
Abstract
Lipid accumulation, which may be caused by the disturbance in very low density lipoprotein (VLDL) secretion in the liver, can lead to fatty liver disease. VLDL is synthesized in endoplasmic reticulum (ER) and transported to Golgi apparatus for secretion into plasma. However, the underlying molecular mechanism for VLDL transport is still poorly understood. Here we show that hepatocyte-specific deletion of meningioma-expressed antigen 6 (Mea6)/cutaneous T cell lymphoma-associated antigen 5C (cTAGE5C) leads to severe fatty liver and hypolipemia in mice. Quantitative lipidomic and proteomic analyses indicate that Mea6/cTAGE5 deletion impairs the secretion of different types of lipids and proteins, including VLDL, from the liver. Moreover, we demonstrate that Mea6/cTAGE5 interacts with components of the ER coat protein complex II (COPII) which, when depleted, also cause lipid accumulation in hepatocytes. Our findings not only reveal several novel factors that regulate lipid transport, but also provide evidence that Mea6 plays a critical role in lipid transportation through the coordinated regulation of the COPII machinery.
Similar content being viewed by others


Introduction
Hepatic steatosis, especially nonalcoholic fatty liver disease (NAFLD), is one of the most prevalent forms of chronic liver diseases worldwide1. The prevalence rates of hepatic steatosis and NAFLD are 21.4% and 19.0%, respectively in the US2. Fat accumulation in the liver due to defects in lipoprotein assembly or transport can lead to hepatic steatosis or NAFLD3,4,5.
A major type of lipoprotein, the very low density lipoprotein (VLDL), is synthesized and assembled in hepatocytes and secreted into the plasma to be delivered to other organs through the circulation5,6. VLDL assembly and secretion from the liver plays an important role in controlling plasma levels of triglycerides (TGs) and cholesterol, and many proteins and lipids are known to be involved in this process7. VLDL synthesis is initiated in the endoplasmic reticulum (ER) of hepatocytes following the synthesis of the apolipoprotein (Apo) B100 and recruitment of TGs, cholesterol esters, and other apolipoproteins such as ApoE. ApoB mutations affect plasma lipid levels and are associated with familial hypobetalipoproteinemia disease8. Although more and more genes have been found to be involved in lipoprotein assembly and transport, and their disruption is associated with hepatic steatosis, the underlying mechanisms are still not very clear9,10,11,12,13.
Vesicular trafficking factors such as coat protein complex II (COPII), which is composed of small GTPase (Sar1) and COPII coat proteins Sec23, 24, 13 and 31, play a crucial role in transporting ER cargoes to the Golgi apparatus14,15,16. Sar1 binds to the ER membrane in its GTP-bound state and recruits the Sec23/24 heterodimer that forms the inner coat, which in turn recruits the outer coat proteins Sec13 and 31 to form the complex14,15. Sar1B mutations are associated with chylomicron retention disease and hepatic steatosis, likely resulting from retention of lipoproteins in the ER17,18,19. Thus, VLDL secretion likely involves Sar1. Whether other components of COPII play a role in lipid or VLDL transport remains to be explored.
Most COPII vesicles are about 60-90 nm in diameter, which are too small to accommodate large cargoes such as procollagen, chylomicrons, or VLDL that are in the range of 100-400 nm20,21. Specific proteins are likely to control the loading of these cargoes and accordingly regulate the size of COPII vesicles. Expression of meningioma-expressed antigen 6 (Mea6), also known as cutaneous T cell lymphoma-associated antigen 5C (cTAGE5C), is upregulated in different tumor tissues or cell lines, and Mea6 is considered as an oncogene22,23,24. Mea6 has been shown to interact with transport and Golgi organization 1 (TANGO1) to facilitate collagen export from the cell25,26,27,28. Deletion of TANGO1 in mice results in defects in collagen secretion similar to the phenotype associated with defect in COPII function29,30. However, the physiological function of Mea6 is still not clear.
Here, we show that Mea6 expression can be induced substantially in mouse liver by high-fat diet (HFD) and hepatocyte-specific Mea6 knockout mice exhibited severe fatty liver and hypolipemia. Phenotypic and biochemical analyses revealed that Mea6 is involved in lipid transport through interaction with COPII components and regulation of COPII assembly.
Results
HFD induces Mea6 expression and hepatocyte-specific deletion of Mea6 leads to severe fatty liver
To elucidate the physiological function of Mea6, Mea6 conditional knockout (cKO) mice were generated through homologous recombination by genomic insertion of two loxP sites flanking exon 11 and an Frt-Neo-Frt cassette in intron 11 into embryonic stem cells (Figure 1A). Accurate targeting was confirmed by PCR using 5′ and 3′ genomic primers and by enzyme digestion (Figure 1A and Supplementary information, Figure S1A). Crossing mice carrying a floxed Mea6 allele with transgenic Cre mouse lines resulted in the excision of exon 11 of Mea6, causing a reading frame shift in exon 12 (Supplementary information, Figure S1B). Mea6Δ/+ mice were obtained through crossing with zona pellucida 3 (ZP3)-Cre mice31, which led to the deletion of Mea6 in the fertilized egg. Mea6Δ/Δ mice were subsequently generated by intercrossing Mea6Δ/+ mice. All Mea6Δ/Δ mice died between gestational days 8.5 and 9.5 (data not shown), indicating that Mea6 is essential for embryonic development.

Mea6 deficiency leads to severe fatty liver. (A) Gene targeting strategy for establishing Mea6 cKO mice. Following homologous recombination, ES cells had one targeted allele of Mea6 (Mea6fl/+), in which exon 11 was flanked by two loxP sites and an Frt-Neo-Frt cassette was inserted in intron 11; in the presence of the Cre recombinase, exon 11 was excised, generating the Mea6Δ/+ mouse. Exons are indicated by numbers inside boxes. A KpnI restriction site was introduced by cloning; primers F1-F3 and R1-R4 were used for genotyping (see also Supplementary information, Figure S1). (B) HFD induces Mea6 expression. One-month-old mice were fed with HFD for 2 months. Liver protein levels were analyzed by western blotting (n = 4 mice in each group). (C, D) Western blotting (C) and real-time PCR (D) analyses of Mea6 expression in the liver of the indicated groups. In D, Ctrl, Mea6fl/+ mice; cKO, Mea6fl/Δ; Alb-Cre mice. Data represent means ± SEM (n = 4). (E) Mea6 expression was analyzed by immunohistochemistry in P14 livers. (F) Representative images of livers from P14 mice. (G) An enrichment of lipid droplets in cKO livers at P1, P7 and P35 was observed by the Oil Red O staining.
Given the embryonic lethality of Mea6Δ/Δ mice, various organs were examined for Mea6 expression to determine a target organ for conditional knockout. Results from quantitative PCR and western blotting revealed that Mea6 mRNA and protein levels were highest in the liver and pancreas (Supplementary information, Figure S1C-S1D).
Mea6 protein has been shown to be induced by 14-fold upon LDL treatment in THP-1 cells32. We therefore fed mice with HFD and found that Mea6 protein levels were induced substantially in the liver (Figure 1B). This implicates that Mea6 may play a role in lipid transportation or metabolism. Mea6 was therefore specifically deleted in hepatocytes by crossing Mea6Δ/+;albumin (Alb)-Cre and Mea6fl/fl mice, which generated four different lines, Mea6fl/Δ;Alb-Cre, Mea6fl/+;Alb-Cre, Mea6fl/+ and Mea6fl/Δ. Mea6 protein expression was lost in Mea6fl/Δ;Alb-Cre mice, which were referred to as cKO mice hereafter (Figure 1C). Mea6fl/+ mouse line was used as control (Ctrl). Real-time PCR and immunohistochemistry further confirmed that Mea6 expression was lost in the liver of Mea6 cKO mice (Figure 1D-1E).
Mea6 cKO mice were grossly normal, although they were smaller than their littermates 1 month after birth (Supplementary information, Figure S2A). Livers from 2-week-old cKO mice were enlarged and pale in color (Figure 1F), despite the liver structure is largely normal (Supplementary information, Figure S2B). However, Oil Red O staining revealed many lipid droplets, a typical indicator of fatty liver, in most liver cells as early as postnatal day 1 (P1) and through adulthood (Figure 1G). In addition, livers of 30% 1-year-old cKO mice developed nodular morphology with lipid storage disease-like structures (Supplementary information, Figure S2C-S2D). These results indicate that Mea6 deficiency in hepatocytes causes fatty liver in mice.
Mea6 deficiency causes hepatic lipid accumulation and hypolipemia
Given that loss of Mea6 in hepatocytes resulted in fatty liver, total cholesterol (TC) and TG levels in the liver were assessed in these mice. Significantly higher TC and TG levels were detected in P35 cKO mice fed chow normally (Fed) or fasted for 16 h (Fasted) (Figure 2A and 2B), while accumulated liver TG level but not TC level was found in fasted P90 cKO mice (Figure 2C). The Oil Red O staining analysis indicates lipid droplet accumulation in cKO mouse liver (Figure 1G). To confirm the cell type specificity of lipid droplet accumulation, primary cultured hepatocytes were stained with the lipid marker Bodipy493. Both the size and number of lipid droplets were increased in hepatocytes derived from cKO mice compared with those from wild-type (WT) mice (Figure 2D).

TC/TG accumulation in livers in cKO mice under both normal and fasting conditions. (A, B) Levels of TC (A) and TG (B) in the liver in P35 mice fed with chow diet (Fed) or fasted for 16 h (Fasted). (C) Increased liver TG level in P90 cKO mice after 16 h fasting. Data shown are means ± SEM (student's t-test). *P < 0.05, **P < 0.01. (D) Primary hepatocytes from Mea6fl/+ (Control) and Mea6 cKO mice were stained with Bodipy493 and labeled with an antibody against Mea6.
We next analyzed the total plasma TC and TG levels. Both plasma TC and TG levels were markedly reduced in P35 and P90 cKO mice either fed normally or under fasting condition compared to control mice (Figures 3A, 3B and Supplementary information, Figure S3A). However, the P35 and P90 cKO mice had largely normal Alb and glucose levels under fasting conditions (Supplementary information, Figure S3B, S3C and data not shown). Similar results were obtained from both young and old animals, suggesting that fatty liver accompanied by hypolipemia in cKO mice is a chronic ailment (Figures 2C and Supplementary information, Figures S2C-S2D, S3A).

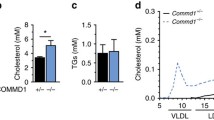
Mea6 knockout leads to reduced VLDL secretion and hypolipaemia. (A, B) Levels of TC (A) and TG (B) in the plasma of P35 mice fed chow normally (Fed) or fasted for 16 h (Fasted). (C) Relative increase of plasma TG levels in both P90 control and cKO mice treated with Triton WR-1339 (Tyloxapol) for 3 h. (D) ApoB-100, ApoB-48 and ApoE levels in the plasma in P90 mice treated with Triton WR-1339 for 3 h were analyzed by western blot (n = 3 mice in each group). Membrane stained with ponceau was used as loading control. (E) 300 μl plasma from 3 mice (either controls or Mea6 cKO mice) fasted for 10 h were combined for FPLC analysis. TG and TC levels in VLDL, LDL and HDL fractions were quantified using corresponding detection kits. ApoB levels in fractions 2-10 (VLDL fractions) were analyzed by western blot. (F) Primary hepatocytes were pulse-labeled with 3H-glycerol for 4 h and then washed three times. The culture medium was collected 0, 2 and 4 h later for detection of secreted 3H-lipid. (G) Primary hepatocytes from WT and Mea6 cKO mice (n = 3) were incubated with [35S] methionine for 1 h, and ApoB-100/-48 were immunoprecipitated from the medium, separated by SDS-PAGE and exposed to phosphorimager. Levels of ApoB-100 were quantified in the right panel. Data are means ± SEM (student's t-test). *P < 0.05, **P < 0.01.
The above results suggest that VLDL secretion in the liver is defective in Mea6 cKO mice. To exclude the possibility that the reduction in plasma TC and TG levels was caused by enhanced degradation of VLDL, 3-month-old mice were fasted for 13 h, and then injected intravenously with Triton WR-1339 (Tyloxapol), which blocks VLDL degradation and clearance by suppressing the activity of lipoprotein, endothelial, and hepatic lipases33; under this condition, plasma TG is mainly derived from VLDL secreted by hepatocytes. Three hours after Triton WR-1339 injection, plasma TG levels were increased significantly in both Ctrl and cKO mice, however, the increase was markedly smaller in cKO animals (Figure 3C). Similarly, the plasma levels of ApoB-48 and ApoE, which may come from other organs such as the guts, were induced significantly in both Ctrl and cKO mice by the treatment (Figure 3D). To our surprise, ApoB-100 which plays an essential role in VLDL assembly, was hardly detectable in both Triton-treated and -untreated cKO mice. We next separated the plasma into VLDL, LDL and HDL fractions by fast protein liquid chromatography (FPLC). As shown in Figure 3E, the levels of TG, ApoB-100 and ApoB-48 in the VLDL fractions from cKO mice were dramatically lower than those from WT littermates. Similarly, the levels of TC in VLDL, LDL and HDL fractions were also substantially lower in Mea6 cKO mice (Figure 3E, bottom). These results suggest that hepatic lipid accumulation and hypolipemia in cKO mice were due to a defect in VLDL secretion.
The reduction in VLDL-TG and ApoB-100 secretion was further determined by pulse-labeling in cultured primary hepatocytes. 3H-glycerol was used to pulse-label newly synthesized glycerolipids. The rate of 3H-glycerolipid secretion was significantly reduced in hepatocytes from cKO mice (Figure 3F), although the level of 3H-glycerolipid synthesis was similar in the two groups 4 h after the pulse-labeling (Supplementary information, Figure S3D). Primary hepatocytes from WT and Mea6 cKO mice were incubated with [35S] methionine for 1 h, and ApoB-100/-48 levels in the medium were determined 2 and 4 h later. As shown in Figure 3G, both secreted ApoB-100 and ApoB-48 levels were significant reduced in cKO hepatocytes. In order to test whether protein secretion is affected in general, we performed general protein export assay and found that Mea6 knockout did not significantly affect the total amount of proteins secreted by primary hepatocytes (Supplementary information, Figure S3E). Taken together, these results indicate that Mea6 is required for the secretion of lipids and selective proteins.
Lipid accumulation in the cKO liver might be due to an increase in lipid biosynthesis. In order to exclude this possibility, the expression of genes involved in lipogenesis (acetyl-CoA carboxylase, fatty acid synthase (FAS), elongation of very long chain fatty acids 6, and sterol regulatory element-binding protein 1c), fatty acid oxidation (carnitine palmitoyltransferase 1A and 2), and VLDL assembly and delivery (ApoB and ApoE), and genes encoding lipid droplet-associated proteins (adipose differentiation-related protein, cell death-inducing DFFA-like effector b, and fat-specific protein 27), was examined by real-time PCR at P1 and P35. There were no significant differences in the expression of these genes between WT and cKO mice at P1 (Supplementary information, Figure S4A), but at P35, FAS expression was markedly reduced in cKO mice (Supplementary information, Figure S4B). These results suggest that increased lipid synthesis is not the cause of the lipid accumulation observed in Mea6-deficient mice, in agreement with the in vitro pulse-labeling results from primary hepatocytes (Supplementary information, Figure S3D).
To more closely examine the function of Mea6 in lipid metabolism, a quantitative lipidomic analysis was performed with P5 livers. The levels of neutral lipids, including cholesteryl esters (CE), TGs and diacylglycerides (DAG), were elevated substantially in the liver of cKO mice (Figure 4A); levels of sphingolipids, such as ceramides (Cer) and the ganglioside GM3, were also higher, whereas that of sphingomyelins (SM) was lower in cKO mice (Figure 4B). However, there was no significant difference in liver phospholipid content between WT and cKO mice (Figure 4C). The opposite lipid profile was observed in the plasma, with levels of CE, free cholesterols, TG and DAG (Figure 4D), as well as levels of SM, Cer and glucosylceramides (Figure 4E) markedly reduced in cKO mice relative to controls. Interestingly, plasma levels of phospholipids, including lysophosphatidylcholine, phosphatidylcholine, phosphatidylethanolamine and phosphatidylinositol, were also decreased in cKO mice (Figure 4F).

Lipidomic analysis in WT and Mea6 cKO mice. Levels of various lipids were measured in the liver (A-C) and plasma (D-F) of P5 WT (Ctrl) and cKO mice. CE, cholesteryl esters; Chol, free cholesterols; TG, triacylglycerides; DAG, diacylglycerides; SM, sphingomyelins; Cer, ceramides; GluCer, glucosylceramides; GM3, GM3 ganglioside; LBPA, lysobisphosphatidic acids; LPC, lysophosphatidylcholines; PC, phosphatidylcholines; PA, phosphatidic acid; PE, phosphatidylethanolamines; PG, phosphatidylglycerols; PI, phosphatidylinositols; PS, phosphatidylserines. Data represent means ± SEM (student's t-test). *P < 0.05, **P < 0.01.
Mea6 interacts with different subunits of COPII complex
To investigate the mechanistic basis of hypolipaemia and fatty liver development in Mea6 cKO mice, quantitative mass spectrometry (iTRAQ) analysis was performed on liver tissues from P5 animals. The levels of several classes of proteins were altered in cKO mice, including, notably, those involved in lipid transport, vesicle trafficking and ER stress (Table 1). Specifically, protein level of ApoE, which is secreted by hepatocytes and plays an important role in lipid transport, was confirmed to be elevated dramatically in the liver, but decreased in the plasma of cKO mice at P35 (Figure 5A), and similar results were also obtained at P1 (Figure 5B). More importantly, ApoB-100, which is essential for VLDL assembly, was also accumulated in the liver, but significantly reduced in the plasma of cKO mice at P1 (Figure 5B), and was hardly detectable in the plasma of cKO mice at P90 (Figure 3D). Interestingly, we also confirmed the iTRAQ results (Table 1) that Sec31A level was reduced in cKO livers (Figure 5D). In addition, we analyzed the expression of other COPII components and found that the level of Sec23A was also decreased in cKO livers (Figure 5C). Mea6 has been implicated to interact with Sec23A/24C in yeast two-hybrid system25. We therefore speculated that Mea6 may regulate lipid transport through interaction with COPII components. To test this possibility, we adopted BioID method to analyze protein interaction in vivo34. BirA*-Sar1B was used as a bait and proteins in close proximity to Sar1B, such as COPII components, will be biotinylated by BirA and precipitated by streptavidin. As shown in Figure 6A, Mea6 was detected in the complex in control but not in cKO hepatocytes. As expected, Sar1B, Sec23A and Sec31A, but not actin, were also detected in the complex, indicating that Mea6 exists in COPII complex under physiological conditions (Figure 6A).

Abnormal levels of apolipoproteins and COPII subunits in the Mea6 cKO mice. (A) Altered levels of ApoE in cKO livers and plasma at P35. Actin and albumin (Alb) served as loading controls (n = 4 mice in each group). (B) Protein levels of ApoB-100/-48 and ApoE, as well as Mea6, were assessed in liver and plasma of WT (Ctrl) and cKO mice at P1. GAPDH and Alb served as loading controls (n = 2 mice in each group). (C) Reduced levels of Sec23A in the livers of cKO mice at P35 (n = 2 mice in each group). (D) Reduced levels of Sec31A but not Sar1B in the livers of cKO mice at P1 and P35. ERGIC53 and GAPDH served as the loading control (n = 3 mice in each group).

Mea6 interacts with COPII components. (A) Primary hepatocytes infected with GFP-BirA-Flag-Sar1B adenovirus for 24 h were supplemented with 20 μM biotin for 24 h. Cells were lysed and immunoprecipitated with Dynabeads. Beads were collected and the bound proteins were analyzed by western blot. (B-D) The interaction of Mea6 with Sar1, Sec23A and Sec24D was assessed by co-IP. Constructs expressing Mea6 and various COPII subunits were transfected alone or in combination into HEK 293T cells; 36 h later, cell lysates were precipitated with Flag agarose beads and the immune complex was probed with Flag or Myc antibody. (E) The interaction between endogenous Sec23A and Mea6 in the mouse liver was assessed by immunoprecipitating with anti-Mea6 and probed with anti-Sec23A antibodies. (F) Schematic representation of Mea6 constructs encoding the full-length (WT) and mutant proteins with truncations in the coiled-coil domain (C), proline-rich domain (PRD, N) or TM domain (ΔN). (G, H) Sec23A or Sec24D construct was transfected into HEK 293T cells individually or in conjunction with WT or mutant Mea6 constructs, and cell lysates were precipitated with Flag agarose beads and probed for the indicated proteins.
To further explore the relationship between Mea6 and COPII complex, constructs encoding Mea6 and various COPII subunits were transfected into HEK 293T cells individually or in combination, and co-immunoprecipitation (co-IP) experiments revealed that Mea6 interacts with Sar1, Sec23A and Sec24D (Figure 6B-6D), but apparently not with Sec13, Sec24C or Sec31A (Figures 7A and Supplementary information, S5A-S5B). An interaction between endogenous Sec23A and Mea6 was also detected (Figure 6E). For a more detailed characterization, Sec23A and Sec24D constructs were transfected individually or in conjunction with WT or deletion mutants of Mea6 (Figure 6F). Only the full-length Mea6 interacted with Sec23A and Sec24D, although a very weak interaction between Sec24D and the Mea6 transmembrane (TM) deletion mutant (Mea6ΔN) was detected (Figure 6G-6H). These results indicate that both the TM and proline-rich domains (PRD) are required for the interaction between Mea6 and Sec23A/24D.

Coordinated interactions between Mea6 and COPII subunits. (A) Mea6 does not interact with Sec13. Mea6, Sec23A and Sec13 constructs were transfected into HEK 293T cells individually or in combination, and cell lysates were precipitated with Flag agarose beads and probed for the indicated proteins. (B) Schematic representation of interactions between Mea6 and COPII subunits. (C-E) Interactions among Mea6 and COP II subunits. Constructs expressing Mea6 and various COPII subunits were transfected into HEK 293T cells individually or in combination as indicated and cell lysates were precipitated with Flag agarose beads and probed for the indicated proteins. (F) Mea6 knockdown leads to reduced interaction between Sec23 and Sec31, which can be rescued by expression of mouse Mea6. siMea6 or siControl was transfected into Bel7402 cells 24 h before the transfection of Sec23A, Sec31A and mouse Mea6 individually or in combination as indicated. Another 24 h later, cell lysates were precipitated with Flag agarose beads and probed for the indicated proteins.
Mea6 and COPII subunits coordinate with each other to form a complex
The outer coat subunit of COPII, Sec31A, interacts with the inner coat subunits, Sec23A and Sec24D35. We have shown above that Mea6 interacts with Sec23A and Sec24D, which interact with each other35 (Supplementary information, Figure S5C), but it did not interact with Sec13 or Sec31A (Figure 7A, Supplementary information, Figure S5B). We therefore hypothesized that Mea6 might interact with Sec13 and Sec31A via Sec23A or Sec24D (model in Figure 7B). To test this hypothesis, we transfected constructs of Mea6, Sec23A, Sec24D, Sec13 and Sec31A into HEK 293T cells individually or in combination, and performed co-IP. Mea6 was able to interact with Sec31A, but not Sec13, when co-expressed with either Sec23A or Sec24D (Figure 7A, 7C-7E, and Supplementary information, Figure S5B). More importantly, Mea6 enhanced the interaction between Sec23A and Sec31A (Figure 7D). These results suggest that Mea6, Sec23A, Sec24D and Sec31A coordinate with each other in the formation of a protein complex.
We show above that co-expression of Mea6 enhances the interaction between Sec23A and Sec31A (Figure 7D). We went on to test whether Mea6 is required for such interaction. We knocked down the expression of Mea6 in Bel7402 cells and inspected the interaction between Sec23A and Sec31A in cells with or without expression of mouse Mea6. As shown in Figure 7F, the interaction between Sec23A and Sec31A was weaker in Mea6-knockdown cells and it could be rescued by overexpression of mouse Mea6. This indicates that Mea6 plays an important role in the interaction between COPII components.
The ultrastructure is disturbed in the Mea6 cKO hepatocytes
Since dilatation of ER structure has been found in fibroblasts from cranio-lenticulo-sutural dysplasia (CLSD) patients with F382L mutation of Sec23A30, we investigated whether the ER structure of hepatocytes would also be affected by Mea6 deletion. Liver specimens were examined by transmission electron microscopy (TEM). The bile canaliculus between adjacent hepatocytes was normal in the liver of control (Figure 8A) and cKO mice, although many larger lipid droplet-like structures were present in the cytosol of cKO hepatocyte (Figure 8B). WT livers had well-stacked ER (Figure 8C), while in the cKO, the ER was distended, and more intriguingly, particles with density similar to lipid droplets were observed in the lumen (Figure 8D). Moreover, small particles were also found within membrane-bound secretory vesicles of the Golgi apparatus in WT mice (Figure 8E), but this was not the case in cKOs (Figure 8F).

The abnormal ultrastructure in the Mea6-KO hepatocytes. Transmission electron micrographs of WT (A, C, E) and cKO (B, D, F) hepatocytes at P14. Compared with WT hepatocytes (A), the Mea6-KO hepatocyte has roughly normal structure with tight junctions adjacent to the bile canaliculus (BC), but lipid droplet (LD) accumulation is observed (B). (C, D) The ER is arranged in stacks of flat cisternae throughout the cytoplasm in WT hepatocytes (C), while in Mea6-KO hepatocytes, the ER is distended with concomitant retention of LD-like structures in the cytoplasm and ER lumen (D, arrows). (E, F) Many VLDL-size particles (arrows) are present in Golgi (G) vesicles in WT (E), but only a few in Mea6-KO cells (F). Instead, many LDs (arrows) are detected in the cytosol (F). Nu, nucleus; M, Mitochondrion.
Knockdown of COPII subunits or Mea6 leads to accumulation of lipid droplets
Given that various COPII subunits interact with Mea6 and coordinate with each other to form a complex, we postulated that they are also involved in lipid transport. To test this hypothesis, we screened shRNA constructs from an shRNA library in Bel7402 cells and found shRNAs that were able to efficiently suppress the expression of Sec23A and Sec24D, but not that of Mea6 and Sec31A (Supplementary information, Figure S6A and data not shown). A significant accumulation of lipid droplets was detected in cells depleted of Sec23A or Sec24D with different sets of shRNAs (Figure 9A and Supplementary information, Figure S6B). For Mea6 and Sec31A, we adopted siRNAs to knockdown their expression (Supplementary information, Figure S6C). Consistent with data from Mea6 cKO animals, Mea6 knockdown also resulted in lipid droplet accumulation in Bel7402 cells, and very similar results were obtained with knockdown of Sec31 (Figure 9B-9C). Interestingly, when the mouse Mea6 cDNA was overexpressed in Bel7402 cells transfected with siMea6, the number of lipid droplets returned back to the control level (Figure 9D and Supplementary information, Figure S6D). Taken together, our findings demonstrate that Mea6, Sec23A, Sec24D and Sec31A are required for lipid secretion.

Requirement of Mea6 and COPII components for lipid transportation in hepatocytes. (A) Numbers of lipid droplets (LDs) in control, Sec23A- or Sec24D-knockdown cells. Bel7402 cells were cotransfected with mCherry expressing vector and shRNAs targeting Sec23A or Sec24D. These cells were then stained with Bodipy493 and DAPI to visualize LDs and nuclei 48 h after transfection. LDs in mCherry-positive cells were counted (representative images are shown in Supplementary information, Figure S6B). Data represent means ± SEM (student's t-test). **P < 0.01. More than 100 transfected cells were counted in each set. (B, C) Bel7402 cells were transfected with siRNAs targeting Mea6 or Sec31A; 48 h later, cells were stained with Bodipy493 and DAPI to visualize LDs and nuclei, respectively. LDs were counted (B). Data represent means ± SEM (student's t-test). **P < 0.01. More than 100 transfected cells were counted in each set. Representative images are shown in C. Scale bar, 5 μm. (D) Bel7402 cells were transfected with siCtrl or siMea6; 24 h later, cells were transfected with pCAGIG-GFP or pCAGIG-Mea6 (mouse). Another 24 h later, cells were stained with LipidTOX™ Red neutral lipid stain to visualize LDs. Among Mea6-knockdown cells, those transfected with Mea6 (white arrows) had decreased LD number compared with those without Mea6 expression (yellow arrows). Scale bar, 5 μm. Right panel: average number of LDs in the positive cells of each treatment. Data represent means ± SEM (student's t-test). **P < 0.01, *P < 0.05. More than 100 cells were counted in each set. (E) Model of lipid transport through cooperation of Mea6 with TANGO1 and COPII subunits. Mea6 at the ER membrane binds to TANGO1 through one of the coiled-coil domains and the COPII subunits Sar1 and Sec23A/24D through the proline-rich domain to facilitate the transport of VLDL from the ER to Golgi vesicles. Mea6 also interacts indirectly with Sec31 via Sec23A/24D. In the absence of Mea6, VLDL cannot be transported to the Golgi apparatus efficiently due to the failed formation of large COPII vesicle, leading to the accumulation of lipid droplets in the cytosol.
Discussion
Mea6 is involved in collagen secretion in vitro and has potential oncogenic function25. The present study demonstrates that Mea6, along with the COPII subunits Sec23A, Sec24D and Sec31A, has an essential role in lipid transport. Mea6 deletion in hepatocytes resulted in obvious fatty liver in mice, characterized by an accumulation of various types of lipids and proteins, including ApoB and ApoE, in the liver. This was accompanied by an evident hypolipemia, which could explain the leaner body mass of the mutant mice. Based on these observations and biochemical studies, a model for the biological function of Mea6 is proposed (Figure 9E). Mea6 is localized at the ER membrane25, where it binds TANGO1 and the COPII subunits Sar1 and Sec23A/24D, through its PRD. Mea6 also interacts indirectly with Sec31A via Sec23A/24D to coordinate the formation of a lattice complex required for the transport of VLDL from the ER to Golgi vesicles (Figures 7B and 9E). In the absence of Mea6, VLDL cannot be efficiently transported to the Golgi apparatus, resulting in the accumulation of VLDL in the ER and lipid droplets in the cytosol, leading to fatty liver and hypolipemia.
It has been reported that VLDL-size lipid particles are transported from the ER to the Golgi apparatus36. Similar particles, but larger in size, were observed in the ER lumen in the Mea6 cKO hepatocytes and in Golgi transport vesicles in WT hepatocytes by TEM (Figure 8). These results, together with evidence from pulse-labeling experiments, indicate that lipid transport from the ER to the Golgi apparatus is disrupted in the absence of Mea6. The resultant accumulation of lipids in the liver is unlikely due to increased lipid synthesis, since the expression of 11 genes involved in lipid metabolism was unchanged or even reduced in the cKO mice (Supplementary information, Figure S4). Multiple proteins and factors have been shown to regulate the size and number of LDs1,37,38. The accumulation of various sizes of LDs in Mea6-deficient hepatocytes is likely to be a secondary effect due to defect in lipid transportation.
ApoE and ApoB are lipid-associated proteins that are required for VLDL assembly39,40,41,42,43. The elevated levels of both proteins in Mea6 cKO livers (Figures 3 and 5) correlated with lipid accumulation, suggesting that VLDL is assembled normally. In contrast, levels of Sec23A and Sec31A were reduced in mutant livers. It is possible that Mea6 deficiency affects the formation of COPII complex, leading to decreased Sec23A and Sec31A levels and consequent lipid accumulation. Another possibility is that Mea6 deficiency-induced lipid accumulation affects the ER structure and quantity, and therefore the levels of Sec23A and Sec31A. Unlike lipids, the secretion of Alb, the major protein secreted by hepatocytes, was unaffected (Supplementary information, Figure S3B), indicating that the transport of proteins mediated by Mea6 is cargo-selective. In addition to ApoE, ApoB and VLDL, several other classes of proteins in the liver (e.g., blood coagulation-related proteins) may also be transported by this mechanism because they are deregulated in cKO livers (Table 1). It would be interesting to examine in the future the different biological functions of Mea6, such as its roles in regulating blood coagulation and ER stress.
TANGO1 and Mea6 have structural similarities including a TM domain, a proline-rich domain (PRD) and two coiled-coil domains, through one of which the two proteins interact25,44. TANGO1 and Mea6 are ER resident chaperone proteins essential for collagen secretion25, while the COPII components Sar1, Sec23/24 and Sec13/31 are required for protein transport from the ER to the Golgi apparatus15. Consistent with previous reports of the interaction of TANGO1 or Mea6 with Sec23A/24C25, in the present study, Mea6 was found to bind Sar1, Sec23A/24D and indirectly with Sec31A but not Sec13 (Figures 6 and 7). The interaction between Mea6 and Sec23A/24D was abolished by deletion of the PRD. In addition, the TM domain was also required to correctly target Mea6 to the ER membrane where it could interact with vesicular transport proteins (data not shown). More interestingly, co-expression of Mea6 promoted the interaction between Sec31A and Sec23A, and expression of Sec23A/24D enabled the interaction of Mea6 with Sec31A (Figure 7). These results suggest that Mea6, Sec23A, Sec24D and Sec31A likely coordinate with each other in the formation of a large protein complex to facilitate the transport of large cargoes, such as VLDL, from the ER to Golgi, and Mea6 plays an important role in the organization of COPII coat.
The depletion of Mea6, Sec23A, Sec24D or Sec31A in human hepatoma Bel7402 cells resulted in lipid accumulation (Figure 9). This result is consistent with the lipid transport model that we propose, which involves both Mea6 and the COPII complex (Figure 9E). Of note, the potential effects of COPII component deficiency on lipolysis could not be completely ruled out, and the exact role of COPII components in VLDL secretion needs to be examined using animal models. Nevertheless, Mea6 cKO and Sar1B mutations cause hepatic steatosis, likely resulting from retention of lipoprotein in the ER17,18,19. This provides more evidence that Mea6 regulates lipid transport through interaction with COPII subunits. Deletion of Tango1 in mice results in defects in collagen secretion and skeletal development29. We have found that knockdown of TANGO1 led to lipid accumulation in Bel7402 cells (data not shown). It would be intriguing to investigate in the future whether liver-specific knockout of Tango1 would result in fatty liver, similar to what was observed in Mea6 cKO mice.
Mea6 has been implicated as a shorter spliced variant of Mia2, a TANGO1-like protein with similar domains45. Nearly complete loss of Mia2 expression with no change of Mea6 expression was discovered in the Mia2cpto/cpto mice which have a point mutation in the N-terminus of Mia2 (not within Mea6). Mia2cpto/cpto mice have reduced cholesterol and TG levels in the plasma45. Different from Mea6 cKO mice which showed severe fatty liver and serious hypolipaemia, the ApoB and ApoE levels are not reduced in the plasma in Mia2cpto/cpto mice, nor are the substantially increased levels of ApoB, ApoE and lipid in the liver of these mice. More importantly, expression of mouse Mea6 could fully reverse lipid droplet accumulation and rescue reduced interaction between Sec23A and Sec31A in Bel7402 cells after Mea6 knockdown (Figures 9 and 7F). Therefore, Mea6 is likely to play a role in lipid transport independent of Mia2.
In summary, the results presented here reveal several novel components of the lipid transport and secretion machinery in the liver, which includes Mea6, Sec23/24 and Sec31. These findings provide insights into mechanisms underlying this basic cellular process, as well as potential biomarkers for screening NAFLD and related conditions.
Materials and Methods
Materials
Oil red, Triton WR-1339, antibody for Mea6 (cTAGE5), β-tubulin, β-actin, Flag, HA and Flag agarose beads were purchased from Sigma; ApoE, ApoB, Sec23A, Sec24C, Sec24D antibodies from Cell Signaling; Myc antibody from MBL Ltd; Bodipy 493/503 from Invitrogen.
Plasmids
Human MEA6 cDNA was amplified from HEK293 cell lines by RT-PCR using primers: 5′-GAGAGCAGCTTTGGCGCTATGGAG-3′ and 5′-GGTTTCTTGCTGTGGTTCTGGATG-3′ and cloned into sites of EcoRI and SalI to generate pCMV-Tag 3B-MEA6 (Myc-MEA6). pCMS.EGFP.Flag-MEA6 (Flag-MEA6), pCMS.EGFP.Flag-MEA6 ΔN (Flag-MEA6 ΔN), pCMS.EGFP.Flag-MEA6 N (Flag-MEA6 N) and pCMS.EGFP.Flag-MEA6 C (Flag-MEA6 C) were also subcloned from pCMV-Tag 3B-MEA6 as well. Primers for Flag-MEA6 ΔN are: 5′-TATGTGGGACGAGAG-3′ and 5′-GGTTTCTTGCTGTGGTTCTGGATG-3′; primers for Flag-MEA6 N are: 5′-GAGAGCAGCTTTGGCGCTATGGAG-3′ and 5′-TGAGCCTCTTCCTCC-3′; primers for Flag-MEA6 C are: 5′-CGGAATGCTGAAAG-3′ and 5′-GGTTTCTTGCTGTGGTTCTGGATG-3′. Mouse Mea6 cDNA was amplified from C57 mouse brain by RT-PCR using primers: 5′-CATGTCGAATTCATGGAATTGGAAGGTCTGGGTG-3′ and 5′-ACGTCAGCGGC CGCTCAGGCCTCCTGTGTTTCTGGA-3′ and cloned into sites of EcoRI and NotI of pCAGIG (Addgene plasmid # 11159) to generate pCAGIG-Mea6 (Ms-Mea6). Constructs for the expression of Sar1, Sec23A, Sec24C, Sec24D, Sec13 and Sec31A were kind gifts from Dr Sidney Yu from Chinese University of Hong Kong. Primers for qRT-PCR are listed in Supplementary information, Table S1. siRNA and shRNA sequences are listed in Supplementary information, Table S2.
High-fat diet mice
Four-week-old C57BL/6 mice were fed with a high-fat diet (45% of kilocalories as fat, product D12451, Research Diets) for 2 months.
Generation of Mea6 knockout mice
Procedures for the generation of Mea6-deficient mice were based on the methods reported previously46. All mice were maintained in a C57BL/6;129/SvEv mixed background. DNA isolated from tail biopsies was used for genotyping. All experimental procedures involved were performed according to protocols approved by the Institutional Animal Care and Use Committee at Institute of Genetics and Developmental Biology, Chinese Academy of Sciences.
Genotyping, RNA extraction, semi-quantitative real-time PCR
Genomic DNA was isolated from mouse tails and genotyped using primer pairs for floxP fragment: F2: 5′-GACACTTGACCCCTCCTCTCC-3′ and R2: 5′-AACGGCTCATGCTTGCTAACC-3′; for deleted fragment: F2: 5′-GACACTTGACCCCTCCTCTCC-3′ and R4: 5′-AAACTACACCTTAAAAGCAATCAA-3′; for Cre: 5′-CGATGCAACGAGTGATGAGG-3′ and 5′-GCATTGCTGTCACTTGGTCGT-3′.
Total RNA was isolated using the TRIzol reagent (Invitrogen). The cDNAs was synthesized from 1.5 μg of total RNA with oligo-(dT). Real-time PCRs were performed with Bio-rad SYBR GREEN PCR Master Mix and the CFX96TM real-time system (Bio-Rad) according to the manufacturer's instructions. Results were normalized to β-Actin levels.
Transmission electron microscopy and histological analysis
Liver tissues were fixed over night at 4 °C with 2.5% glutaraldehyde in 0.1 M phosphate buffer, pH7.4. Post-fixation was in 1% osmium tetroxide for 2 h. Tissues were dehydrated in serial acetone incubations and embedded in EPON812 resin. Ultrathin sections were cut on a Leica ultramicrotome. Following sequential staining with uranyl acetate and lead citrate, the sections were photographed on a transmission electron microscope (JEM-1400, JEOL, Japan) operated at 80 KV and images were captured with a CCD camera (Gatan).
For histological analysis, liver tissues were dissected from mutant and control mice immediately after euthanasia and fixed in 4% paraformaldehyde for up to 24 h, then dehydrated in a graded series of ethanol and embedded in paraffin. Five-micrometer-thick sections were cut with a Leica RM2255 rotary microtome. After deparaffinization, sections were stained with hematoxylin and eosin (H&E), observed under a light microscope with a CCD camera.
Primary hepatocyte isolation
Anaesthetized mouse was perfused with solution 1 (1 × EBSS without Ca2+ and Mg2+, and 0.5 mM EGTA, pH7.4) followed by solution 2 (1 × EBSS with Ca2+ and Mg2+, and 0.01 M Hepes, pH7.4) and then solution 3 (1 × EBSS with Ca2+ and Mg2+ and 0.01 M Hepes, 0.25 mg/ml collagenase IV, pH7.4) for 15 min until the liver showed a “reticulated appearance”. Liver capsule was disrupted with forceps in 5 ml of solution 3 and the hepatocytes were washed out by swirling the tissue around. The cells were collected and filtered through a nylon mesh into 10 ml of DMEM with serum. The cells were then washed 3 times and placed into culture dishes in DMEM containing 10% fetal bovine serum (FBS).
Cell culture, western blot, Co-IP and immunofluoresence staining
Bel7402 and HEK293 cells were cultured and transfected, western blotting and co-IP were performed as we described previously47,48. Immunofluorescence staining was performed as described previously49. Briefly, cells cultured on cover-slips were fixed in 4% paraformaldehyde/PBS solution for 30 min at room temperature, followed by permeabilization with 25 μg/ml digitonin/PBS for 15 min. Intracellular lipids were visualized with 20 μg/ml Bodipy 493/503 in 150 mM NaCl. Cells were imaged on an LSM Zeiss confocal microscope.
Metabolic labeling of lipids and ApoB in primary cultured hepatocytes
Cells were labeled with [3H] glycerol (4 mCi/ml, 1 ml/dish) in DMEM containing 10% FBS for 4 h. Total lipids were extracted from cells and medium, separately. The radioactivity associated with [3H] glycerolipid was quantified by scintillation counting. ApoB and total protein secretion was examined by pulse-chase analysis.
Regular and mass spectrometry analysis of liver and plasma lipids
TC, TG, albumin and glucose were quantified using cholesterol, TC, albumin and Glu detection kits according to manufacturer's instructions (Kehua Bio-engineering Co., Ltd. China). For lipoprotein analysis, 300 μl of plasma from 3 mice was fractionated by FPLC using a Superose 6 column. TG and TC in each fraction was quantified using the kits and ApoB-100 levels were detected by western blot.
Mass spectrometry analysis of lipids. Lipid extractions from plasma and liver samples were carried out using a modified version of the Bligh and Dyer's protocol as previously described50. Extracted organic fractions were pooled and dried in a miVac (Genevac, UK). Samples were stored in −80 °C until further analyses. Lipids were analyzed using an Agilent 1260 HPLC coupled with a triple quadrupole/ion trap mass spectrometer (4500Qtrap, ABSciex). Individual lipid species were quantified by referencing to spiked internal standards, which include PC-14:0/14:0, LPC-C20, PE-14:0/14:0, PS-14:0/14:0, PA-17:0/17:0, PG-14:0/14:0, C14-LBPA, C8-GluCer, C17-Cer, C12-SM and 4 ME 16 Diether DG from Avanti Polar Lipids and LIPID MAPS (Alabaster, AL); 16:0-PI from Echelon Biosciences, Inc. (Salt Lake City, UT); as well as d6-C18 CE, d6-Cho and d5-TAG from CDN isotopes (Quebec, Canada). The detailed analytical conditions have been previously described elsewhere51.
Quantitative proteome analysis of liver proteins
The lysates (100 μg protein for each sample) were reduced with 10 mM DTT at 37 °C for 1 h, alkylated with 55 mM iodoacetamide at room temperature for 1 h in the dark. The denaturing buffer of the lysates was then replaced with the iTRAQ dissolution buffer using the Microcon YM-30 centrifugal filter units (EMD Millipore Corporation, MA, USA). The lysates were digested with sequencing grade trypsin (1:50 (w:w)) at 37 °C overnight. The resulting tryptic peptides were collected and labeled with iTRAQ® Reagents (AB Sciex Inc., MA, USA) according to manufacturer's manual. The labeled samples were mixed together with equal ratios in amount, and 20 μg of the mixture was then desalted and fractionated into 6 fractions with C18-strong cation exchange (SCX) stop-and-go-extraction tips (StageTips)52. The peptides in each fraction were analyzed by a TripleTOF 5600 mass spectrometer (AB SCIEX) coupled online to an Eksigent NanoLC Ultra in Information Dependent Mode and with iTRAQ reagent collision energy adjustment on53. The ProteinPilot 4.5 software (AB SCIEX) with the Paragon database search algorithm was applied to identify and quantify peptides and proteins. The UniProt proteome sequences for Mus musculus (including canonical and isoforms) were used for the database search. The confidence of quantitation for differentially expressed proteins was analyzed with the ProteinPilot Descriptive Statistics Template (PDST) (beta v3.07p, AB SCIEX).
Affinity capture of biotinylated proteins
Based on the method of Roux et al.34, primary hepatocytes were infected with GFP-BirA-Sar1B expressing adenovirus. After 24 h, cells were supplemented with 20 μM biotin for 24 h. After three PBS washes, cells were lysed at 25 °C in 1 ml lysis buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 0.4% SDS, 5 mM EDTA, 1 mM DTT, and 1× Complete protease inhibitor (Roche)) and sonicated. Supernatants were incubated with 60 μl dynabeads (MyOne Steptavadin C1, Invitrogen) overnight. Beads were collected and washed. Bound proteins were removed from the magnetic beads with 50 μl of SDS-sample buffer saturated with biotin at 98 °C for western blot analysis.
Statistical analysis
All data were analyzed using Prism software (GraphPad Software, Inc, CA). Values listed were means ± SEM. Comparisons were made using Student's t-test. *P < 0.05, **P < 0.01 for all statistics herein.
Author Contributions
YW, FG and ZX designed the experiments; YW, LL, HZ and JF performed the experiments; FZ, MY, LS, LY, SML and HW provided help for the experiments; XC, YW, GS and ZX analyzed the data; GS and FG contributed to manuscript preparation; YW, LL and ZX wrote the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
Goh VJ, Silver DL . The lipid droplet as a potential therapeutic target in NAFLD. Semin Liver Dis 2013; 33:312–320.
Lazo M, Hernaez R, Eberhardt MS, et al. Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Epidemiol 2013; 178:38–45.
Cohen JC, Horton JD, Hobbs HH . Human fatty liver disease: old questions and new insights. Science 2011; 332:1519–1523.
Kawano Y, Cohen DE . Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol 2013; 48:434–441.
Yao Z, Zhou H, Figeys D, Wang Y, Sundaram M . Microsome-associated lumenal lipid droplets in the regulation of lipoprotein secretion. Curr Opin Lipidol 2013; 24:160–170.
Tiwari S, Siddiqi SA . Intracellular trafficking and secretion of VLDL. Arterioscler Thromb Vasc Biol 2012; 32:1079–1086.
Sundaram M, Yao Z . Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr Metab (Lond) 2010; 7:35.
Schonfeld G . Familial hypobetalipoproteinemia: a review. J Lipid Res 2003; 44:878–883.
Jornayvaz FR, Shulman GI . Diacylglycerol activation of protein kinase Cepsilon and hepatic insulin resistance. Cell Metab 2012; 15:574–584.
Go GW, Srivastava R, Hernandez-Ono A, et al. The combined hyperlipidemia caused by impaired Wnt-LRP6 signaling is reversed by Wnt3a rescue. Cell Metab 2014; 19:209–220.
Chen XW, Wang H, Bajaj K, et al. SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. Elife 2013; 2:e00444.
Ye J, Li JZ, Liu Y, et al. Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab 2009; 9:177–190.
Anstee QM, Day CP . The genetics of nonalcoholic fatty liver disease: spotlight on PNPLA3 and TM6SF2. Semin Liver Dis 2015; 35:270–290.
Kuehn MJ, Herrmann JM, Schekman R . COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature 1998; 391:187–190.
Barlowe C, Orci L, Yeung T, et al. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 1994; 77:895–907.
Adams EJ, Chen XW, O'Shea KS, Ginsburg D . Mammalian COPII coat component SEC24C is required for embryonic development in mice. J Biol Chem 2014; 289:20858–20870.
Jones B, Jones EL, Bonney SA, et al. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet 2003; 34:29–31.
Gusarova V, Brodsky JL, Fisher EA . Apolipoprotein B100 exit from the endoplasmic reticulum (ER) is COPII-dependent, and its lipidation to very low density lipoprotein occurs post-ER. J Biol Chem 2003; 278:48051–48058.
Fryer LG, Jones B, Duncan EJ, et al. The endoplasmic reticulum coat protein II transport machinery coordinates cellular lipid secretion and cholesterol biosynthesis. J Biol Chem 2014; 289:4244–4261.
Malhotra V, Erlmann P . Protein export at the ER: loading big collagens into COPII carriers. EMBO J 2011; 30:3475–3480.
Gillon AD, Latham CF, Miller EA . Vesicle-mediated ER export of proteins and lipids. Biochim Biophys Acta 2012; 1821:1040–1049.
Guinn BA, Gilkes AF, Mufti GJ, Burnett AK, Mills KI . The tumour antigens RAGE-1 and MGEA6 are expressed more frequently in the less lineage restricted subgroups of presentation acute myeloid leukaemia. Br J Haematol 2006; 134:238–239.
Usener D, Schadendorf D, Koch J, Dubel S, Eichmuller S . cTAGE: a cutaneous T cell lymphoma associated antigen family with tumor-specific splicing. J Invest Dermatol 2003; 121:198–206.
Comtesse N, Niedermayer I, Glass B, et al. MGEA6 is tumor-specific overexpressed and frequently recognized by patient-serum antibodies. Oncogene 2002; 21:239–247.
Saito K, Yamashiro K, Ichikawa Y, et al. cTAGE5 mediates collagen secretion through interaction with TANGO1 at endoplasmic reticulum exit sites. Mol Biol Cell 2011; 22:2301–2308.
Saito K, Chen M, Bard F, et al. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell 2009; 136:891–902.
Aridor M, Weissman J, Bannykh S, Nuoffer C, Balch WE . Cargo selection by the COPII budding machinery during export from the ER. J Cell Biol 1998; 141:61–70.
Saito K, Yamashiro K, Shimazu N, Tanabe T, Kontani K, Katada T . Concentration of Sec12 at ER exit sites via interaction with cTAGE5 is required for collagen export. J Cell Biol 2014; 206:751–762.
Wilson DG, Phamluong K, Li L, et al. Global defects in collagen secretion in a Mia3/TANGO1 knockout mouse. J Cell Biol 2011; 193:935–951.
Boyadjiev SA, Fromme JC, Ben J, et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet 2006; 38:1192–1197.
Lewandoski M, Wassarman KM, Martin GR . Zp3-cre, a transgenic mouse line for the activation or inactivation of loxP-flanked target genes specifically in the female germ line. Curr Biol 1997; 7:148–151.
Kang JH, Kim HT, Choi MS, et al. Proteome analysis of human monocytic THP-1 cells primed with oxidized low-density lipoproteins. Proteomics 2006; 6:1261–1273.
Borensztajn J, Rone MS, Kotlar TJ . The inhibition in vivo of lipoprotein lipase (clearing-factor lipase) activity by triton WR-1339. Biochem J 1976; 156:539–543.
Roux KJ, Kim DI, Raida M, Burke B . A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 2012; 196:801–810.
Stagg SM, LaPointe P, Razvi A, et al. Structural basis for cargo regulation of COPII coat assembly. Cell 2008; 134:474–484.
Raabe M, Veniant MM, Sullivan MA, et al. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J Clin Invest 1999; 103:1287–1298.
Fei W, Yang H . Genome-wide screens for gene products regulating lipid droplet dynamics. Method Cell Biol 2012; 108:303–316.
Skinner JR, Harris LA, Shew TM, Abumrad NA, Wolins NE . Perilipin 1 moves between the fat droplet and the endoplasmic reticulum. Adipocyte 2013; 2:80–86.
Reddick RL, Zhang SH, Maeda N . Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arterioscler Thromb 1994; 14:141–147.
Kuipers F, Jong MC, Lin Y, et al. Impaired secretion of very low density lipoprotein-triglycerides by apolipoprotein E- deficient mouse hepatocytes. J Clin Invest 1997; 100:2915–2922.
Mensenkamp AR, Jong MC, van Goor H, et al. Apolipoprotein E participates in the regulation of very low density lipoprotein-triglyceride secretion by the liver. The J Biol Chem 1999; 274:35711–35718.
Farese RV Jr, Ruland SL, Flynn LM, Stokowski RP, Young SG . Knockout of the mouse apolipoprotein B gene results in embryonic lethality in homozygotes and protection against diet-induced hypercholesterolemia in heterozygotes. Proc Natl Acad Sci USA 1995; 92:1774–1778.
Huang LS, Voyiaziakis E, Markenson DF, Sokol KA, Hayek T, Breslow JL . Apo B gene knockout in mice results in embryonic lethality in homozygotes and neural tube defects, male infertility, and reduced HDL cholesterol ester and apo A-I transport rates in heterozygotes. J Clin Invest 1995; 96:2152–2161.
Heckel D, Brass N, Fischer U, et al. cDNA cloning and chromosomal mapping of a predicted coiled-coil proline-rich protein immunogenic in meningioma patients. Hum Mol Genet 1997; 6:2031–2041.
Pitman JL, Bonnet DJ, Curtiss LK, Gekakis N . Reduced cholesterol and triglycerides in mice with a mutation in Mia2, a liver protein that localizes to ER exit sites. J Lipid Res 2011; 52:1775–1786.
Liu P, Jenkins NA, Copeland NG . A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 2003; 13:476–484.
Liu S, Yu M, He Y, et al. Melittin prevents liver cancer cell metastasis through inhibition of the Rac1-dependent pathway. Hepatology 2008; 47:1964–1973.
Xu Z, Kukekov NV, Greene LA . POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J 2003; 22:252–261.
Yang T, Sun Y, Zhang F, et al. POSH localizes activated Rac1 to control the formation of cytoplasmic dilation of the leading process and neuronal migration. Cell Rep 2012; 2:640–651.
Lam SM, Tong L, Reux B, et al. Lipidomic analysis of human tear fluid reveals structure-specific lipid alterations in dry eye syndrome. J Lipid Res 2014; 55:299–306.
Lam SM, Wang Y, Duan X, et al. The brain lipidomes of subcortical ischemic vascular dementia and mixed dementia. Neurobiol Aging 2014; 35:2369–2381.
Rappsilber J, Mann M, Ishihama Y . Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2007; 2:1896–1906.
Tambor V, Hunter CL, Seymour SL, Kacerovsky M, Stulik J, Lenco J . CysTRAQ – A combination of iTRAQ and enrichment of cysteinyl peptides for uncovering and quantifying hidden proteomes. J Proteomics 2012; 75:857–867.
Acknowledgements
We would like to thank Dr Sidney Yu for providing constructs, Drs WZ Jin, X Huang, BL Song and P. Li for their constructive comments, Miss Xiahe Huang at IGDB for help with mass spectrometry analysis. This work was supported in part by grants from the NSF (China) (31430037/31471132/31270826/31271156) and the MOST (China) “973” program (2014CB942801/2012CB517904).
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Characterization of Mea6 6cKO cKO mouse. (PDF 164 kb)
Supplementary information, Figure S2
Body weight and liver histological analysis. (PDF 312 kb)
Supplementary information, Figure S3
Reduced VLDL secretion but normal albumin and glucose secretion, normal lipid synthesis and normal total protein secretion in Mea6 knockout mice. (PDF 84 kb)
Supplementary information, Figure S4
mRNA levels of different genes involved in lipid metabolism in P1 and P35 livers. (PDF 87 kb)
Supplementary information, Figure S5
Mea6 does not interact with Sec24C but interacts indirectly with Sec31A. (PDF 97 kb)
Supplementary information, Figure S6
Knockdown of Mea6, Sec23A, Sec24D and Sec31A caused LDs accumulation in Bel7402 cells. (PDF 210 kb)
Supplementary information, Table S1
Primers for real-time analysis (PDF 177 kb)
Supplementary information, Table S2
Sequences of siRNA or shRNA (PDF 465 kb)
Rights and permissions
About this article

Cite this article
Wang, Y., Liu, L., Zhang, H. et al. Mea6 controls VLDL transport through the coordinated regulation of COPII assembly. Cell Res 26, 787–804 (2016). https://doi.org/10.1038/cr.2016.75
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2016.75
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Unlocking the mysteries of VLDL: exploring its production, intracellular trafficking, and metabolism as therapeutic targets
Lipids in Health and Disease (2024)
-
Coordinated reprogramming of renal cancer transcriptome, metabolome and secretome associates with immune tumor infiltration
Cancer Cell International (2023)
-
Upregulation of WDR6 drives hepatic de novo lipogenesis in insulin resistance in mice
Nature Metabolism (2023)
-
Methamphetamine-Induced Cognitive Deficits and Psychiatric Symptoms Are Associated with Serum Markers of Liver Damage
Neurotoxicity Research (2020)