
Abstract
The msh homeobox 1 (Msx1) and msh homeobox 2 (Msx2) coordinate in myoblast differentiation and also contribute to muscle defects if altered during development. Deciphering the downstream signaling networks of Msx1 and Msx2 in myoblast differentiation will help us to understand the molecular events that contribute to muscle defects. Here, the proteomics characteristics in Msx1- and Msx2-mediated myoblast differentiation was evaluated using isobaric tags for the relative and absolute quantification labeling technique (iTRAQ). The downstream regulatory proteins of Msx1- and Msx2-mediated differentiation were identified. Bioinformatics analysis revealed that these proteins were primarily associated with xenobiotic metabolism by cytochrome P450, fatty acid degradation, glycolysis/gluconeogenesis, arginine and proline metabolism, and apoptosis. In addition, our data show Acta1 was probably a core of the downstream regulatory networks of Msx1 and Msx2 in myoblast differentiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Skeletal muscle differentiation is a vital stage of development (Casadei et al. 2009; Muntoni et al. 2002). In early myogenesis, mesodermal cells differentiate into myogenic progenitors and, then, myoblasts. Later, myoblasts differentiate into multinucleated myotubes, which are controlled by the transcriptional regulatory system (Gredinger et al. 1998; Muntoni et al. 2002). However, a number of myoblasts are quiescent in adult muscle as satellite cells. When muscle cells are damaged by physical exercise stress or muscle lesions, satellite cells are activated and proliferate to form new myofibers, which can repair damaged muscle cells (Cooper et al. 1999; Murach et al. 2018; Schultz and McCormick 1994).
Decades of developmental research have provided a profile of skeletal muscle formation. The myogenesis progression is involved in the coordination of a set of transcription factors, including Pax3 which plays a key role in the initiation of myogenesis, and muscle regulatory factors (MRFs) consisting of Myf5, Myod1, MRF4 and myogenin (Berkes and Tapscott 2005; Pownall et al. 2002; Rudnicki et al. 1993; Tapscott 2005). In addition, homeoproteina Six1 and Six4 were recently found important in the regulation of myogenesis and muscle development (Grifone et al. 2005; Relaix et al. 2013; Wang et al. 2016). The program is also controlled by a network of signaling pathways, for example, Shh and Wnt signals are crucial for the induction of myogenesis (Munsterberg et al. 1995; Munsterberg and Lassar 1995). Hepatocyte growth factor (HGF) signaling pathway is indispensable for myoblasts migration (Andermarcher et al. 1996; Bladt et al. 1995). Insulin signaling is found to promote myoblast fusion and differentiation (Charge and Rudnicki 2004; van der Velden et al. 2006). In contrast, fibroblast growth factor (FGF) signaling pathway inhibits myoblast differentiation while promoting proliferation (Itoh et al. 1996; Milasincic et al. 1996).
Homeobox is one of the vital gene families that regulate embryogenesis (Hobert and Westphal 2000; Hu et al. 2001). The Msx homeobox (Msx) gene family contains three members, two of which (Msx1 and Msx2) express in undifferentiated precursors during development (Cornelison et al. 2000; Wang and Abate-Shen 2012). For example, Msx1 and Msx2 are both expressed at early embryonic developmental stage, mainly expressed in migrating and proliferating progenitor cells, and are downregulated upon differentiation. (Bendall et al. 1999; Houzelstein et al. 1999; Wang and Abate-Shen 2012). Forced expression of Msx1 and Msx2 in myoblasts inhibits their differentiation (Kuwajima et al. 2004; Wang et al. 2011; Zhou et al. 2021). It has been reported that Msx1 promotes C2C12 cell proliferation. As the homologous gene of Msx1, Msx2 may also play the same role as Msx1 in C2C12 cells (Kodaka et al. 2015). Although previous studies have explored how Msx1 and Msx2 inhibit the myoblast differentiation during development, the molecular mechanisms by which Msx1 and Msx2 coordinating myoblasts differentiation remain largely unknown.
Molecular characterization of the proteome is necessary for a complete understanding of biological systems. iTRAQ is widely employed, with a proven value in quantitative proteomics (Noirel et al. 2011; Wu et al. 2018). The proteomics landscape of Msx1- and Msx2-mediated inhibiting myoblasts differentiation has not been reported. Hence, we used C2C12 myoblasts to force the expression of Msx1 and Msx2, and we profiled protein expression changes correlated to the downstream signaling networks of Msx1 and Msx2, via a quantitative proteomics approach. Our analysis showed the proteomics characteristics in Msx1- and Msx2-mediated myoblasts differentiation.
Results
Proteomics Identification of Msx1- and Msx2-Mediated Myoblast Differentiation
To identify key proteins involved in the downstream signaling networks of Msx1 and Msx2 in skeletal muscle development, we firstly chose C2C12 to over-express Msx1 or Msx2 and induced them to differentiate with DMEM containing 2% horse serum (Lechner et al. 1996; Li and Johnson 2006). C2C12 is a primary line of murine myoblasts, rendering it as a popular model of skeletal muscle development (Burattini et al. 2004; Yamamoto et al. 2008). We found that both over-expression of Msx1 and Msx2 inhibited the formation of myotubes relative to the control, as evident from the appearance of myotubes (Fig. 1a). Furthermore, we conducted western blotting to measure the maker, myosin heavy chain (MHC), of terminal muscle differentiation (Orii et al. 2002; Rauch and Loughna 2005). After induced differentiation, MHC was only expressed in the C2C12 cells without the expression of Msx1 and Msx2 (Fig. 1b). Taken together, we modeled the Msx1- and Msx2-mediated myoblast differentiation.

Forced expression of Msx1 and Msx2 inhibits C2C12 cells differentiation. a Myotube formation assay. b Western blotting against MHC, Msx1 and Msx2. The scale bars represent 50 µm
We collected six groups of samples at different differentiation stages as shown in Fig. 2a, and the whole proteome analysis was performed on each replicate. After screening based on a false discovery rate of 1%, we found that a total of 8044 peptides corresponding to 1545 proteins were identified, of which 1535 proteins have quantitative information (Table S1). The reliability of quantitative information was evaluated by principal component analysis (PCA), in which the three biological replicates of each sample showed a tendency of being clustered together, indicating good reproducibility (Fig. 2b). In addition, the Pearson correlation analysis was performed on the three replicates of each sample, and the results showed that the correlation coefficient was about 0.8, indicating that there is a good correlation among different replicates (Fig. 2c).

Sample collection and the proteomics analysis a The workflow of quantitative proteomics. b The principal component analysis (PCA) of different samples. c Correlation analysis of different samples. d Vocano plot of differentially expressed proteins from our study
Msx1-Mediated Myoblast Differentiation have Distinct Protein Profiles with that of Msx2
To accurately present the differences of protein expression among samples, we conducted differential expression analysis for proteomics data between Msx over-expressed samples and the wild-type samples before and after the induced differentiation. We identified 71, 45, 27, and 38 differentially expressed proteins (DEPs) (p-value ≤ 0.05 and fold change ≥ 1.5 or ≤ 0.83), respectively (Fig. 2d, Tables S3–S5). The overlapped DEPs lists are shown in Fig. S1a. Before the induced differentiation stage, there are common 32 downstream regulatory proteins for Msx1 and Msx2, 39 specific downstream regulatory proteins for Msx1, while 13 specific proteins for Msx2 (Fig. 3b). After the induced differentiation stage, there are 17 commom downstream regulatory proteins for Msx1 and Msx2, 10 specific downstream regulatory proteins for Msx1, while 21 specific proteins for Msx2 (Fig. 3b). Heatmaps of the Msx1 and Msx2 common regulatory proteins are shown in Fig. 3d, e, respectively. Totally, the DEPs have partial overlap, and also have their specificity, which is consistent with the previous knowledge that Msx1 and Msx2 have redundancy in their biological functions (Han et al. 2007; Kodaka et al. 2015; Lallemand et al. 2005; Le Bouffant et al. 2011; Ogi et al. 2005; Zhuang et al. 2009).

Venn and Heatmap analysis of DEPs a Overlap of all DEPs. b Venn of DEPs before induced differentiation. c Venn of DEPs after induced differentiation. d Heatmap of common DEPs before induced differentiation. e Heatmap of common DEPs after induced differentiation
Functional Classification Annotation Analysis of DEPs Involved in Msx1- and Msx2-Mediated Myoblast Differentiation
To further investigate the downstream signaling networks of Msx1 and Msx2 in Myoblast differentiation, GO analysis of the DEPs were conducted. Before the induced differentiation stage, translation, protein folding, rRNA processing, etc. were enriched in Msx2-mediated differentiation (Fig. 4b, Table S7), while Msx1-mediated differentiation was involved in mRNA processing, angiogenesis, inflammatory response, cell adhesion and G protein-coupled receptor signaling pathway (Fig. 4a, Table S6). Cellular components and molecular function showed that the DEPs from Msx1- and Msx2-mediated differentiation enriched in many common terms such as extracellular exosome, membrane, cytoplasm, nucleus, zinc ion binding, GTPase activity, DNA binding, and structural constituent of ribosome (Fig. 4a, b, Table S6, S7). After the induced differentiation, Msx1-mediated differentiation was primarily associated with regulation of muscle contraction, transition between fast and slow fiber, and so on (Fig. 4c, Table S8), while regulation of transcription, apoptotic mitochondrial changes were associated with Msx2-mediated differentiation (Fig. 4d, Table S9). Similarly, cellular components were involved in the cytoplasm, extracellular exosome, membrane, mitochondrion in Msx1-mediated differentiation. Molecular functions were primarily enriched in GTPase activity, ATP binding, DNA binding, and ubiquitin-protein ligase binding in Msx2-mediated differentiation (Fig. 4c, d, Table S8, S9).

Gene ontology analysis a GO analysis of DEPs for Msx1_B compared to WT_B. b GO analysis of DEPs for Msx2_B compared to WT_B. c GO analysis of DEPs for Msx1_A compared to WT_A. d GO analysis of DEPs for Msx2_A compared to WT_A
Protein functions should be presented through pathway analysis. We mapped all DEPs from our study to the KEGG pathway database. Before the induced differentiation, the pathways are related to endocytosis, PI3K-AKt signaling pathway, glycolysis/glouconeogenesis, fatty acid degradation. While, after the induced differentiation, the pathways are related to tyrosine metabolism, adrenergic signaling in cardiomyocytes, chemical carcinogenesis elongation, systemic lupus erythematosus, tight junction, and so on (Fig. 5a, Table S10). Taken together, biological processes and KEGG enrichment analysis results were similar for Msx1 and Msx2 samples, both before and after induced differentiation, indicating that the downstream signaling networks of Msx1 and Msx2 in myoblast differentiation are highly overlapping.

KEGG analysis and PPIs networks a KEGG pathway analysis of all DEPs. b PPIs networks of DEPs for Msx1_B compared to WT_B. c PPIs networks of DEPs for Msx2_B compared to WT_B. d PPIs networks of DEPs for Msx1_A compared to WT_A. e PPIs networks of DEPs for Msx2_A compared to WT_A
PPIs Networks of DEPs Involved in Msx1- and Msx2-Mediated Myoblast Differentiation
To provide insight into the possible cellular mechanisms, comprehensive protein–protein interactions (PPIs) were used to depict numerous biological aspects of complex regulatory networks and signaling pathways. Hence, we used a multi-omics data analysis tool, OmicsBean, to construct PPI networks (Huang et al. 2020; Lu et al. 2020b; Szklarczyk et al. 2016) for DEPs. Before the induced differentiation stage, we found that the PPIs from Msx1-mediated differentiation might play important roles in amino sugar and nucleotide sugar metabolism, arginine and proline metabolism, glycolysis/gluconeogenesis (Fig. 5b, Table S11). In contrast, the highly clustered pathways for the PPIs from Msx2-mediated differentiation were related to fatty acid degradation, arginine and proline metabolism, glycolysis/gluconeogenesis (Fig. 5c, Table S12). After induced differentiation, the PPIs from Msx1-mediated differentiation might play important roles in the Metabolism of xenobiotics by cytochrome P450, while PPIs from Msx2-mediated differentiation in Apoptosis (Fig. 5d, e, Tables S13, S14). These results were similar to those found in the biological processes (Fig. 4) and KEGG enrichment (Fig. 5a) analyses for DEPs, further demonstrating that these processes and pathways may play important roles in Msx1- and Msx2-mediated signaling networks during myoblast differentiation.
Discussion
Proteins are the embodiment of biological functions. From genes to proteins, they need to undergo transcriptional regulation, translational regulation and post-translational modification regulation. Proteomics, as an important field of functional genomics, can make up for the defect that the transcriptome cannot reflect biological information (Hanash 2003; Pandey and Mann 2000). Although it has been known that myoblast differentiation is regulated by Msx1 and Msx2 (Hu et al. 2001; Lee et al. 2006; Wang et al. 2011), the molecular mechanism of Msx1 and Msx2 coordinating myoblast differentiation is still largely unknown. Based on the present study by iTRAQ labeling quantitative quantification technology, the downstream regulatory proteins of Msx1 and Msx2 in skeletal muscle development can be measured simultaneously in a single MS assay.
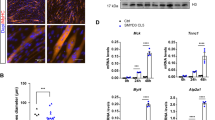
Our data show that the downstream regulatory proteins of Msx1- and Msx2-mediated inhibiting myoblasts differentiation overlap partly, and also have their specificity. Among these, in the undifferentiated stage, the higher expression of Hsp90 suggests Hsp90-dependent serine-threonine kinase may play an important role in Msx1- and Msx2-mediated myoblast differentiation (Yun and Matts 2005). During muscle development, Histone H4 plays a vital role in the methylation of SIX1 (Wei et al. 2018). Our data indicate Histone H4 has up-regulated expression in Msx1- and Msx2-mediated differentiation suggests Msx1 and Msx2 may associate with the SIX1 pathway in muscle development. In the differentiated stage, the high expression of Aqp1 suggests that Msx1- and Msx2-mediated myoblast differentiation by regulating Aqp1 expression because Aqp1 is involved in myotube Atrophy (Oelkrug et al. 2015). In addition, PPIs analysis showed that Acta1 was a highly concentrated cluster, suggesting that Acta1 was probably the core of the downstream regulatory network of Msx1 and Msx2 mediated myoblast differentiation. In addition, we conducted RT-qPCR to compare the relative transcript amounts of Acta1 and demonstrated that Acta1 is significantly down-regulated in Msx1/2 over-expressed C2C12 cells (Msx1/2_OE) compared to that in C2C12 wild types (WT) (Fig. S1). In conclusion, our study systematically showed the proteomic landscape of Msx1- and Msx2-mediated myoblast differentiation.
Methods and Materials
Cell Culture
Mouse C2C12 cells were cultured in DMEM media containing 10% fetal bovine serum (FBS) in humidified atmosphere with 5% CO2 at 37 °C. For induced C2C12 differentiation, C2C12 cells were cultured in DMEM media containing 2% horse serum for 72 h (Lee et al. 2006). Retroviral packaging pLZRS-IRES-GFP plasmids derivatives were transfected using Lipofectamine3000 reagent (Invitrogen) into Phoenix cells (ATCC) (Wang and Abate-Shen 2012).
The Overexpression of Msx1 and Msx2
We used retroviral transfer system for the overexpression of Msx1 and Msx2. Replication-defective retroviruses were produced by transfecting Phoenix retroviral packaging cells with the relevant pLZRS-IRES-GFP plasmid derivatives expressed Msx1 or Msx2 using lipofectamine 2000 reagent (Life Technologies). The packaging cells transfected were selected with 5 mg/mL puromycin. The C2C12 cells were infected using the collected viral supernatants.
Peptide Preparation and iTRAQ Reagent Labeling
Peptide Preparation was performed previously (Lu et al. 2020a). Briefly, after sonicating the cells with lysis buffer. The supernatant was reduced at 37 °C for 1 h with 10 mM DTT, and then alkylated at 25 °C with 30 mM Iodoacetamide. The protein concentration was measured by Bradford method. 200 μg proteins was taken from each sample and digested at 37 °C for 12 h with trypsin at a ratio 1–20 (trypsin to protein). 100 g peptides were labelled from each samples according to the manufacturer’s protocol (Lu et al. 2020a). 113-iTRAQ tag for C2C12 wild-type cells before induced differentiation (WT_B). 114-iTRAQ tag for C2C12 wild-type cells after induced differentiation (WT_A). 115-iTRAQ tag for the overexpression of Msx1 in C2C12 cells before induced differentiation (Msx1_B). 116-iTRAQ tag for the overexpression of Msx1 in C2C12 cells after induced differentiation (Msx1_B). 117-iTRAQ tag for the overexpression of Msx2 in C2C12 cells before induced differentiation (Msx2_B). 118-iTRAQ tag for the overexpression of Msx2 in C2C12 before induced differentiation (Msx2_A).
LC–MS/MS and Proteomic Data Analysis
Mass Spectrometry Analysis was first conducted in a nanoflow EASY-nLC 1000 system (Thermo Fisher Scientific) as previously reported (Lu et al. 2020a). Briefly, the peptides were separated using a prepMap100 C18 Column and then examined on an PrepMap RSLC C18 Column. For MS/MS analysis, 15 of the most intense peptide ions (> 5000 counts) were fragmented using HCD fragmentation.
The proteomics data were processed in Mascot search engine (Matrix Science, London, UK) (Lu et al. 2020b) based on the mouse database. Peptides scored by Mascot software greater than the 99% confidence interval are considered to be successfully identified. When C2C12 differentiated, the ratio of proteins were acquired from the comparison of WT_A/WT_B, while C2C12 cells undifferentiated, the ratio of proteins were acquired from the comparison of Msx1_A/Msx1_B, Msx2_A/Msx2_B, among which the ratios that more than 1.5 or less than 0.83 were considered as the differential expression of protein.
Bioinformatics Analysis
Gene ontology information of the differentially expressed protein was obtained from DAVID database (Dennis et al. 2003; Lu et al. 2020b) (http://david.abcc.ncifcrf.gov/) and processed using GraphPad Prism 8 software. Pathway analysis with the differentially expressed protein was performed using KEGG database (https://www.kegg.jp/). PPIs networks were conducted on omicsbean platform (www.omicsbean.cn). The differentially expressed proteins (DEPs) were analyzed using fold change method. The proteins with p-value ≤ 0.05 and fold change ≥ 1.5 were defined as up-regulated proteins, while the proteins with p-value ≤ 0.05 and fold change ≤ 0.83 were defined as down-regulated proteins.
RT-qPCR
The C2C12 wild types and Msx1/2 over-expressed C2C12 cells were homogenized using TRIzol (Life Technologies). RNA was extracted using RNeasy Mini Kit (Qiagen). Reverse transcription was performed using HiScript III RT SuperMix for qPCR (+ gDNA wiper) (Vazyme). RT-qPCR reactions were set up using SYBR® Green Realtime PCR Master Mix (TOYOBO). The Ct values for genes of interest were normalized to GAPDH, and expressions of genes are represented as 2−[∆Ct] or 2−[∆∆Ct] for fold change over control condition. All primers used for qRT-PCR are listed in Table S15.
Availability of Data and Material
Data are available via ProteomeXchange with identifier PXD026944.
References
Andermarcher E, Surani MA, Gherardi E (1996) Co-expression of the HGF/SF and c-met genes during early mouse embryogenesis precedes reciprocal expression in adjacent tissues during organogenesis. Dev Genet 18:254–266. https://doi.org/10.1002/(SICI)1520-6408(1996)18:3
Bendall AJ, Ding J, Hu G, Shen MM, Abate-Shen C (1999) Msx1 antagonizes the myogenic activity of Pax3 in migrating limb muscle precursors. Development 126:4965–4976. https://doi.org/10.1242/dev.126.22.4965
Berkes CA, Tapscott SJ (2005) MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol 16:585–5951. https://doi.org/10.1016/j.semcdb.2005.07.006
Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C (1995) Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 376:768. https://doi.org/10.1038/376768a0
Burattini S, Ferri P, Battistelli M, Curci R, Luchetti F, Falcieri E (2004) C2C12 murine myoblasts as a model of skeletal muscle development: morpho-functional characterization. Eur J Histochem 223–234:1
Casadei L, Vallorani L, Gioacchini AM, Guescini M, Burattini S, D’Emilio A, Biagiotti L, Falcieri E, Stocchi V (2009) Proteomics-based investigation in C2C12 myoblast differentiation. Eur J Histochem EJH 53:1. https://doi.org/10.4081/ejh.2009.e31
Charge SB, Rudnicki MA (2004) Cellular and molecular regulation of muscle regeneration. Physiol Rev 84:209–238. https://doi.org/10.1152/physrev.00019.2003.10.1152/physrev.00019.2003
Cooper R, Tajbakhsh S, Mouly V, Cossu G, Buckingham M, Butler-Browne G (1999) In vivo satellite cell activation via Myf5 and MyoD in regenerating mouse skeletal muscle. J Cell Sci 112:2895–2901. https://doi.org/10.1242/jcs.112.17.2895
Cornelison D, Olwin BB, Rudnicki MA, Wold BJ (2000) MyoD−/− satellite cells in single-fiber culture are differentiation defective and MRF4 deficient. Dev Biol 224:122–137. https://doi.org/10.1006/dbio.2000.9682
Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA (2003) DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 4:1–11. https://doi.org/10.1186/gb-2003-4-9-r60
Gredinger E, Gerber AN, Tamir Y, Tapscott SJ, Bengal E (1998) Mitogen-activated protein kinase pathway is involved in the differentiation of muscle cells. J Biol Chem 273:10436–10444. https://doi.org/10.1074/jbc.273.17.10436
Grifone R, Demignon J, Houbron C, Souil E, Niro C, Seller MJ, Hamard G, Maire P (2005) Six1 and Six4 homeoproteins are required for Pax3 and Mrf expression during myogenesis in the mouse embryo. Development 132:2235–2249. https://doi.org/10.1242/dev.01773.10.1242/dev.01773
Han J, Ishii M, Bringas P Jr, Maas RL, Maxson RE Jr, Chai Y (2007) Concerted action of Msx1 and Msx2 in regulating cranial neural crest cell differentiation during frontal bone development. Mech Dev 124:729–745. https://doi.org/10.1016/j.mod.2007.06.006
Hanash S (2003) Disease proteomics. Nature 422:226–232. https://doi.org/10.1038/nature01514
Hobert O, Westphal H (2000) Functions of LIM-homeobox genes. Trends Genet 16:75–83. https://doi.org/10.1016/S0168-9525(99)01883-1
Houzelstein D, Auda-Boucher G, Chéraud Y, Rouaud T, Blanc I, Tajbakhsh S, Buckingham ME, Fontaine-Pérus J, Robert B (1999) The homeobox gene Msx1 is expressed in a subset of somites, and in muscle progenitor cells migrating into the forelimb. Development 126:2689–2701. https://doi.org/10.1242/dev.126.12.2689
Hu G, Lee H, Price SM, Shen MM, Abate-Shen C (2001) Msx homeobox genes inhibit differentiation through upregulation of cyclin D1. Development 128:2373–2384. https://doi.org/10.1242/dev.128.12.2373
Huang J, Wu Z, Zhang X (2020) Short-term mild temperature-stress-induced alterations in the C. elegans phosphoproteome. Int J Mol Sci 21:6409. https://doi.org/10.3390/ijms21176409
Itoh N, Mima T, Mikawa T (1996) Loss of fibroblast growth factor receptors is necessary for terminal differentiation of embryonic limb muscle. Development 122:291–300. https://doi.org/10.1242/dev.122.1.291
Kodaka Y, Tanaka K, Kitajima K, Tanegashima K, Matsuda R, Hara T (2015) LIM homeobox transcription factor Lhx2 inhibits skeletal muscle differentiation in part via transcriptional activation of Msx1 and Msx2. Exp Cell Res 331:309–319. https://doi.org/10.1016/j.yexcr.2014.11.009
Kuwajima T, Taniura H, Nishimura I, Yoshikawa K (2004) Necdin interacts with the Msx2 homeodomain protein via MAGE-D1 to promote myogenic differentiation of C2C12 cells. J Biol Chem 279:40484–40493. https://doi.org/10.1074/jbc.M404143200
Lallemand Y, Nicola M-A, Ramos C, Bach A, Saint Cloment C, Robert B (2005) Analysis of Msx1; Msx2 double mutants reveals multiple roles for Msx genes in limb development. Development 132:3003–3014. https://doi.org/10.1242/dev.01877
Le Bouffant R, Souquet B, Duval N, Duquenne C, Hervé R, Frydman N, Robert B, Habert R, Livera G (2011) Msx1 and Msx2 promote meiosis initiation. Development 138:5393–5402. https://doi.org/10.1242/dev.068452
Lechner C, Zahalka MA, Giot J-F, Møller N, Ullrich A (1996) ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. Proc Natl Acad Sci 93:4355–4359. https://doi.org/10.1073/pnas.93.9.4355
Lee H, Quinn JC, Prasanth KV, Swiss VA, Economides KD, Camacho MM, Spector DL, Abate-Shen C (2006) PIAS1 confers DNA-binding specificity on the Msx1 homeoprotein. Genes Dev 20:784–794. https://doi.org/10.1101/gad.1392006
Li J, Johnson SE (2006) ERK2 is required for efficient terminal differentiation of skeletal myoblasts. Biochem Biophys Res Commun 345:1425–1433. https://doi.org/10.1016/j.bbrc.2006.05.051
Lu J, Fu Y, Li M, Wang S, Wang J, Yang Q, Ye J, Zhang X, Ma H, Chang F (2020a) Global quantitative proteomics studies revealed tissue-preferential expression and phosphorylation of regulatory proteins in Arabidopsis. Int J Mol Sci 21:6116. https://doi.org/10.3390/ijms21176116
Lu J, Fu Y, Li M, Wang S, Wang J, Yang Q, Ye J, Zhang X, Ma H, Chang F (2020b) Global quantitative proteomics studies revealed tissue-preferential expression and phosphorylation of regulatory proteins in Arabidopsis. Int J Mol Sci 21:1. https://doi.org/10.3390/ijms21176116
Milasincic DJ, Calera MR, Farmer SR, Pilch PF (1996) Stimulation of C2C12 myoblast growth by basic fibroblast growth factor and insulin-like growth factor 1 can occur via mitogen-activated protein kinase-dependent and -independent pathways. Mol Cell Biol 16:5964–5973. https://doi.org/10.1128/mcb.16.11.5964
Munsterberg AE, Kitajewski J, Bumcrot DA, McMahon AP, Lassar AB (1995) Combinatorial signaling by Sonic hedgehog and Wnt family members induces myogenic bHLH gene expression in the somite. Genes Dev 9:2911–2922. https://doi.org/10.1101/gad.9.23.2911.10.1101/gad.9.23.2911
Munsterberg AE, Lassar AB (1995) Combinatorial signals from the neural tube, floor plate and notochord induce myogenic bHLH gene expression in the somite. Development 121:651–660. https://doi.org/10.1101/gad.9.23.2911
Muntoni F, Brown S, Sewry C, Patel K (2002) Muscle development genes: their relevance in neuromuscular disorders. Neuromuscul Disord 12:438–446. https://doi.org/10.1016/s0960-8966(01)00326-1
Murach KA, Englund DA, Dupont-Versteegden EE, McCarthy JJ, Peterson CA (2018) Myonuclear domain flexibility challenges rigid assumptions on satellite cell contribution to skeletal muscle fiber hypertrophy. Front Physiol 9:635. https://doi.org/10.3389/fphys.2018.00635
Noirel J, Evans C, Salim M, Mukherjee J, Yen Ow S, Pandhal J, Khoa Pham T, Biggs A (2011) Methods in quantitative proteomics: setting iTRAQ on the right track. Curr Proteomics 8:17–30. https://doi.org/10.2174/157016411794697408
Oelkrug C, Horn K, Makert GR, Schubert A (2015) Novel in vitro platform to investigate myotube atrophy. Anticancer Res 35:2085–2091
Ogi H, Suzuki K, Ogino Y, Kamimura M, Miyado M, Ying X, Zhang Z, Shinohara M, Chen Y, Yamada G (2005) Ventral abdominal wall dysmorphogenesis of Msx1/Msx2 double-mutant mice. Anat Record Discov Mol Cell Evolut Biol 284:424–430. https://doi.org/10.1002/ar.a.20180
Orii H, Ito H, Watanabe K (2002) Anatomy of the planarian Dugesia japonica I. The muscular system revealed by antisera against myosin heavy chains. Zool Sci 19:1123–1131. https://doi.org/10.2108/zsj.19.1123
Pandey A, Mann M (2000) Proteomics to study genes and genomes. Nature 405:837–846. https://doi.org/10.1038/35015709
Pownall ME, Gustafsson MK, Emerson CP Jr (2002) Myogenic regulatory factors and the specification of muscle progenitors in vertebrate embryos. Annu Rev Cell Dev Biol 18:747–783. https://doi.org/10.1146/annurev.cellbio.18.012502.105758
Rauch C, Loughna PT (2005) Static stretch promotes MEF2A nuclear translocation and expression of neonatal myosin heavy chain in C2C12 myocytes in a calcineurin-and p38-dependent manner. Am J Physiol Cell Physiol 288:C593–C605. https://doi.org/10.1152/ajpcell.00346.2004
Relaix F, Demignon J, Laclef C, Pujol J, Santolini M, Niro C, Lagha M, Rocancourt D, Buckingham M, Maire P (2013) Six homeoproteins directly activate myod expression in the gene regulatory networks that control early myogenesis. Plos Genet 9:e1003425. https://doi.org/10.1371/journal.pgen.1003425
Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R (1993) MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 75:1351–135910. https://doi.org/10.1016/0092-8674(93)90621-v
Schultz E, McCormick KM (1994) Skeletal muscle satellite cells. Rev Physiol Biochem Pharmacol 123:213–257. https://doi.org/10.1007/BFb0030904
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P (2016) The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw937
Tapscott SJ (2005) The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 132:2685–2695. https://doi.org/10.1242/dev.01874
van der Velden JLJ, Langen RCJ, Kelders MCJM, Wouters EFM, Janssen-Heininger YMW, Schols AMWJ (2006) Inhibition of glycogen synthase kinase-3 beta activity is sufficient to stimulate myogenic differentiation. Am J Physiol-Cell Ph 290:C453–C462. https://doi.org/10.1152/ajpcell.00068.2005
Wang HH, Li XX, Liu HH, Sun LL, Zhang RP, Li L, Wangding MC, Wang JW (2016) Six1 induces protein synthesis signaling expression in duck myoblasts mainly via up-regulation of mTOR. Genet Mol Biol 39:151–161. https://doi.org/10.1590/1678-4685-Gmb-2015-0075
Wang J, Abate-Shen C (2012) The MSX1 homeoprotein recruits G9a methyltransferase to repressed target genes in myoblast cells. PLoS ONE 7:e37647. https://doi.org/10.1371/journal.pone.0037647
Wang J, Kumar RM, Biggs VJ, Lee H, Chen Y, Kagey MH, Young RA, Abate-Shen C (2011) The Msx1 homeoprotein recruits polycomb to the nuclear periphery during development. Dev Cell 21:575–588. https://doi.org/10.1016/j.devcel.2011.07.003
Wei D, Li A, Zhao C, Wang H, Mei C, Khan R, Zan L (2018) Transcriptional regulation by CpG sites methylation in the core promoter region of the bovine SIX1 gene: roles of histone H4 and E2F2. Int J Mol Sci 19:213. https://doi.org/10.3390/ijms19010213
Wu S, Li J, Jin X (2018) iTRAQ-based quantitative proteomic analysis reveals important metabolic pathways for arsenic-induced liver fibrosis in rats. Sci Rep 8:1–9. https://doi.org/10.1038/s41598-018-21580-x
Yamamoto DL, Csikasz RI, Li Y, Sharma G, Hjort K, Karlsson R, Bengtsson T (2008) Myotube formation on micro-patterned glass: intracellular organization and protein distribution in C2C12 skeletal muscle cells. J Histochem Cytochem 56:881–892. https://doi.org/10.1369/jhc.2008.951228
Yun B-G, Matts RL (2005) Hsp90 functions to balance the phosphorylation state of Akt during C2C12 myoblast differentiation. Cell Signal 17:1477–1485. https://doi.org/10.1016/j.cellsig.2005.03.006
Zhou G, Yang Y, Zhang X, Wang J (2021) Msx1 cooperates with Runx1 for inhibiting myoblast differentiation. Protein Expr Purif 179:105797. https://doi.org/10.1016/j.pep.2020.105797
Zhuang F, Nguyen MP, Shuler C, Liu Y-H (2009) Analysis of Msx1 and Msx2 transactivation function in the context of the heat shock 70 (Hspa1b) gene promoter. Biochem Biophys Res Commun 381:241–246. https://doi.org/10.1016/j.bbrc.2009.02.016
Funding
This work was supported by the grant from the National Natural Science Foundation Grant of China to Jingqiang Wang (31972885).
Author information
Authors and Affiliations
Contributions
GZ: methodology, investigation, formal analysis, visualization, data curation, and original draft. SM: methodology and resources. MY: methodology, and data curation. YY: conceptualization, project administration, and reviewing and editing of the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
43657_2022_49_MOESM1_ESM.tif
Fig. S1 RT-qPCR assay to valuate Acta1 expression in Msx1/2 over-expressed myoblast cells. Values are the means ± SD. ***p < 0.001 from unpaired student t tests. (TIF 153 kb)
Rights and permissions
About this article

Cite this article
Zhou, G., Ma, S., Yang, M. et al. Global Quantitative Proteomics Analysis Reveals the Downstream Signaling Networks of Msx1 and Msx2 in Myoblast Differentiation. Phenomics 2, 201–210 (2022). https://doi.org/10.1007/s43657-022-00049-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43657-022-00049-y