
Abstract
Emerging studies document the roles of long non-coding RNAs (LncRNAs) in regulating gene expression at chromatin level but relatively less is known how they regulate DNA methylation. Here we identify an lncRNA, Dum (developmental pluripotency-associated 2 (Dppa2) Upstream binding Muscle lncRNA) in skeletal myoblast cells. The expression of Dum is dynamically regulated during myogenesis in vitro and in vivo. It is also transcriptionally induced by MyoD binding upon myoblast differentiation. Functional analyses show that it promotes myoblast differentiation and damage-induced muscle regeneration. Mechanistically, Dum was found to silence its neighboring gene, Dppa2, in cis through recruiting Dnmt1, Dnmt3a and Dnmt3b. Furthermore, intrachromosomal looping between Dum locus and Dppa2 promoter is necessary for Dum/Dppa2 interaction. Collectively, we have identified a novel lncRNA that interacts with Dnmts to regulate myogenesis.
Similar content being viewed by others

Introduction
Although only a small number of functional long non-coding RNAs (lncRNAs) have been well characterized to date, they seem to control major biological processes impacting cell differentiation and development1. Specifically, recent work suggested that a large number of lncRNAs function to epigenetically modulate gene expression through interacting with chromatin modifiers or remodelers. They may act as signals, guides or scaffolds to the chromatin to regulate expression of target genes2,3,4. In some cases, the lncRNA and the target genes are transcribed from the same locus or in a very close distance. In other cases, however, an lncRNA could act over a long distance on a distal target gene. This may involve three-dimensional folding of chromatin which brings the target locus and the lncRNA in a close proximity.
The genesis of skeletal muscle during embryonic development and postnatal life serves as a paradigm for stem and progenitor cell maintenance, lineage specification and terminal differentiation. During injury-induced regeneration, muscle stem cells (satellite cells) are activated, proliferate, differentiate and fuse to form myofibers to repair the damage5. The process of myoblast differentiation is a powerful system for investigating the biological functions of lncRNAs because of the availability of an in vitro C2C12 murine myoblast cell line; it can faithfully recapitulate the myogenesis in culture and the transcriptional networks coordinating gene expressions are extensively studied using this cell line. Specification and differentiation of skeletal muscle stem cells (myoblasts) into myotubes are driven by a family of muscle-restricted basic Helix-Loop-Helix transcription factors (TFs), MyoD, Myf5, myogenin and MRF4, which activate the differentiation program by inducing the transcription of muscle-specific genes such as myosin heavy chain (MyHC), alpha actin (α-actin) and troponin isoforms. It is becoming increasingly clear that a complex network of transcription factors, epigenetic regulators and non-coding RNAs is pivotal for myogenesis6. The establishment and maintenance of DNA methylation patterns resulting in modulation of gene expression is one of the key steps in epigenetic regulation during muscle cell differentiation. This modification is mediated by the members of the DNA methyltransferase (DNMT) family, conventionally classified as de novo (DNMT3a and DNMT3b) and maintenance (DNMT1) Dnmts. DNA methylation is thought to repress many muscle gene loci. For example, it has been shown that DNA methylation helps to restrict myogenin activation and demethylation of myogenin promoter appears necessary for the differentiation program to proceed7. However, the precise mechanisms regulating methylation/demethylation in myogenesis is still far from being understood and it remains unclear whether such events are general or specific to a subset of genes. More importantly, although much has been done on how lncRNAs modulate chromatins, very little is known in terms of how they are involved in DNA methylation. The best characterized example is Kcnq1ot1 lncRNA which was shown to interact with DNMT1 to maintain methylation of ubiquitously imprinted genes both in developmental stages and somatic tissues8. Very recently, Ruscio AD et al.9 also reported the interaction between DNMT1 and a novel lncRNA, ecCEBPA, which interestingly leads to DNMT1 sequestration and prevents CEBPA gene locus methylation. However, it is unknown whether other DNMT enzymes (DNMT3a, DNMT3b) may also interact with lncRNAs, through which the patterns of de novo DNA methylation can be modulated.
In this study, we identified an lncRNA named Dum (developmental pluripotency-associated 2 (Dppa2) Upstream binding Muscle lncRNA) in myoblast cells from the de novo assembly of high-throughput RNA-sequencing data. The expression of Dum is tightly associated with in vitro and in vivo myogenesis processes and induced by MyoD upon myoblast differentiation. Functional studies demonstrated that it acts as a pro-myogenic factor in both myoblast differentiation and muscle regeneration in vivo. Further mechanistic investigation revealed that Dum functions by in cis silencing of its neighboring gene Dppa2 through interacting and recruiting multiple Dnmts (Dnmt1, Dnmt3a and Dnmt3b) to its promoter regions; and intrachromosomal looping is necessary for Dum/Dppa2 interaction. Altogether, our studies have uncovered a novel functional lncRNA that modulates DNA methylation.
Results
A lncRNA Dum is associated with skeletal myogenesis
In an attempt to discover the functional lncRNAs associated with myogenesis, we applied an ab initio identification pipeline10 to the RNA-seq data from differentiating C2C12 mouse myoblast cells. Among all the identified lncRNAs, Dum is a known transcript transcribed from mouse chromosome 16. An evident Pol II peak that marked its promoter region and an H3K4me3-H3K36me3 domain which typically defines lincRNA locus11 was also identified. It is annotated in UCSC with three isoforms and in Refseq as 5330426P16Rik (Figure 1A). Through rapid amplification of cDNA ends we demonstrated that 5330426P16Rik is abundantly expressed in C2C12 cells. It contains two exons with a full length of 1 817 bp (Figure 1B and Supplementary information, Figure S1A) as confirmed by northern blotting analysis (Figure 1C and Supplementary information, Figure S1B) and RT-PCR amplification (Figure 1D). A Dppa2 gene is located in its distant upstream region and transcribes from the opposite strand (Figure 1B). Dppa2 is highly expressed in pluripotent stem cells and involved in the maintenance of pluripotency of embryonic stem (ES) cells12. By RNA-fluorescence in situ hybridization (RNA-FISH), Dum was found to be distributed in both nucleus and cytoplasm of C2C12 myoblasts, distinct from the nuclear transcript U1 which was mainly found in the nucleus (Figure 1E and Supplementary information, Figure S1C). Consistently, by cellular fractionation assay, almost equal amounts of Dum transcripts were found in nuclear and cytoplasmic extracts (Figure 1F); well-known lncRNAs, Xist, Malat1 and U1, on the other hand, were mainly detected in the nuclei; and Yam-1, as we recently showed13, was found in both fractions with almost equal amounts. Using our recently developed coding potential predication software iSeeRNA14, Dum is predicated to be a non-coding RNA (data not shown) which folds into extensive stem-loop structures (Supplementary information, Figure S1D). Results from in vitro translation assay also confirmed its non-coding nature (Supplementary information, Figure S1E). Interestingly, a human ortholog was predicted on chromosome 3; evidence of its expression was found in various tissues and cells through exploring GENCODE data (Supplementary information, Figure S1F).

Dum is a novel lncRNA associated with skeletal myogenesis. (A) Genomic snapshot of mouse Dum generated in UCSC (blue), Refseq (black), MyoD ChIP-seq (green), RNA-seq (pink), Pol II, H3K4me3 and H3K36me3 ChIP-seq (green) tracks. (B) Schematic illustration of the genomic location and structure of mouse Dum locus. A MyoD-binding site is shown as green diamond. (C, D) Detection of full-length Dum by northern blotting (C) or RT-PCR (D) in C2C12 myoblasts. (E) Visualization of Dum or U1 in C2C12 myoblasts by RNA-FISH. Scale bar, 50 μm. (F) The expression of Dum, Yam-1, Xist, Malat1 and U1 in nuclear or cytoplasmic fraction of C2C12 myoblasts. (G) The binding of MyoD on the Dum promoter as shown by ChIP-PCR. (H) Knockdown of MyoD by siRNA oligos decreased Dum. (I) Overexpression of a MyoD plasmid in 10T1/2 cells increased Dum expression as shown. (J) The levels of MyoD, myogenin, MyHC and troponin mRNAs were increased in cells from I. (K-P) The expression of Dum was detected by qRT-PCR. The expression of Dum in differentiating C2C12 (K); in differentiating satellite cells freshly isolated from mouse limb muscles (L); during CTX-induced regeneration (M); in muscles of postnatal mice at the indicated ages (N); in muscles from wild-type or dystrophic mdx mice at the indicated ages (O); and in mature mouse skeletal muscle tissue or isolated primary myoblasts (P). All PCR data were normalized to GAPDH mRNA and represent mean ± SD of three independent experiments. *P < 0.05, **P < 0.01. See also Supplementary information, Figure S1.
To investigate its relevancy in myogenesis, we first examined whether it is regulated by MyoD, reasoning that a functional lncRNA may be under regulation of the master myogenic transcription factor. By analyzing publically available MyoD ChIP-seq data from C2C12 cells15, a potential binding peak was identified −26 bp upstream of transcriptional start site (Figure 1A and 1B). Results from ChIP-PCR assay further confirmed the MyoD binding not only in proliferating myoblasts (MBs) but also in differentiating myotubes (MTs) (Figure 1G and Supplementary information, Figure S1G). Consistently, knockdown of MyoD with an siRNA oligo in C2C12 cells significantly decreased Dum expression by 35% as compared to negative control (siNC) (Figure 1H); overexpression of a MyoD plasmid in 10T1/2 fibroblast cells, on the other hand, upregulated Dum expression (Figure 1I) along with the expression of several myogenic genes myogenin, MyHC and troponin (Figure 1J).
The regulation of Dum by MyoD suggested that it is likely a functional lncRNA in skeletal muscle cells. To gain more insights, we first examined its temporal and spatial expression patterns in several myogenesis systems in vitro and in vivo. During C2C12 cell differentiation, Dum was found to be robustly upregulated during the early stage from proliferating myoblasts (-24 h) to 12 h in differentiation medium (DM) but gradually decreased afterwards (24 and 48 h) (Figure 1K), suggesting that it can be a pro-myogenic factor during the early differentiation. Consistently, during the differentiation of freshly isolated satellite cells (SCs), Dum expression significantly increased in the early stage (Figure 1L). To further examine its expression dynamics during myogenesis in vivo, we employed a widely-used muscle regeneration model in which the intramuscular injection of cardiotoxin (CTX) results in muscle injury and in turn induces muscle regeneration16. Dum was found to be highly induced during the early regeneration stage when satellite cells became activated, proliferated and started to differentiate, but gradually downregulated later on when the newly formed fibers matured and regeneration was completed in about 10 days (Figure 1M). Consistently, high levels of Dum were observed in limb muscles of newborn mice (at the age of 3 days, 5 days and 2 weeks) which displayed active myogenesis, but the level of Dum decreased as the neonatal myogenesis ceased after about 2 weeks and remained low as the mice aged (Figure 1N). Moreover, when compared to normal muscles from wild-type mice, higher levels of Dum were detected in dystrophic muscles from mdx mice which were featured by a pathologically active degeneration and regeneration17 (Figure 1O). The above results strongly suggested that Dum is associated with active myogenesis in vitro and in vivo. In addition, Dum expression is highly enriched in the activated satellite cells or primary myoblasts freshly isolated from mouse limb muscle compared to the whole muscle itself (Figure 1P), suggesting that its function is related to satellite cell activities but not mature muscle homeostasis.
Dum is a pro-myogenic factor during myoblast differentiation
The early induction of Dum expression during C2C12 differentiation suggested that it may be a pro-myogenic factor during myoblast differentiation. To test this notion, we knocked down Dum using an siRNA oligo, siDum#1. Successful knockdown (Figure 2A and Supplementary information, Figure S2A) significantly delayed C2C12 differentiation as assessed by examination of several myogenic markers, myogenin, MyHC and troponin, at both RNA (Figure 2B) and protein (Figure 2C) levels during a 4-day differentiation course. The results were also strengthened by immunofluorescence staining for MyHC and troponin proteins in differentiating myotubes on day 2 (Figure 2D); the number of positive myotubes was reduced by siDum treatment by 46% and 23%, respectively (Figure 2E). Furthermore, co-transfection of siDum together with a myogenin or troponin luciferase reporter significantly inhibited their luciferase activities as compared to negative control oligos (Figure 2F). To confirm the above results, a second siRNA oligo, siDum#2, was used and the same inhibitory effect was observed by assaying the myogenic RNA expression (Supplementary information, Figure S2B) and reporter activities (Supplementary information, Figure S2C). In addition, since it is still questionable whether siRNAs can efficiently delete nuclear resident lncRNAs, we repeated the above experiments using an optimized phosphorothioate-modified antisense oligodeoxynucleotide (ASO) against Dum. Knockdown of Dum by the ASO also delayed myogenic differentiation (Figure 2G-2J). Surprisingly, in gain-of-function assay using a Dum overexpression plasmid, no impact on myogenic differentiation was observed despite repeated efforts (Supplementary information, Figure S2D-S2G).

Dum functions to promote muscle cell differentiation. (A) Knockdown of Dum by siRNA oligos in C2C12 cells decreased the expression of Dum during a 4-day differentiation course. (B) The indicated myogenic genes, myogenin, MyHC and troponin were downregulated in cells from A. (C) Knockdown of Dum decreased the levels of the indicated proteins during a 4-day differentiation course. (D) The cells in A were visualized on day 2 in differentiation medium (DM). Immunofluorescence staining for MyHC or troponin was performed. (E) The number of positively stained cells in D was quantified. (F) Knockdown of Dum in C2C12 cells decreased the luciferase activities of the indicated reporters. (G) Knockdown of Dum by an ASO oligo in C2C12 cells decreased the expression of Dum and the indicated myogenic genes, myogenin, MyHC and troponin. (H) The cells from G were visualized on day 2 in DM. Immunofluorescence staining for MyHC or troponin was performed. (I) The number of positively stained cells in H was quantified. (J) Knockdown of Dum by ASO in C2C12 cells decreased the luciferase activities of the indicated reporters. (K) Knockdown of Dum in freshly isolated satellite cells decreased the myogenic differentiation. (L, M) Knockdown of Dum in single fibers decreased the satellite cell differentiation as shown by immunofluorescence staining for myogenin. All PCR data were normalized to GAPDH mRNA and represent mean ± SD of three independent experiments. All luciferase activity data were normalized to Renillia protein and represent mean ± SD of three independent experiments. *P < 0.05, **P < 0.01. See also Supplementary information, Figure S2.
To extend our findings in C2C12 cells to a more physiologically relevant setting, we tested the function of Dum in freshly isolated satellite cells. In keeping with its pro-myogenic function in C2C12 cells, knockdown of Dum by siRNA oligos impaired the myogenic differentiation as shown by the reduced levels of myogenin and MyHC mRNAs (Figure 2K). These findings were further confirmed in satellite cells associated with freshly isolated single myofiber which serves as an excellent ex vivo model. Transfection of siDum oligos impaired satellite cell differentiation capacity as shown by a 33% decrease in the number of myogenin-positive cells per fiber compared to siNC treatment (Figure 2L and 2M).
Loss of Dum delayed CTX-induced muscle regeneration in vivo
The above findings underscored the role of Dum in satellite cell function, leading us to believe that it exerts a role in muscle regeneration in vivo. To test this notion, we depleted Dum in mouse limb muscles during injury-induced regeneration using intramuscular injection of siRNA oligos as described before17,18,19. As illustrated in Figure 3A, the injection of siDum or negative control oligos was performed three times on days 1/4, 2 and 4 post CTX injection and muscles were harvested at the designated times for analyses. Results indicated that injection of siDum oligos led to a significant loss of Dum expression along the regeneration course (Figure 3B). Accordingly, the mRNA levels of Pax7, MyoD, myogenin and embryonic MyHC (eMyHC, a marker for newly formed fibers) were all significantly decreased (Figure 3B); and the protein levels of Pax7, MyoD and mygenin were also reduced (Figure 3C, 43%, 45% and 42%), suggesting a delay of myogenic program. Consistently, by immunofluorescence staining of the muscle sections, the numbers of cells positively stained for Pax7, MyoD, myogenin and eMyHC were evidently reduced (Figure 3D-3G, 60%, 40%, 33% and 44%). Altogether, the above results demonstrated that loss of Dum caused a significant delay of injury-induced muscle regeneration in vivo.

Dum knockdown in vivo impaired the injury-induced muscle regeneration. (A) Injection scheme for siNC or siDum oligos into CTX-injured muscles. n = 4 mice for each group. (B) Dum siRNA injection into CTX-injured muscles decreased the levels of the indicated RNAs at multiple time points after CTX injection. (C) Upper panels: Dum siRNA injection decreased the levels of the proteins in two representative mice. Lower panel: α-tubulin was used as normalization for the quantification of the western blot band intensity. (D-G) Immunofluorescence staining for Pax7, MyoD and myogenin was performed on the injected muscles in A on day 3 and eMyHC on day 6. Positively stained cells were quantified. All PCR data were normalized to GAPDH mRNA and represent mean ± SD of three independent experiments. *P < 0.05, **P < 0.01.
Dum promotes myogenesis through in cis regulation of Dppa2 transcription
To probe into the mechanisms underlying the pro-myogenic function of Dum, we considered its potential regulation on neighboring genes because many lncRNAs are believed to function in cis20. Indeed, knockdown of Dum in C2C12 cells led to a significant change in the expression of 17 out of 23 neighboring genes examined, ranging from 0.1 to 13 folds (Figure 4A and Supplementary information, Figure S3A). Interestingly, the majority (11/17) were upregulated. This finding demonstrated that Dum is a strong in cis regulator.

Dum regulates Dppa2 gene expression in cis. (A) RNAs were isolated from C2C12 cells transfected with siNC or siDum for qRT-PCR measurement of the expression of Dum neighboring genes. Expression folds are shown with respect to siNC cells where normalized copy numbers were set to 1. (B) Expression of Dppa2 and Oct4 in the transfected cells from A. (C) Expression of Dppa2 and Oct4 in regenerating muscle injected with siNC or siDum oligos. (D, E) Expression of Dpppa2 or Oct4 during C2C12 (D)or primary myoblast (E) differentiation. (F, G) C2C12 cells (F) or primary myoblasts (G) were transfected with the indicated siRNA oligos and the myogenic reporter. Luciferase activities were determined at 48 h post transfection. Relative luciferase unit (RLU) is shown with respect to siNC cells where normalized luciferase values were set to 1. (H) The indicated siRNA oligos were injected into the CTX-induced regenerating muscle and the expression of the myogenic markers were meansured 3 days after the injection. The data represent mean ± SD of three independent experiments. All PCR data were normalized to GAPDH mRNA and represent mean ± SD of three independent experiments. All luciferase acitivity data were normalized to Renillia protein and represent mean ± SD of three independent experiments. *P < 0.05, **P < 0.01. See also Supplementary information, Figure S3.
Among all the affected genes, Dppa2 caught our attention because it was reported to regulate Oct4 which inhibits muscle cell differentiation21. Consistently, knockdown of Dum by siRNA (Figure 4B) or ASO oligos (Supplementary information, Figure S3B) in C2C12 cells led to an induction of both Dppa2 and Oct4 expression. Furthermore, loss of Dum in CTX-injured muscles upregulated the expressions of Dppa2 and Oct4 through the regeneration course (Figure 4C). In contrast to the upregulation of Dum levels, Dppa2 and Oct4 expression was gradually downregulated during the early differentiation of both C2C12 cells (Figure 4D) and freshly isolated satellite cells (Figure 4E), suggesting the anti-myogenic nature of Dppa2. At a functional level, indeed, knockdown of Dppa2 by siRNA oligos upregulated the myogenic reporter activities. Furthermore, co-transfection of siDppa2 oligos significantly reverted the inhibitory effect of siDum (Figure 4F), suggesting that Dum function is probably dependent on Dppa2. This was validated in primary myoblasts. siDum oligos inhibited the primary myoblast differentiation, whereas siDppa2 promoted it (Figure 4G). Furthermore, in the regenerating muscles, siDppa2 treatment was able to rescue the suppressive effect of siDum on regeneration (Figure 4H).
Dum silences Dppa2 expression through recruiting Dnmts to its promoter CpG sites
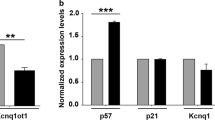
The above results demonstrated that Dum suppresses Dppa2 transcription upon differentiation. To further elucidate the regulatory mechanisms at the molecular level, we asked how Dum modulates Dppa2 transcription. Although it is still unclear how Dppa2 transcription is regulated, Ruau et al.22 reported that its level is increased by 5′-Aza-2′-deoxycytidine (5-Aza) treatment in neurosphere cells, leading us to hypothesize that Dppa2 may be under control by DNA methylation and Dum likely silences its expression through regulating the promoter methylation. To confirm that Dppa2 transcription is regulated by DNA methylation in myoblasts, we treated C2C12 cells with 5-Aza. Expectedly, the treatment induced Dppa2 expression (Figure 5A); as a positive control, myogenin gene was also induced as previously reported23. Several CpG sites were identified in two regions of Dppa2 promoter by computational prediction (EMBOSS Cpgplot; http://www.ebi.ac.uk) (Figure 5B). Using Bisulfate Genomics Sequencing (BGS) assay, we uncovered that 8 CpG sites were indeed methylated to various degrees upon C2C12 differentiation (Figure 5B). As expected, knockdown of Dum reduced the degree of methylation at these CpG sites. Interestingly, the promoters of the other 10 genes (RP124, Dppa4, Nfkbiz, Cd47, Cb1b, Senp7 V234, Penp, Cdc54, IFT57 and Jo3rik) that were upregulated upon Dum knockdown (Figure 4A) were not subjected to methylation by BGS assay (data not shown), suggesting a unique regulatory mechanism of Dum on Dppa2.

Dum interacts with Dnmts to induce Dppa2 promoter methylation. (A) C2C12 cells were treated with or without 5-Aza; the expression of Dppa2 or myogenin was decreased as shown by qRT-PCR. (B) CpG sites were identified in two regions (region 1, −1 308 to −1 166 and region 2, −185 to −47) of Dppa2 promoter and the methylation was measured using bisulfate genomic sequencing in siNC- or siDum-treated cells. The degree of methylation was decreased by siDum as shown by percentage of the methylated cytosines from 10 randomly sequenced colonies. (C) Dnmt1, Dnmt3a and Dnmt3b bound to Dum but not Xist in C2C12 cells as revealed by RIP assay. (D) In vitro-generated biotin-labeled full-length or the deletion fragments were used to pull down Dnmts. No binding to Ezh2 and YY1 was detected (negative controls). (E) Knockdown of Dum by siRNA oligos in C2C12 cells decreased the enrichment of Dnmts on Dppa2 promoters by ChIP-PCR assay. (F) Dnmt binding to Dum was detected in CTX-induced regenerating muscle by in vivo RIP assay. (G) Knockdown of Dum in regenerating muscle decreased the enrichment of Dnmts on Dppa2 promoters by in vivo ChIP-PCR assay. (H) Transfection of the in vitro-transcribed Dum transcripts led to successful overexpression of Dum as compared to transfection of in vitro-transcribed GFP transcripts. It, however, had no effect on Dppa2 expression. (I) The ectopic Dum or control GFP transcripts from H were not detected on Dppa2 promoter. (J) Dum ChIRP with both even and odd antisense oligos retrieved a significant amount of genomic DNAs corresponding to Dppa2 promoter regions 1 and 2 as defined in B but not in a third region (−1 589 to −1 411) and GAPDH locus. LacZ ChIRP retrieved no signal. (K, L) The enrichment of Rad21 or Nipbl on Dum locus (promoter or coding regions) or Dppa2 promoter region was detected by CHIP-PCR assay. A negative control (NC) region (20 kb upstream of Dum locus) showed no enrichment for the above factors. (M, N) Knockdown of Rad21 or Nipbl by siRNA oligos increased the expression of Dppa2 in C2C12 cells as revealed by qRT-PCR. (O) Double DNA FISH of Dum (red) and Dppa2 (green) genomic loci in C2C12 cells. White stars indicate the co-localized loci from a tetraploid cell. All PCR data were normalized to GAPDH mRNA and represent mean ± SD of three independent experiments. *P < 0.05, **P < 0.01. See also Supplementary information, Figure S4.
The above results demonstrated that Dum indeed modulates Dppa2 promoter methylation, leading us to speculate that Dum is involved in targeting or association with DNMTS. It is known that de novo enzymes Dnmt3a and Dnmt3b could form complexes with the major maintenance enzyme Dnmt1 to cooperate in establishing and maintaining genomic methylation patterns24. To test whether Dum interacts with this complex, we first performed RNA immunoprecipitation (RIP) assay using antibodies against Dnmt1, Dnmt3a or Dnmt3b. Notably, all three antibodies retrieved significant amounts of Dum RNAs, with Dnmt1 pulling down the highest level (6-fold increase compared to IgG control); as a negative control, Xist RNAs were not retrieved (Figure 5C), indicating a specific interaction between Dnmts and Dum. To strengthen this finding, we further performed RNA pull-down assay using in vitro-generated biotinylated full-length (FL) Dum transcripts. Consistently, as shown in Figure 5D, Dum transcripts pulled down substantial amounts of Dnmts. As negative controls, beads alone or GFP transcripts did not retrieve Dnmts. To further map the binding domain, a series of deletion mutants of Dum were generated and tested for the binding with Dnmts. Interestingly, deletion of the 3′ fragment (F1+2) did not affect the binding efficiency with Dnmts; further deletion of the middle domain (F1) resulted in reduced binding with Dnmt1 and 3a and loss of binding with Dnmt3b; and the middle (F2) or 3′ domain (F3) alone could not bind with any Dnmt, indicating that both the 5′ and the middle domains are required for effective binding with Dnmts. Furthermore, we performed ChIP-PCR assays to show that knockdown of Dum significantly impaired the binding of Dnmts to the above identified two CpG regions (Figure 5E and Supplementary information, Figure S4A), confirming that Dum is critical for the association of Dnmts with Dppa2 promoter. The above findings were further validated in regenerating muscles by performing in vivo RIP and ChIP assays. The association of Dnmt3a and Dnmt1 with Dum was indeed detected on Dppa2 promoter in the muscles in vivo (Figure 5F), and the occupancy of Dnmts on Dppa2 promoter was markedly decreased by intramuscular siDum treatment (Figure 5G).
Altogether, our data suggested that Dum mainly functions in cis to silence Dppa2 expression upon myogenic differentiation, which explains why ectopic expression of Dum had no detectable impact on myogenic differentiation (Supplementary information, Figure S2). To further confirm its cis acting nature, the in vitro-transcribed Dum full-length transcripts were transfected into C2C12 cells. Successful overexpression of Dum, however, did not change the expression level of Dppa2 (Figure 5H); consistently, no exogenous Dum transcripts were detected on the endogenous Dppa2 promoter by pulling down biotin-labeled Dum transcripts (Figure 5I). Interestingly, a substantial degree of complementarity was found between Dum sequence (1 285-1 414) and Dppa2 promoter (Supplementary information, Figure S4B), suggesting that the endogenous Dum probably associates with Dppa2 promoter through direct RNA:DNA interaction (Supplementary information, Figure S4C). Indeed, using Chromatin Isolation by RNA Purification (ChIRP) assay25 with both odd and even tiling oligos against Dum, we were able to specifically retrieve substantial amount of endogenous Dum transcripts from the above tested Dppa2 promoter regions 1 and 2 but not a nearby region 3 (Figure 5J and Supplementary information, Figure S4D).
To further explore how Dum orchestrates Dppa2 promoter activity over such a long distance (∼2 Mb), we thought of chromatin looping that has been demonstrated between transcriptional regulatory elements and promoters26. As evident from the literature and argued by Taberlay et al.27, the available genome-wide assays for chromatin conformation analyses are unreliable for interpreting connectivity at distances >100 kb. However, the chromatin occupancies of the cohesin complex, which facilitates enhancer-promoter looping, may infer such interactions27,28,29. Therefore, we assessed the occupancy of Rad21, a member of cohesion complex, and Nipbl, a cohesin-loading factor. We found significant enrichment of both factors on Dum locus and Dppa2 promoter (Figure 5K and 5L), suggesting that the long-range chromatin interaction may occur between Dum and Dppa2 loci. Furthermore, knockdown of Rad21 or Nipb1 by siRNA oligos led to an induction of Dppa2 expression (Figure 5M and 5N), confirming the involvement of cohesin complex in Dppa2 silencing. To further strengthen the above results, we visualized the localization of the Dum locus and Dppa2 promoter by double DNA FISH using two probes that recognize either the Dum or Dppa2 promoter. As expected, we found that these two loci were spatially colocalized in ∼60% cells examined (Figure 5O). Meanwhile, the centromere region recognized by a negative control probe showed no colocalization with Dum locus in all cells examined (Supplementary information, Figure S4E).
Discussion
In this study, we unraveled the novel functional roles of a lncRNA, Dum, in regulating skeletal muscle cell differentiation and muscle regeneration. As illustrated in Figure 6, we demonstrated that Dum promotes myogenic differentiation by silencing Dppa2 expression in cis. Mechanistically, it promotes DNA methylation of Dppa2 promoter by binding to its promoter and recruiting Dnmts.

Proposed model of Dum regulation of Dppa2 expression. The model depicts the role of the MyoD-Dum-Dppa2 regulatory axis in myogenic differentiation and regeneration. Dum expression is induced by MyoD upon myoblast differentiation. The intrachromosomal looping between Dum and Dppa2 loci juxtaposes Dum transcripts to Dppa2 promoter; subsequently, Dum interacts with and recruits Dnmts to Dppa2 promoter, leading to CpG site hypermethylation and gene silencing.
Although a large number of lncRNAs are pervasively identified from mammalian genomes, only a minority have been understood at the functional level4. The existent studies from about 100 characterized lncRNAs support a major role in epigenetic regulation through interacting with histone modifiers. Dum represents one of the few lncRNAs that modulate DNA methylation and the first identified to interact with both de novo methylation and maintenance Dnmts. Our findings suggest a novel model of silencing Dppa2 locus in which Dum recruits Dnmts through intrachromosomal looping. Dum appears to be a strong in cis regulating factor since a range of neighboring genes were all changed by Dum knockdown. However, Dppa2 seems to be the only one regulated by Dum through modulating its promoter methylation. The other genes may be controlled through other mechanisms or indirectly through other factors. It is still unclear to us whether Dum is necessary for the intrachromosomal looping formation between Dum locus and Dppa2 promoter. Most likely, it is not involved in the looping formation because ectopic expression of Dum had no effect on Dppa2 promoter repression, suggesting that Dum acts co-transcriptionally when it is in close contact with Dppa2 promoter; Dum expression is probably induced by the pre-existing looping. Further studies are needed to shed light on this aspect. This also sets Dum apart from Kcnqu1 ot1 lncRNA which functions as a molecular hinge to link the Kcnq1 promoter and KvDMR1 DNAs together and a scaffold for a long-range intrachromosomal loop30. Our results from ChIRP assay also indicated that Dum anchors to Dppa2 CpG regions through directly binding to the two regions identified, suggesting that it acts as a tethering molecule to recruit Dnmts. This can be related to the recently identified ecCEBPA which anchors DNMT1 to the locus9, suggesting that lncRNA-Dnmt interaction may be a general phenomenon. However, in the case of ecCEBPA, DNMT1 is sequestrated not targeted to the locus. Furthermore, unlike ecCEBPA which interacts with Dnmt1 only, Dum interacts with both maintenance and de novo DNMTs, ie, Dnmt1, Dnmt3a and Dnmt 3b. This suggests that Dum is possibly involved in the cooperation between the DND methyltransferases probably by functioning as a scaffolding molecule. All three Dnmts seem to bind the 1-1 167 domain of Dum; further deletion of the middle domain abolished Dnmt3b binding while retained binding to Dnmt1 and Dnmt3b, suggesting that this fragment is critical for Dnmt3b interaction. In the future it will be interesting to map the exact interacting interface even structural element of Dum with each Dnmt.
Much of our understanding of epigenetic regulatory network in skeletal myogenesis is focused on the level of chromatin and relatively less is known on the DNA methylation. Our study underscores the importance of promoter CpG methylation in skeletal myogenesis; it is also the first to show that Dppa2 is a regulator of myogenesis. Dppa2 was previously known to be highly enriched in pluripotent cells with decreased expression in differentiated cells12. Our results indicate that analogous to its function in ES cells, Dppa2 might play a role in maintenance of the undifferentiated state and proliferation of myoblast cells. Nothing was known how Dppa2 expression is downregulated in differentiated cells; our findings thus provide the first line of evidence to show DNA methylation is responsible for its silencing. This mechanism may work beyond myoblasts cells to explain how Dppa2 is downregulated during ES cell differentiation or in other cell types. In addition to its function in the myoblast differentiation, it is likely that Dum also plays a role in the other steps of satellite cell function. In vivo treatment of siDum in the injured muscles decreased the Pax7 and MyoD expression and the number of Pax7- and MyoD-positive satellite cells (Figure 3). This strongly suggested that knockdown of Dum may also impact satellite cell activation/proliferation or self-renewal capacity. Future exploration of these aspects will lead to a more comprehensive picture of how Dum functions in muscle stem cells and muscle regeneration.
Materials and Methods
Cell lines
Mouse C2C12 myoblasts and 10T1/2 cells were obtained from ATCC and cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg of streptomycin (1% Pen/Strep) at 37 °C in 5% CO2. For myogenic differentiation experiment, cells were seeded in 100 mm plates and when reaching 90% confluence they were shifted to DMEM containing 2% horse serum (HS). 10T1/2 cells were cultured in DMEM supplemented with 10% FBS and induced to myogenic differentiation after MyoD transfection by shifting to DMEM containing 2% HS. For 5-Aza treatment, C2C12 cells were treated with 10 mM 5-Aza (Zymo research, CA, USA) for 72 h. Total RNAs were then extracted for RNA analysis.
Primary myoblast isolation
Primary myoblasts were isolated from ∼1-week-old mouse muscles as described before16,17. Briefly, total hind limb muscles (3-6 mice per group) were digested with 5 mg/ml type IV collagenase (Life Technologies, Carlsbad, CA, USA) and 1.4 mg/ml dispase II (Life Technologies) for 0.5 h, and cell suspensions were filtered through 70 and 40 μm cell strainer, respectively, then pre-plated for 1 h. Non-adherent cells were centrifuged and cultured on gelatin-coated plates (Iwaki, Japan) in F10 medium (Life Technologies) supplemented with 20% FBS and basic fibroblast growth factor (bFGF) (Life Technologies, 25 ng/ml). After removing fibroblasts by pre-plating, primary myoblast cells were cultured in F10/DMEM medium (1:1) supplemented with 20% FBS and bFGF.
Cell fractionation
For fractionation assay, cytoplasmic and nuclear RNAs were extracted from C2C12 myoblasts as previously described13. Briefly, cells were harvested after trypsinization and washed with PBS twice. Cell pellet was then resuspended in RSB buffer (10 mM Tris, pH 7.4, 10 mM NaCl, 3 mM MgCl2) and incubated on ice for 3 min followed by centrifugation at 4 °C. The pellet was then resuspended in RSBG40 buffer (10 mM Tris, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 10% glycerol, 0.5% Noidet P-40, 0.5 mM dithiothretol and 100 U/ml rRNasin) followed by centrifugation. The supernatant was transferred to a new tube as cytoplasmic fraction; the pellet was resuspended in RSGB40 buffer with 1/10 volume of detergent (3.3% sodium deoxycholate and 6.6% Tween 40) followed by centrifugation. The supernatant was saved as cytoplasmic fraction. The pellet was used as nuclear fraction. RNAs were extracted from both fractions using Trizol.
Single-fiber isolation and use
Single-fiber isolation was performed as previously described31. Briefly, two of extensor digitorum longus (EDL) muscles were excised from C57BL/6 mice and digested in 1 ml of DMEM containing 500 U/ml collagenase II, 10% HS, 1% Pen/Strep at 37 °C with gentle agitation for 75 min. The digestion solution is then transferred into 20 ml of pre-warmed DMEM containing 10% HS, 1% Pen/Strep, 20 mM HEPES, pH 7.3, in HS-precoated 100 mm Petri dish. Single fibers were liberated by gently triturating the digested EDL muscles against the edge of Petri dish using a fire-polished Pasteur pipet with wide tip. Once around 100 fibers have fallen off, the dishes were placed back to incubator. Individual, healthy (non-shrinking) fibers were transferred to a new HS-coated 100 mm dish using the HS-coated P1000 tips every 15-25 min and the transfer was repeated three times to remove debris and the interstitial cells from fibers. Finally, 50 single fibers were transferred to each 35 mm dish with 1 ml of Ham's F10 medium containing 10% HS, 0.05% chick embryo extract, and cultured in suspension. Transfection was performed on the same day. 50 pmol siRNA or 2 μg plasmid is mixed with 10 μl Lipofectamine 2000 in 50 μl Opti-MEM I and incubated for 20 min before adding to myofibers and incubated at 37 °C overnight. Generally, every 24 h, 50% of the medium was replaced with Ham's F10 medium with 20% FBS. For differentiation, 24 h after transfection, 50% of the medium was replaced with DMEM containing 2% HS and incubated for 3 days. For immunofluorescence staining, fibers were fixed with 2% paraformaldehyde in medium and stained using anti-Pax7 or anti-myogenin antibodies. The number of Pax7- or myogenin-positive cells was quantified from at least 20 fibers.
DNA constructs
To generate a Dum expression plasmid, the full-length coding region of Dum was amplified and cloned into pcDNA3.1(+) vector (Invitrogen) using NheI and KpnI sites. The expression vectors for the truncated mutants were generated by amplifying the corresponding fragment and cloned into the pcDNA3.1(+) vector. For in vitro transcription, the DNA was subcloned into pBluescript vector using BamHI and Kpa1 sites. Sequences of the primers used can be found in Supplementary information, Table S1. Myogenin, MyHC and troponin luciferase reporters (MyoG-Luc, MyHC-Luc and Troponin-Luc) were used as described32. Renilla luciferase reporter was obtained from Promega and used according to the manufacturer's instructions.
Sodium bisulfite modification and genomic sequencing
Genomic DNA was modified by sodium bisulfate as described previously33,34. Genomic DNA was extracted from cultured cells, using the DNA-easy kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. 2 μg of DNA was bisulfate modified at 50 °C overnight. Bisulfate-modified DNA was purified with the Wizard DNA Clean-up system (Promega) and eluted in water. To get the bisulfate-specific genomic PCR products, 1 μl (∼50 ng) bisulfate-modified DNA was applied in a 25 reaction volume by HotStarTaq DNA Polymerase kit (Qiagen). DNA fragments were gel purified with the QIAquick Gel Extraction kit (Qiagen) and sequenced (BGI).
Double DNA FISH
Double DNA FISH was modified as described35. Position-specific probes were obtained from BAC clones of RP23-361N24 for Dppa2 and RP24-264D19 for Dum (http://www.chori.org/bacpac/). DNA probes were synthesized with nick translation kit (Abbotte, IN, USA) and spectrum green or orange dUTP for direct labeling. Mouse chromosome 16 centromeric probe labeled with FITC was purchased from ID Labs Inc (London, UK). Dum and Dppa2 DNA FISH probes were labeled with Alexa Fluor 488 (green) and 594 (red) by Nick Translation, respectively. C2C12 cells were treated with colcemid (50 ng/ml, Invitrogen) for 3 h. The trypsinized single cells were then swollen by 75 mM KCl treatment for 20 min at 37 °C and fixed by cold methanol/acetic acid (v/v, 3:1) for 5 min three times. Cells were denatured at 75 °C for 5 min in prewarmed 2× SSC and 70% deionized formamide (pH 7.0). Next, cells were hybridized overnight at 37 °C with denatured DNA probes (73 °C for 5 min). After hybridization, two washes of 10 min at 73 °C with 0.4× SSC/50% deionized formamide, pH 7.0, followed by two washes of 5 min at ambient temperature with 2× SSC/50% deionized formamide. Slides were then mounted with ProLong Gold antifade reagent with DAPI. Pictures were taken with a Zeiss microscope with 100 oil lens. Colocalization signals were analyzed in double-positive cells.
ChIP assay
ChIP assays using chromatins from C2C12 myoblasts or myotubes were performed as previously described16,36 using 5 μg of antibodies against MyoD (Santa Cruz Biotechnology, TX, USA), DNMT3A/3B (Abcam, Cambridge, UK), DNMT1 (Abcam), Rad21 (Abcam) and NIPBL (Bethyl, TX, USA) or isotype IgG (Santa Cruz Biotechnology) used as a negative control. Genomic DNA pellets were resuspended in 20 μl of water. qRT-PCR was performed with 1 μl of immunoprecipitated material with SYBR Green Master Mix (Life Technologies). Relative enrichment is calculated as the amount of amplified DNA normalized to input and relative to values obtained after normal IgG immunoprecipitation, which were set as 1. Primers used are listed in Supplementary information, Table S1.
Oligonucleotides
siRNA oligos were obtained from Ribobio (Guangzhou, China). The 19-nucleotide siRNA duplexes against mouse MyoD coding region (siRNA, 5′-GCCUGAGCAAAGUGAAUGA-3′) or coding region (siRNA, 5′-CAGCAGACGACUUCUAUGA-3′), Dum coding region (siRNA, 5′-GAATGAUCGUCCCAUGUUA-3′) or coding region (siRNA, 5′-GAAAGAGAAUCCAAGGUAA-3′) or coding region (siRNA, 5′-GAGAGAAACUGGUAGAUAU-3′), Dppa2 coding region (siRNA, 5′-GCAGAUGCCUGUCUUACAA-3′) or coding region (siRNA, 5′-CGGAGACACUCCUAUUCUA-3′), Rad21 coding region (siRNA 5′-GCAGCUUAUAAUGCCAUUA-3′) or coding region (siRNA, 5′-CCAGUACAAAGAUGACAAU-3′) or coding region (siRNA, 5′-GCGGUAUAUUAGAUGACAA-3′), NIPBL coding region (siRNA, 5′-GCAGAUGCCUGUCUUACAA-3′) or coding region (siRNA, 5′-GCAUCCGAGUCUAAUGUUU-3′) or coding region (siRNA, 5′-GCACCAAUGCUCGGAACAA-3′) and scrambled oligos were obtained from Ribobio. In each case, 50 μM oligos were used for transient transfections into cells. 2′-o-methylated and phosphorothioate-modified ASO oligo against Dum or scrambled oligo were synthesized at Ribobio. 100 nM oligo was introduced into C2C12 cells with Lipofectamine 2000 (Life Technologies).
RT-PCR and northern blotting analysis
Total RNAs from cells were extracted using TRIzol reagent (Life Technologies) according to the manufacturer's instructions and cDNAs were prepared using M-MLV (Moloney Murine Leukemia Virus) Reverse Transcriptase (Life Technologies) and Oligo (dT) 20 primer. Analysis of mRNA expression was performed with SYBR Green Master Mix (Life Technologies) as described on an ABI PRISM 7900HT Sequence Detection System (Life Technologies) using GAPDH for normalization37. For northern blotting analysis, sense RNA probe was synthesized by MAXIscript T7 kit (Ambion) with pBluescript-Dum DNA linearized with KpnI while antisense RNA probe was synthesized by MAXIscript T3 kit (Ambion) with BamHI-linearized DNA. All RNA probes were labeled by UTP [α-32P] (Perkin Elmer).
RNA FISH
In situ hybridyzation was performed following the protocol from Dr Prasanth KV38. Biotin-labeled sense and antisense RNA probes were synthesized with linearized pBluscript-Dum DNAs (KpnI/BamHI digested) and MAXIscript T7/T3 In vitro transcription kit, respectively (Invitrogen, Carlsbad, CA, USA), and Biotin RNA labeling Mix (Roche, Rotkreuz, Germany). Cells were fixed with 2%–4% freshly prepared paraformaldehyde (in 1× PBS, pH 7.2) for 15 min at room temperature (RT). The cells were permeabilized with 0.2%–0.5% Triton X-100, 2 mM VRC (NEBiolabs) on ice for 5-10 min and then washed 2 times in 2× SSC for 10 min. 200 g or more of the probe and yeast tRNA (20 μg; Sigma) were lyophilized and redissolved in 10 μl deionized formamide (Ambion) and denatured at 75–100 °C for 10 min and immediately chilled in ice for 3–5 min. 10 μl of hybridization buffer was added to each reaction to make a final hybridization cocktail of 20 μl per coverslip. Coverslips were incubated at 37 °C overnight in a humidified chamber. After hybridization, coverslips were washed in 2× SSC, 50% formamide (pH 7.2) for 3× 5 min at 42 °C, 2× SSC (pH 7.2) for 3 5 min at 42 °C, 1× SSC (pH 7.2) for 3× 5 min at 42 °C and 4× SSC for 2× 10 min at RT. After washing with 0.2× SSC at 65 °C for 1 h, Cy3-labeled streptavidin was added to cells for 1 h at RT. After washing three times with PBS, cells were then mounted with prolong Gold antifade reagent with DAPI (Invitrogen, Carlsbad, CA, USA) and images were captured with Zeiss microscope with 63× oil lens.
RNA pull-down assay
RNA pull-down and deletion mapping were performed as described39. Briefly, biotinylated RNAs were prepared using MAXIscript T7/T3 In vitro transcription kit (Life Technologies) and Biotin RNA labeling Mix. The above RNAs were denatured at 90 °C for 2 min and then denatured with RNA structure buffer (Life Technologies) at RT for 20 min. 5 × 106 C2C12 cell pellets were treated with 20% nuclear isolation buffer (1.28 M sucrose; 40 mM Tris-HCl, pH 7.5; 20 mM MgCl2; 4% Triton X-100) with 1× Complete Protease Inhibitor Cocktail (PIC, Roche). Nuclei were collected by 2 500× g centrifugation for 15 min. Nuclear pellet was resuspended in 1 ml RIP buffer (150 mM KCl, 25 mM Tris, pH 7.4, 0.5 mM DTT, 0.5% NP-40, 1 mM PMSF and 1× PIC) and sonicated with three cycles (30 s interval, 30 s sonication) using Bioruptor (Diagenode, Liege, Belgium). After centrifugation at 13 000 RPM for 10 min to remove nuclear membrane and debris, 1 mg of C2C12 nuclear extract was then mixed with 3 μg of denatured RNA, and incubated at RT for 1 h. 30 μl washed streptavidin agarose beads (Life Technologies) were added to each pull-down reaction and further incubated at RT for 1 h. Beads were pelleted and washed for five times in Handee spin columns (Pierce, IN, USA) using RIP buffer. The resulting beads were boiled in loading buffer to retrieve the proteins which were then detected by standard western blotting technique. For in vivo pull-down, the muscle tissue was collected in nuclear isolation buffer and biotin IPs were performed as above described.
RIP assay
RIP was also performed as described39. In short, C2C12 cells were crosslinked with 1% formaldehyde and collected for lysis by RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% NP-40, and 0.5% sodium deoxycholate, 0.5 mM DTT, 1 mM PMSF, 1× proteinase inhibitor cocktail and 1% RNaseOut). The lysate was incubated with specific antibodies or IgG control overnight. The RNA/protein complex was recovered with protein G Dynabeads and washed with RIPA buffer several times. After reverse crosslink with proteinase K at 45 °C for 45 min, RNA was recovered with Trizol and analyzed by RT-PCR.
RNA-biotin-based pull-down assays for the detection of RNA-targeted genomic regions
RNA pull-down assay for detecting of genomic DNA was also performed as described40. In short, biotinylated RNAs were prepared using MAXIscript T7 In vitro transcription kit (Life Technologies) and Biotin RNA labeling Mix (Roche). The resultant transcripts were DNase treated and dephosphorylated by using Antarctic phosphatase (New England BioLabs). C2C12 cells were transfected with the biotinylated RNAs (70-100 nM of final concentration) with Lipofectamine 2000 (Life Technologies). 24 h following transfection, the treated cultures were crosslinked by adding formaldehyde directly to tissue culture media to a final concentration of 1% for 10 min and stopped by the addition of glycine to a final concentration of 0.125 M. The extracted genomic DNA (200 μl eluent) is then used to detect promoter-specific transcripts with the biotin-linked pull-down assay. Genomic DNA was isolated and exposed to avidin magnetic beads. The final elutes, following the binding of the biotin linked RNAs with the avidin magnetic beads and washing, then were PCR amplified.
Chromatin isolation by RNA purification
CHIRP was performed as described25. The antisense probes (20 nucleotides in length) were designed and divided into odd and even pools. C2C12 cells were harvested and cross-linked by 1% glutaraldehyde and sonicated for 4 h by Bioruptor sonicator. Probe pool and cell lysates were then incubated at 37 °C for 4 h. Magnet beads were added to pull down probes and separated with magnet strip. RNAs isolated with Trizol were reverse transcribed into cDNA by M-MLV reverse transcriptase. RNA retrieval was calculated by the percentage of pull-down RNA over input. DNA isolated by phenol-chloroform-ethanol precipitation was used for qRT-PCR. DNA binding enrichment was represented by the percentage of the pull-down DNA over input DNA.
Immunoblotting and immunostaining
For western blot analysis, total cell extracts were prepared and used as previously described32,41,42. The following dilutions were used for each antibody: Pax 7 (Developmental Studies Hybridoma Bank; 1:2 000), MyoD (Santa Cruz Biotechnology; 1:2 000), myogenin (Santa Cruz Biotechnology; 1:2 000), troponin (Sigma; 1:2 000), MyHC (Sigma; 1:2 000), Rad21 (Abcam; 1:2 000), Nibpl (Bethyl; 1:2 000), α-tubulin (Sigma; 1:5 000), GAPDH (1:5 000; Santa Cruz Biotechnology). Immunofluorescence of cultured cells and single fibers was performed using the following antibodies: troponin (Sigma; 1:200) and MyHC (Sigma; 1:350). Frozen muscle sections were prepared and stained as previously described. Immunofluorescence on frozen muscle sections was performed using the following antibodies: Pax 7 (Developmental Studies Hybridoma Bank; 1:100), MyoD (Santa Cruz Biotechnology; 1:100), myogenin (Santa Cruz Biotechnology; 1:200) and eMyHC (Novocastra, Leica Microsystems, 1:200). For quantification, counts were performed from a minimum of 20 randomly chosen fields, from 5-6 sections throughout the length of the muscle in 4-6 per group. All fluorescent images were captured with a Zeiss microscope (Zeiss, German).
Animal studies
Mice (C57B/L6) were housed in the animal facilities of The Chinese University of Hong Kong (CUHK) under conventional conditions with constant temperature and humidity and fed a standard diet. Animal experimentation was approved by the CUHK Animal Ethics Committee. For CTX (Latoxan, Valence, France) injection, ∼7-week-old mice were injected with 50 μl of CTX (10 μΜ) into the tibialis anterior (TA) muscles. Oligos were prepared by preincubating 15 μm siRNA oligos with Lipofectamine 2000 for 15 min and injections were made in a final volume of 60 μl in OPTI-MEM (Invitrogen, NY, USA) on days 1/4, 2 and 4. Mice were sacrificed and TA muscles were harvested on days 0, 2, 3 and 6, and total RNAs and proteins were extracted for real-time RT-PCR and western blotting analyses. For immunofluorescene staining of MyoD, myogenin and Pax7, muscle sections were collected on day 3 and day 6 for eMyHC. Five mice were used in each group.
References
Fatica A, Bozzoni I . Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet 2014; 15:7–21.
Brockdorff N . Noncoding RNA and Polycomb recruitment. RNA 2013; 19:429–442.
Mercer TR, Mattick JS . Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol 2013; 20:300–307.
Ulitsky I, Bartel DP . lincRNAs: genomics, evolution, and mechanisms. Cell 2013; 154:26–46.
Buckingham M . Myogenic progenitor cells and skeletal myogenesis in vertebrates. Curr Opin Genet Dev 2006; 16:525–532.
Saccone V, Puri PL . Epigenetic regulation of skeletal myogenesis. Organogenesis 2010; 6:48–53.
Lucarelli M, Fuso A, Strom R, Scarpa S . The dynamics of myogenin site-specific demethylation is strongly correlated with its expression and with muscle differentiation. J Biol Chem 2001; 276:7500–7506.
Mohammad F, Mondal T, Guseva N, Pandey GK, Kanduri C . Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 2010; 137:2493–2499.
Di Ruscio A, Ebralidze AK, Benoukraf T, et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013; 503:371–376.
Sun K, Zhao Y, Wang H, Sun H . Sebnif: an integrated bioinformatics pipeline for the identification of novel large intergenic noncoding RNAs (lincRNAs) — application in human skeletal muscle cells. PLoS One 2014; 9:e84500.
Guttman M, Amit I, Garber M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009; 458:223–227.
Du J, Chen T, Zou X, Xiong B, Lu G . Dppa2 knockdown-induced differentiation and repressed proliferation of mouse embryonic stem cells. J Biochem 2010; 147:265–271.
Lu L, Sun K, Chen X, et al. Genome-wide survey by ChIP-seq reveals YY1 regulation of lincRNAs in skeletal myogenesis. EMBO J 2013; 32:2575–2588.
Sun K, Chen X, Jiang P, Song X, Wang H, Sun H . iSeeRNA: identification of long intergenic non-coding RNA transcripts from transcriptome sequencing data. BMC Genomics 2013; 14 Suppl 2:S7.
Cao Y, Yao Z, Sarkar D, et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell 2010; 18:662–674.
Lu L, Zhou L, Chen EZ, et al. A novel YY1-miR-1 regulatory circuit in skeletal myogenesis revealed by genome-wide prediction of YY1-miRNA network. PLoS One 2012; 7:e27596.
Wang L, Zhou L, Jiang P, et al. Loss of miR-29 in myoblasts contributes to dystrophic muscle pathogenesis. Mol Ther 2012; 20:1222–1233.
Zhou L, Wang L, Lu L, Jiang P, Sun H, Wang H . A novel target of microRNA-29, Ring1 and YY1-binding protein (Rybp), negatively regulates skeletal myogenesis. J Biol Chem 2012; 287:25255–25265.
Zhou L, Wang L, Lu L, Jiang P, Sun H, Wang H . Inhibition of miR-29 by TGF-beta-Smad3 signaling through dual mechanisms promotes transdifferentiation of mouse myoblasts into myofibroblasts. PLoS One 2012; 7:e33766.
Guil S, Esteller M . Cis-acting noncoding RNAs: friends and foes. Nat Struct Mol Biol 2012; 19:1068–1075.
Lang KC, Lin IH, Teng HF, et al. Simultaneous overexpression of Oct4 and Nanog abrogates terminal myogenesis. Am J Physiol Cell Physiol 2009; 297:C43–C54.
Ruau D, Ensenat-Waser R, Dinger TC, et al. Pluripotency associated genes are reactivated by chromatin-modifying agents in neurosphere cells. Stem Cells 2008; 26:920–926.
Hupkes M, Jonsson MK, Scheenen WJ, et al. Epigenetics: DNA demethylation promotes skeletal myotube maturation. FASEB J 2011; 25:3861–3872.
Kim G, Ni J, Kelesoglu N, Roberts R, Pradhan S . Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J 2002; 21:4183–4195.
Chu C, Qu K, Zhong FL, Artandi SE, Chang HY . Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 2011; 44:667–678.
Sanyal A, Lajoie BR, Jain G, Dekker J . The long-range interaction landscape of gene promoters. Nature 2012; 489:109–113.
Taberlay PC, Kelly TK, Liu CC, et al. Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell 2011; 147:1283–1294.
Phillips-Cremins JE, Sauria ME, Sanyal A, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 2013; 153:1281–1295.
Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010; 467:430–435.
Zhang H, Zeitz MJ, Wang H, et al. Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus. J Cell Biol 2014; 204:61–75.
Cheung TH, Quach NL, Charville GW, et al. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 2012; 482:524–528.
Wang H, Hertlein E, Bakkar N, et al. NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol Cell Biol 2007; 27:4374–4387.
Yang Y, Zhou L, Lu L, et al. A novel miR-193a-5p-YY1-APC regulatory axis in human endometrioid endometrial adenocarcinoma. Oncogene 2013; 32:3432–3442.
Zhao Y, Yang Y, Trovik J, et al. A novel wnt regulatory axis in endometrioid endometrial cancer. Cancer Res 2014; 74:5103–5117.
Xiang JF, Yin QF, Chen T, et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res 2014; 24:513–531.
Zhou L, Wang L, Lu L, Jiang P, Sun H, Wang H . A novel target of microRNA-29, Ring1 and YY1-binding protein (Rybp), negatively regulates skeletal myogenesis. J Biol Chem 2012; 287:25255–25265.
Yang Y, Zhou L, Lu L, et al. A novel miR-193a-5p-YY1-APC regulatory axis in human endometrioid endometrial adenocarcinoma. Oncogene 2013; 32:3432–3442.
Tripathi V, Ellis JD, Shen Z, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 2010; 39:925–938.
Tsai MC, Manor O, Wan Y, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010; 329:689–693.
Han J, Kim D, Morris KV . Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proc Natl Acad Sci USA 2007; 104:12422–12427.
Wang H, Garzon R, Sun H, et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 2008; 14:369–381.
Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 1999; 19:5785–5799.
Acknowledgements
We thank Dr Xiaoxing Li for helping bisulfate genomic specific sequencing study. We thank Dr Prasanth KV and Qinyu Hao for their kind help and discussion on FISH experiment. The work described in this paper was substantially supported by General Research Funds (GRF) from the Research Grants Council (RGC) of the Hong Kong Special Administrative Region, China (476310 and 476113 to HW and 473713 to HS), the 973 program of Ministry of Science and Technology of China (2014CB964700 and 2011CB965201), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA01020106).
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Dum is a long non-coding RNA. (PDF 358 kb)
Supplementary information, Figure S2
Knock-down of Dum delayed C2C12 differentiation but over-expression showed no effect. (PDF 62 kb)
Supplementary information, Figure S3
The neighboring genes of Dum. (PDF 151 kb)
Supplementary information, Figure S4
Dum binds to Dppa2 promoter. (PDF 194 kb)
Supplementary information, Table S1
Oligos/Primers used in the study. (PDF 28 kb)
Rights and permissions
About this article

Cite this article
Wang, L., Zhao, Y., Bao, X. et al. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res 25, 335–350 (2015). https://doi.org/10.1038/cr.2015.21
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2015.21
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Molecular regulatory mechanism of key LncRNAs in subclinical mastitic cows with folic acid supplementation
BMC Genomics (2023)
-
A lncRNA from an inflammatory bowel disease risk locus maintains intestinal host-commensal homeostasis
Cell Research (2023)
-
Non-coding RNA regulation of integrins and their potential as therapeutic targets in cancer
Cellular Oncology (2023)
-
ZFX-mediated upregulation of CEBPA-AS1 contributes to acute myeloid leukemia progression through miR-24-3p/CTBP2 axis
Cell Biology and Toxicology (2023)
-
DUBR suppresses migration and invasion of human lung adenocarcinoma cells via ZBTB11-mediated inhibition of oxidative phosphorylation
Acta Pharmacologica Sinica (2022)