2 signaling pathway in the regulation of CCL4 expression by TGF-β1. The precise regulatory mechanism of TGF-β1 on ERK-dependent CCL4 suppression, which probably involves ERK cross-talk, is still unclear and should be investigated in future research.
It has been reported that TGF-β1 has the potential to slow the progress of IVDD in animal models.36 In our previous study, we demonstrated that TGF-β1 exerts anti-catabolic effects by inhibiting Syndecan-4 expression and promoting MMP3 expression in NP cells.34, 40 To further elucidate the role of TGF-β1 in IVDD, we performed an in vivo study. Disc-puncture animal models have been used extensively in IVDD research.41 We successfully established an IVDD model in rats in our previous study.22 As in that study, we observed degenerative changes between 2 and 8 weeks after surgery, including decreased signal intensities on MR imaging and reduced NP sizes and disc heights on H&E staining. Interestingly, after TGF-β1 injection, we observed significantly increased MR signal intensities, as well as significantly increased disc heights and NP content. CCL4 expression levels in NP cells decreased significantly after TGF-β1 injection. These findings indicate that TGF-β1 has protective effects on IVDD, effects that may be associated with CCL4 suppression.
Inflammatory cytokines are highly expressed within some degenerative discs, and inflammation-like reactions may sensitize entrapped nerves, resulting in pain-signal transmission after minimal mechanical stimulation in animal models.42 In our previous study, we demonstrated that both inflammatory cytokine expression and posterior AF rupture were associated with pain-behavior changes in the IVDD rat model.22 In the degenerative-disc model, inflammatory factors were highly expressed in the DRG, a phenomenon to which pain was attributed.22, 43 In this study, the posterior disc-puncture model was established as previously described. Interestingly, after TGF-β1 intradiscal injection, pain behavior and TNF-α and IL-1β immunoreactivity in the DRG were attenuated, suggesting that TGF-β1 can suppress inflammatory cytokine expression in the DRG and alleviate inflammation-related pain.
Some researchers recently reported that LBP is caused by a combination of inflammatory pain and neuropathic pain associated with a long-lasting inflammatory state.44 Saika et al.45 suggested that CCL4 is a novel key mediator and that the peripheral CCL4-CCR5 axis participates in neuropathic pain. It also has been reported that peripheral nerve injury elicits CCL3 and CCR1 up-regulation, leading to pain.19 On the basis of our results, we propose that TGF-β1 alleviates pain in part by suppressing CCL3 and CCL4 expression.
There were several limitations to this study. First, we did not examine the relationship between CCL3/4 and pain development or the regulatory mechanism underlying this relationship. Second, we did not evaluate the TGF-β1-CCL3/4 axis receptor, nor did we elucidate the exact mechanism by which the receptor participates in IVDD or pain development. Further research is needed to elucidate the details of the mechanisms underlying these phenomena.
On the basis of our findings, we propose that TGF-β1 down-regulates CCL4 expression in NP cells through activation of the ERK signaling pathway. Furthermore, we suggest that TGF-β1 can prevent degenerative processes, inhibit inflammatory responses in the DRG and prevent pain development in the IVDD rat model. The results of this study may indicate that TGF-β1 is a therapeutic target for the control of inflammation-related pain associated with IVDD.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Maher C, Underwood M, Buchbinder R . Non-specific low back pain. Lancet 2016; 16: 30970–30979.
Balagué F, Mannion AF, Pellisé F, Cedraschi C . Non-specific low back pain. Lancet 2012; 379: 482–491.
Luoma K, Riihimaki H, Luukkonen R, Raininko R, Viikari-Juntura E, Lamminen A . Low back pain in relation to lumbar disc degeneration. Spine 2000; 25: 487–492.
Freemont AJ . The cellular pathobiology of the degenerate intervertebral disc and discogenic back pain. Rheumatology 2009; 48: 5–10.
Maidhof R, Alipui DO, Rafiuddin A, LevineM, Grande DA, Chahine NO . Emerging trends in biological therapy for intervertebral disc degeneration. Discov Med 2012; 14: 401–411.
Moriguchi Y, Alimi M, Khair T, Berlin C, Bonassar LJ, Härtl R et al. Biological treatment approaches for degenerative disk disease: a literature review of in vivo animal and clinical data. Global Spine J 2016; 6: 497–518.
Walsh AJ, Bradford DS, Lotz JC . In vivo growth factor treatment of degenerated intervertebral discs. Spine 2004; 29: 156–163.
Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA 1993; 90: 770–774.
Masuda K . An HS. Growth factors and the intervertebral disc. Spine J 2004; 4: 330S–340S.
Nishida K, Kang JD, Gilbertson LG, Moon SH, Suh JK, Vogt MT et al. Modulation of the biologic activity of the rabbit intervertebral disc by gene therapy: an in vivo study of adenovirus-mediated transfer of the human transforming growth factor beta 1 encoding gene. Spine 1999; 24: 2419–2425.
Wang SL, Yu YL, Tang CL, Lv FZ . Effects of TGF-β1 and IL-1β on expression of ADAMTS enzymes and TIMP-3 in human intervertebral disc degeneration. Exp Ther Med 2013; 6: 1522–1526.
Tsuji T, Chiba K, Imabayashi H, Fujita Y, Hosogane N, Okada Y et al. Age-related changes in expression of tissue inhibitor of metalloproteinases-3 associated with transition from the notochordal nucleus pulposus to the fibrocartilaginous nucleus pulposus in rabbit intervertebral disc. Spine 2007; 32: 849–856.
Pattison ST, Melrose J, Ghosh P, Taylor TK . Regulation of gelatinase-A (MMP-2) production by ovine intervertebral disc nucleus pulposus cells grown in alginate bead culture by transforming growth factor-beta(1) and insulin like growth factor-I. Cell Biol Int 2001; 25: 679–689.
Yang H, Cao C, Wu C, Yuan C, Gu Q, Shi Q et al. TGF-βl suppresses inflammation in cell therapy for intervertebral disc degeneration. Sci Rep 2015; 5: 13254.
Haringman JJ, Ludikhuize J, Tak PP . Chemokines in joint disease: the key to inflammation? Ann Rheum Dis 2004; 63: 1186–1194.
Gerard C, Rollins BJ . Chemokines and disease. Nat Immunol 2001; 2: 108–115.
Wang J, Tian Y, Phillips KL, Chiverton N . Haddock G, Bunning RA, et al. Tumor necrosis factor α- and interleukin-1β-dependent induction of CCL3 expression by nucleus pulposus cells promotes macrophage migration through CCR1. Arthritis Rheum 2013; 65: 832–842.
Li Z, Wang X, Pan H, Zheng Z, Liu H, Wang J et al. Resistin promotes CCL4 expression through toll-like receptor-4 and activation of the p38-MAPK and NF-κB signaling pathways:implications for intervertebral disc degeneration. Osteoarthritis Cartilage 2016; 16: 30310–30317.
Kiguchi N, Kobayashi Y, Maeda T, Saika F, Kishioka S . CC-chemokine MIP-1alpha in the spinal cord contributes to nerve injury-induced neuropathic pain. Neurosci Lett 2010; 484: 17–21.
Kiguchi N, Maeda T, Kobayashi Y, Fukazawa Y, Kishioka S . Macrophage inflammatory protein-1alpha mediates the development of neuropathic pain following peripheral nerve injury through interleukin-1beta up-regulation. Pain 2010; 149: 305–315.
Saika F, Kiguchi N, Kobayashi Y, Fukazawa Y, Kishioka S . CC-chemokine ligand4/macrophage inflammatory protein-1beta participates in the induction of neuropathic pain after peripheral nerve injury. Eur J Pain 2012; 16: 1271–1280.
Li Z, Liu H, Yang H, Wang J, Wang H, Zhang K et al. Both expression of cytokines and posterior annulus fibrosus rupture are essential for pain behavior changes induced by degenerative intervertebral disc: An experimental study in rats. J Orthop Res 2014; 32: 262–272.
Tolonen J, Grönblad M, Vanharanta H, Virri J, Guyer RD, Rytömaa T et al. Growth factor expression in degenerated intervertebral disc tissue. An immunohistochemical analysis of transforming growth factor beta, fibroblast growth factor and platelet-derived growth factor. Eur Spine J 2006; 15: 588–596.
Specchia N, Pagnotta A, Toesca A, Greco F . Cytokines and growth factors in the protruded intervertebral disc of the lumbar spine. Eur Spine J 2002; 11: 145–151.
Tolonen J, Grönblad M, Virri J, Seitsalo S, Rytömaa T, Karaharju E . Transforming growth factor beta receptor induction in herniated intervertebral disc tissue: an immunohistochemical study. Eur Spine J 2001; 10: 172–176.
Li W, Liu T, Wu L, Chen C, Jia Z, Bai X et al. Blocking the function of inflammatory cytokines and mediators by using IL-10 and TGF-β: a potential biological immunotherapy for intervertebral disc degeneration in a beagle model. Int J Mol Sci 2014; 15: 17270–17283.
Wang J, Markova D, Anderson DG, Zheng Z, Shapiro IM, Risbud MV . TNF-alpha and IL-1 beta promote a disintegrin-like and metalloprotease with thrombospondin type I motif-5-mediated aggrecan degradation through syndecan-4 in intervertebral disc. J Biol Chem 2011; 46: 39738–39749.
Kawaguchi S, Yamashita T, Katahira G, Yokozawa H, Torigoe T, Sato N . Chemokine profile of herniated intervertebral discs infiltrated with monocytes and macrophages. Spine 2002; 27: 1511–1516.
Phillips KL, Cullen K, Breakwell LM, Bunning RA, Cross AK, Le Maitre CL et al. Potential roles of cytokines and chemokines in human intervertebral disc degeneration: interleukin-1 is a master regulator of catabolic processes. Osteoarthritis Cartilage 2015; 23: 1165–1177.
Cuadrado A, Nebreda A . Mechanisms and functions of p38 MAPK signaling. Biochem J 2010; 429: 403–417.
Tian Y, Yuan W, Fujita N, Wang J, Wang H, Shapiro IM et al. Inflammatory cytokines associated with degenerative disc disease control aggrecanase-1 (ADAMTS-4) expression in nucleus pulposus cells through MAPK and NF-κB. Am J Pathol 2013; 182: 2310–2321.
Wuertz K, Vo N, Kletsas D, Boos N . Inflammatory and catabolic signaling in intervertebral discs: the roles of NF-κB and MAPkinases. Eur Cell Mater 2012; 23: 103–119.
Wei Y, Zhi-Hong W, Gui-Xing Q, Bin Y, Jun C, Yi-Peng W . Extracellular signal-regulated kinase inhibition modulates rat annulus fibrosus cell response to interleukin-1. Spine 2013; 38: E1075–E1081.
Yang H, Gao F, Li X, Wang J, Liu H, Zheng Z . TGF-β1 antagonizes TNF-α induced up-regulation of matrix metalloproteinase 3 in nucleus pulposus cells: role of the ERK1/2 pathway. Connect Tissue Res 2015; 56: 461–468.
Xiao YQ, Malcolm K, Worthen GS . Gardai S, Schiemann WP, Fadok VA, et al. Cross-talk between ERK and p38 MAPK mediates selective suppression of pro-inflammatory cytokines by transforming growth factor-beta. J Biol Chem 2002; 277: 14884–14893.
Yang H, Yuan C, Wu C, Qian J, Shi Q, Li X et al. The role of TGF-β1/Smad2/3 pathway in platelet-rich plasma in retarding intervertebral disc degeneration. J Cell Mol Med 2016; 20: 1542–1549.
Hayashida T, Decaestecker M, Schnaper HW . Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-beta-dependent responses in human mesangial cells. FASEB J 2003; 17: 1576–1578.
Shiou SR, Yu Y, Guo Y, Westerhoff M, Lu L, Petrof EO et al. Oral administration of transforming growth factor-β1 (TGF-β1) protects the immature gut from injury via Smad protein-dependent suppression of epithelial nuclear factor κB(NF-κB) signaling and proinflammatory cytokine production. J Biol Chem 2013; 288: 34757–34766.
Gu AD, Wang Y, Lin L, Zhang SS, Wan YY . Requirements of transcription factor Smad-dependent and -independent TGF-β signaling to control discrete T-cell functions. Proc Natl Acad Sci USA 2012; 109: 905–910.
Yang H, Liu H, Li X, Pan H, Li Z, Wang J et al. TNF-α and TGF-β1 regulate Syndecan-4 expression in nucleus pulposus cells: role of the mitogen-activated protein kinase and NF-kB pathways. Connect Tissue Res 2015; 56: 281–287.
Singh K, Masuda K, An HS . Animal models for human disc degeneration. Spine J 2005; 5: 267S–279S.
Olmarker K . Puncture of a lumbar intervertebral disc induces changes in spontaneous pain behavior: an experimental study in rats. Spine 2008; 33: 850–855.
Murata Y, Onda A, Rydevik B, Takahashi K, Olmarker K . Distribution and appearance of tumor necrosis factor-alpha in the dorsal root ganglion exposed to experimental disc herniation in rats. Spine 2004; 29: 2235–2241.
Miyagi M, Ishikawa T, Kamoda H, Suzuki M, Murakami K, Shibayama M et al. ISSLS prize winner: disc dynamic compression in rats produces long-lasting increases in inflammatory mediators in discs and induces long-lasting nerve injury and regeneration of the afferent fibers innervating discs: a pathomechanism for chronic discogenic low back pain. Spine 2012; 37: 1810–1818.
Saika F, Kiguchi N, Kobayashi Y, Fukazawa Y, Kishioka S . CC-chemokine ligand 4/macrophage inflammatory protein-1β participates in the induction of neuropathic pain after peripheral nerve injury. Eur J Pain 2012; 16: 1271–1280.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no: 81572175) and the Outstanding Young Investigator Support Program of The First Affiliated Hospital, Sun Yat-sen University, awarded to Doctor Jianru Wang.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Zhang, J., Li, Z., Chen, F. et al. TGF-β1 suppresses CCL3/4 expression through the ERK signaling pathway and inhibits intervertebral disc degeneration and inflammation-related pain in a rat model. Exp Mol Med 49, e379 (2017). https://doi.org/10.1038/emm.2017.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.136
- Springer Nature Limited
This article is cited by
-
Involvement of HDAC2-mediated kcnq2/kcnq3 genes transcription repression activated by EREG/EGFR-ERK-Runx1 signaling in bone cancer pain
Cell Communication and Signaling (2024)
-
Serglycin secreted by late-stage nucleus pulposus cells is a biomarker of intervertebral disc degeneration
Nature Communications (2024)
-
Codelivery of TGF-β1 and anti-miR-141 by PLGA microspheres inhibits progression of intervertebral disc degeneration
Journal of Orthopaedic Surgery and Research (2023)
-
Single-cell RNA sequencing in orthopedic research
Bone Research (2023)
-
Recombinant human endostatin combined with radiotherapy promotes cardiomyocyte apoptosis in rats via TGFβ1/Smads/CTGF signaling pathway
BMC Cardiovascular Disorders (2022)