
Abstract
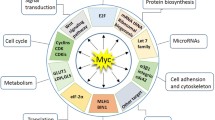
The universally active transcription factor c-Myc is essential for the regulation of global gene expression and has an impact on many biological functions, including cell division, proliferation, and death. Approximately 70% of human malignancies are caused by dysregulation of c-Myc, contributing to tumor initiation and maintenance. As a result, the therapeutic targeting of c-Myc has attracted considerable interest in the development of cancer drugs. Extensive in vivo studies have demonstrated that inhibition of c-Myc leads to substantial anti-proliferative effects and sustained tumor regression, while remaining reversible in healthy tissues. Despite its pivotal role in cancer progression, the lack of druggable binding pockets and complex protein–protein interaction (PPI) interfaces has traditionally deemed c-Myc as an “undruggable” target. Nevertheless, alternative strategies, such as disrupting the Myc/Max complex, inhibiting Myc transcription and/or translation, destabilizing Myc protein, and exploring synthetic lethality associated with Myc overexpression, have been explored to achieve desirable anti-tumor effects. This review provides a comprehensive overview of recent advancements in targeting oncogenic c-Myc, specifically focusing on its potential as a therapeutic target for cancer treatment. We discuss the underlying mechanisms of c-Myc dysregulation, its impact on cellular pathways, and the challenges associated with developing effective pharmacological inhibitors. Furthermore, we summarize emerging strategies and technologies that have shown promise in tackling the complex network of c-Myc interactions, aiming to develop conceptually innovative and efficient anticancer therapies that can be applied to a wide range of tumors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cancer, a phenomenon of fast formation of aberrant cells that develop outside of their defined borders, often invading other organs, is a lethal disease known to humankind. After ischemic heart disease, it is the second most common reason for mortality. The incidence of cancer is likely to reach 21.3 million by 2030 with about 13 million deaths (World Health Organization 2016). In developing countries alone, there is a 57% rise in cancer patients and 65% cancer-related deaths (World Health Organization 2016). According to the estimates of The American cancer society, the projected cases in 2022 itself is around 1,918,030 new cases with 609,360 cancer death concerning 350 deaths per day from lung cancer (Siegel et al. 2022). Cancer is a multistage process that starts with precancerous lesions and progresses to malignancy due to genetic factors or mutations in the individual or exposure to physical carcinogens like ultraviolet and ionizing radiations from sunlight and x-rays respectively, chemical carcinogens like benzene, beryllium, asbestos, vinyl chloride, tobacco smoke, alcohol, aflatoxin (a food contaminant), and arsenic (a drinking water contaminant), or biological carcinogens like Epstein-Barr virus, Human Papillomavirus, Hepatitis B virus that are responsible for liver and cervical cancer cases. (World Health Organization 2022).
Usually, a number of things contribute to onset cancer. As a result of cumulative or sequential DNA damage, many proto-oncogenes, including c-Myc [4] are activated and tumor suppressor oncogene genes are inactivated, altering the DNA repair mechanism and regulating apoptosis. Cell transformation may eventually result from the accumulation of DNA damage (Chen et al. 2014). Myc, Myc-N, and Myc-L are the three proto-oncogenes that constitutes the Myc family. Tumor therapy to focuses on the expression or activity of these proteins. In many human tumors, the expression of Myc proteins is up-regulated and down-regulated (Lorentzen et al. 2011; Kit et al. 2017). Although the exact number is unknown, majority of human tumors have unregulated expression of either Myc, -L, or -N (Wolf and Eilers 2020). The Myc paralogs enforce various altered phenotypes in some organisms, indicating that there are functionally significant distinctions(Kawauchi et al. 2012). A range of mechanisms can contribute to enhanced Myc expression in certain tumors, including changes to the Myc genes themselves, such as translocations that connects potent enhancers to the Myc coding sequence or amplifying factors of Myc family genes. The sequencing of human tumors and genomes has confirmed that approximately 70% of tumors have increased Myc expression (Vickers 2017). Tumorigenesis and sustained tumor growth are some more causes for aberrant c-Myc expression.
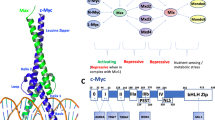
To be carcinogenic, Myc must dimerize with the widely expressed basic helix-loop-helix leucine zipper protein Max (see Fig. 1). This requirement for dimerization may allow for the regulation of Myc activity through the use of small compounds that impede Myc/Max dimerization (Berg et al. 2002). Several variables influence the formation of the Myc-Max heterodimer, including post-translational changes of Myc and Max, as well as the availability of their binding partners. Myc and Max dimerization is required for transcriptional activity because it allows them to bind to DNA and recruit co-activators or co-repressors to regulate gene expression (Blackwood and Eisenman 1991).

Structure of 1NKP, human MYC-MAX heterodimer. PDB DOI: https://doi.org/10.2210/pdb1NKP/pdb NDB: PD0386. Literature Reference (Nair and Burley 2003)
Interference with Myc-Max dimerization has been the main method for finding possible Myc inhibitors (Berg et al. 2002). Since there are no clear binding sites for small molecules on this surface, it is commonly regarded as “undruggable”. The dimerization of Myc with Max can nevertheless be fully stopped by a single amino acid alteration despite the high interaction surface (Nair and Burley 2003). This discovery supports the general idea that interaction disruption can be accomplished by attaching a high-affinity ligand to a portion of the interaction surface. Small compounds intended to target the Myc-Max interface were the first inhibitors of Myc-Max dimerization. The most effective of these could prevent Myc-Max dimerization and Myc-induced oncogenic cell transformation (Yin et al. 2003). Most malignancies have c-Myc deregulation, underscoring the importance of c-Myc/MAX heterodimerization as a cancer therapeutic target. c-Myc and MAX heterodimerization permits c-Myc to bind to the E-box sequence and carry out its regulatory role. The c-Myc/MAX heterodimerization is important in cancer cell metabolism. It affects the expression of genes involved in glucose metabolism, such as GLUT1 and LDHA, which are essential for cancer cell proliferation and survival evolution. Furthermore, the c-Myc/MAX heterodimer regulates genes involved in glycolysis, glutaminolysis, and nucleotide synthesis (Singh et al. 2022). Myc targeting tactics include focusing on it at all functional levels as well as its protein–protein interactors. Some of these approaches have resulted in experimental inhibitors that are now in clinical trials (Ponzielli et al. 2005). Nonetheless, despite more than three decades of R&D, direct suppression of Myc-Max interactions and binding to chromatin, with the goal of obstructing downstream gene expression typical of Myc-dependent tumors, has proven ineffectual to date (Blackwood and Eisenman 1991).
Structure of Myc: cause of dysregulation, impact on cellular pathways
There are three identified members of the Myc oncogene family: Myc c-, N-, and L-. The identification of the c-Myc gene initially stemmed from its resemblance to an avian retroviral transforming gene, v-Myc (Sheiness and Bishop 1979). In the case of human neuroblastomas, amplified N-Myc genes were discovered based on their similarity to the c-MYC gene (Kohl et al. 1983). c-Myc and N-Myc genes possess a three-exon structure, with exons 2 and 3 containing the primary coding domain(Battey et al. 1983). These genes encode nuclear phosphoproteins of comparable size that include highly conserved clusters of amino acids that are believed to play important roles in BHLH/LZ (basic helix-loop-helix leucine zipper), nucleic acid binding, targeting, and in vitro transforming activities. (Adams et al. 1985; DePinho et al. 1986; Land et al. 1983; J. Stone et al. 1987). The initial identification of L-Myc occurred in a specific group of human small cell lung carcinomas (SCLC), where it was observed to be amplified. Independently, L-Myc was also isolated from normal murine and human genomes due to its similarity to N-Myc. When a partial nucleotide sequence analysis was conducted on the human L-Myc gene, it revealed two distinct segments of nucleotide sequence that exhibited significant similarity to conserved sequences found in both the N-Myc and c-Myc genes. This discovery prompted the suggestion that L-Myc could be a new member of the Myc gene family. (Nau et al. 1985; Zimmerman et al. 1986). Morphological evolution is frequently linked to genetic alterations in the control of cell proliferation and differentiation patterns (Atchley and Hall 1991). Early in vertebrate evolution, a gene duplication created the c-Myc lineage and another lineage, which eventually gave rise to the N- and L-Myc lineages by another gene duplication. Myc gene family evolutionary divergence conforms closely to the known branching order of the major vertebrate groupings (Atchley and Fitch 1995). The mosaic evolution of the myc gene family is characterized by variable rates of sequence divergence among conserved motifs (Prendergast and Ziff 1992). Max, a closely comparable dimerization partner protein, has considerably less sequence variability than Myc, indicating that natural selection is at work to preserve dimerization capability with Myc and related genes (Atchley and Fitch 1995). The Myc gene family members differ in terms of tissue and developmental stage expression, relative oncogenicity, and retroviral interactions (Prendergast and Ziff 1992). The Myc gene family is functionally similar in that it has distinct transcriptional activation and DNA-binding domains (Kato et al. 1990). The Myc gene family’s function is drastically altered when conserved motifs are deleted.
The human c-Myc gene is found on chromosome 8 q24.1 (see Fig. 2). Multiple promoters are used in the regulation of c-Myc transcription. Pu27, also known as nuclease hypersensitivity element III1 (NHE III1), is a critical regulatory element that controls 80–90% of the gene’s transcriptional activity (Islam et al. 2014). This G-rich region has a length of 27 base pairs and is located -142 to -115 base pairs proximal of the P1 promoter (see Fig. 3). It has a double helix shape that is transcriptionally active. Furthermore, the G-rich strand has the capability to create an intramolecular G-quadruplex arrangement consisting of repeating sequences that encompasses three or four guanine residues. This G-quadruplex structure suppresses c-Myc transcription, resulting in its silence (Cashman et al. 2008). As a result, addressing this region may offer promise for reducing c-Myc elevated expression in neoplastic cells, inducing significant DNA damage is caused in both telomeric and nontelomeric areas of the genome, resulting in chromosomal abnormalities and telomeric DNA loss (Islam et al. 2014). Pu-27 creates a stable G-quadruplex structure, which adds to its anti-growth actions. Interfering with c-Myc expression is still a feasible option for suppressing telomere replication, and Pu-27 has been demonstrated to interfere with c-Myc transcription. The precise method by which Pu-27 causes c-Myc promoter breakdown is unknown, however there is evidence that c-Myc plays a role in chromosomal rearrangement and remodeling via the telomere (Louis et al. 2005). It suppresses the expression of telomeric shelterin proteins, DNA damage response mediators, and G2 checkpoint regulators while having no effect on DNA repair molecules or telomere maintenance genes (Palm and de Lange 2008). Therefore, targeting G-quadruplex structure formed by Pu-27 maybe of potential therapeutic approach ro reduce c-Myc expression in neoplastic cells.

A Functional domains and interactors in the MYC gene and protein structure (top). MYC locus (middle), MYC gene organisation (bottom). The primary MYC protein product, which has 439 amino acids, is displayed. Ref. paper (Carabet et al. 2018), B Madej T, Lanczycki CJ, Zhang D, Thiessen PA, Geer RC, Marchler-Bauer A, Bryant SH. “MMDB and VAST + : tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014 Jan; 42 (Database issue): D297-303

Exon and promoter locations are depicted in the diagram with arrows. The nuclease hypersensitivity element III1 (NHE III1) is a segment in blend of transcription activators with DNA within the P1 promoter’s upstream region. Ref. paper (Chen et al. 2014)
The nuclear phosphoprotein c-Myc, which weighs 65 kDa, is an element of the BHLH/LZ protein family (Cashman et al. 2008). It performs a crucial function in governing cell differentiation, apoptosis, metabolism, and proliferation. Dysregulation of c-Myc is a highly common anomaly frequently observed in cancer cases (Soucek and Evan 2002). The N-terminal transactivation domain (NTD), a central region, and a C-terminal domain (CTD) are some of the various domains that make up the 439 amino acid (aa) long c-Myc protein. The N-terminal domain encompasses the transcription activation domain (TAD) along with three segments labeled as Myc box (MB)-I, II, and III, each spanning approximately 20 amino acids (aa) in length. (Sakamuro and Prendergast 1999; Delmore et al. 2011). These regions, namely MBI and MBII at amino acids 45–63 and 129–143, respectively, are necessary for the protein’s biological functions, particularly in the processes of transcriptional regulation and transformation. MBI is required for MYC-mediated transcription regulation and transformation. It participates in the activation of target genes and is required for MYC-induced primary fibroblast transformation in collaboration with activated RAS. Furthermore, MBI is required for effective MYC-induced apoptosis irrespective of the p53 pathway. These findings emphasize the significance of MBI in MYC’s numerous biological activities, such as cellular proliferation, cancer, and apoptosis (Zhang et al. 2017). MBII is necessary for all transformations. MBII is required for MYC-dependent tumor start because it promotes connections with acetyltransferase-containing complexes, allowing histone acetylation. Co-expression of the non-transforming MB0 and MBII deletion proteins restores MYC’s complete transforming activity, demonstrating that MBII confers a different molecular function essential for oncogenic MYC activity (Kalkat et al. 2018). On the other hand, the C-terminal domain, which extends from amino acids 360 to 437, is crucial for interacting with proteins that are related with b/HLH/LZ, including Myc-associated factor X (Max). Proliferation, transformation, and apoptosis are all influenced by the formation of a heterodimer between c-Myc and Max (Greenwood 2002). The HLH/LZ domain of a c-Myc protein allows it to create a heterodimer with an additional transcription factor, Max (see Fig. 4). The c-Myc/Max complex subsequently attaches to a particular DNA recognition sequence known as the E-box element, which features a central motif of CAC(G/A) TG. Because they are c-Myc targets, the c-Myc-Max complex may transactivate or inhibit genes that include this Myc E-box motif in their regulatory domains.

Human c-Myc protein domain structure. The domains of transactivation are depicted in grey. Black represents MYC boxes I and II (MBI and MBII). Light grey and checkered patterns represent DNA-binding domains, the basic region (BR), and the helix-loop-helix (HLH) domain, respectively. A black box with white dots represents the dimerization domain of the leucine zipper (LZ). The positions of the amino acids are indicated below the protein. Exon 2 contains the information for the transactivation domain, whereas exon 3 encodes the instruction for the DNA-binding and dimerization domains. Ref. paper (Dudley et al. 2002)
c-Myc has a binary role in tumor development, operating as both a suppressor and a maintainer of tumors (Cartwright et al. 2005). The chief purpose of c-Myc involves stimulating cell growth and impeding the process of cell differentiation. It regulates the cellular metamorphosis from the G1 phase to the subsequent stages of the cell cycle, as well as the function of cyclins and cyclin-dependent kinases (Hermeking et al. 2000; Fernandez et al. 2003). c-Myc can repress cyclin-dependent kinase inhibitors and interacts with transcription factors like MIZ-1 (Staller et al. 2001). Coordinated signaling pathways involving NF-κB and HIFs are crucial for controlling cellular proliferation and differentiation. c-Myc can activate NF-κB, leading to increased cell proliferation, but it can also inhibit transactivation of NF-κB and increased receptiveness to TNF-induced apoptosis [37]. HIFs can influence cell cycle succession and the expression of proteins like cyclin D1, p21, and p27. HIF-1α and HIF-2α are the two components of HIF, have contrasting impacts on cell cycle regulation by interacting with c-Myc (Gordan et al. 2007). MYC has been shown to boost HIF1a activity at the chromatin level. It facilitates RNA polymerase II pausing release by promoting chromatin opening. MYC binding is essential for transcriptional activators or repressors to be recruited to the promoters of HIF1 target genes. In response to hypoxia, HIF1 and HIF2 restrict mitochondrial biogenesis. HIF1 stimulates FOXO3a transcription and activity, which represses a collection of nuclear-encoded mitochondrial genes by directly antagonizing MYC on gene promoters. HIF1 also inhibits PGC1b transcription by antagonizing MYC. Hypoxia-induced suppression of PGC1b and TFAM in human pulmonary endothelial cells is mediated by both HIF1 and HIF2 (Yanping Li et al. 2020). c-Myc dysregulation is always associated with rapidly proliferating B-cell tumors, whereas nuclear factor (NF)- κB addiction is observed in both indolent and diffuse large B-cell lymphomas. Overexpression of c-Myc boosted the proliferation of Epstein-Barr virus-latency III immortalized B cells, a result that was dependent on NF-κB (David et al. 2017).
MYC gene dysregulation, a crucial contributor in cancer, is generated by physiological imbalances caused by mechanisms such as gene amplification, chromosomal translocation, and changes in regulatory elements governing MYC expression. Increased c-MYC expression disturbs normal cellular metabolism, resulting in increased nutrient absorption and altered energy production, both of which are typical characteristics of cancer cells. MYC dysregulation impairs cell differentiation, resulting in an increase of undifferentiated cells and tumor development. Furthermore, MYC dysregulation reduces apoptosis, allowing cancer cells to avoid cell death signals and survive (Sammak et al. 2019). Another factor for dysregulation of Myc genes is nuclear localization meaning that they predominantly reside within the cell nucleus, posing a challenge for monoclonal antibody targeting, as effective binding within the nucleus is hindered. Intracellular localization is vital for MYC’s role in gene expression and cellular processes. This nuclear residence enables interactions with proteins like Max to form transcription factor complexes, regulating target gene expression. Targeting MYC’s nuclear localization could be a potential therapeutic strategy against MYC-driven cancers, recognizing its complexity in cancer development regulation (Lancho and Herranz 2018). c-MYC and its counterparts exhibit intrinsic disorder, lacking a defined three-dimensional structure. This disorder enables diverse interactions with binding partners and involvement in various cellular processes. The absence of a stable structure contributes to the functional adaptability of MYC, allowing different conformations and interactions with proteins and DNA. This intrinsic disorder is crucial for MYC’s role in gene expression and oncogenesis. While challenging for drug development due to lacking well-defined binding sites, recent studies explore targeting specific structural elements, such as G-quadruplexes and c-MYC-Max dimerization, as potential strategies to inhibit MYC function (Kumar et al. 2017). Given that c-MYC lacks enzymatic function, it cannot catalyze cellular chemical processes. In addition, MYC proteins lack well-defined ligand binding sites, making them appear “undruggable” for therapeutic intervention. Recent research, however, investigates alternate techniques, such as interrupting G-quadruplexes in the c-MYC promoter or blocking c-MYC-Max dimerization. Despite the lack of typical enzymatic activity and ligand binding sites, these techniques seek to disrupt MYC connections and functions (Chen et al. 2018; Alexandrova and Podlipnik 2023).
Myc-induced apoptosis is an important intrinsic tumor suppressor mechanism that restricts Myc’s ability to cause cancer. However, the mechanisms that determine whether Myc activation results in cell proliferation or cell death remain unknown (Murphy et al. 2008). Oncogenic Myc activates the ARF/p53 tumor suppressor pathway as well as apoptosis, two powerful tumor suppressor programs that effectively limit Myc’s oncogenic potential (Evan and Littlewood 1998). Because overexpressed Myc may only be tolerated by cells that have already lost their tumor suppressor pathways, low-level unregulated Myc may be a more efficient initiator of oncogenesis. When tumor suppressor pathways are intact early on, selection favors low-level oncogene activity and the relatively indolent clonal growth it provides. Only once the necessary intrinsic tumor suppressor pathways have been destroyed by spontaneous mutation does enhance oncogene activity, together with the increased aggressiveness it confers on tumors, become subject to positive selection (Murphy et al. 2008).
c-MYC overexpression and transformation in tumors
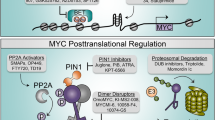
Chromosomal translocations and c-MYC locus amplification are seen in a wide range of spontaneously occurring tumors, which lead to constitutive overexpression of c-Myc (Spencer and Groudine 1991). c-Myc is stabilized and accumulates at high levels following Ras activation (Sears et al. 1999). Two mechanisms support this stabilization: the Raf-MEK-ERK kinase cascade and the PI (3) K-Akt system, which obstructs GSK-3 (Henriksson et al. 1993). These processes phosphorylate certain c-Myc sites, Thr 58 and Ser 62, which inhibit c-Myc degradation via the ubiquitin–proteasome system (Sears et al. 1999). Throughout the early G1 phase, the PI (3) K-Akt cascade is active, blocking GSK-3 and stabilizing c-Myc (Cross et al. 1995). Later in the G1 phase, GSK-3 becomes active and phosphorylates c-Myc on Thr 58, boosting its destruction. c-Myc turnover requires Thr 58 phosphorylation, and its mutation yields a more robust and carcinogenic variant of c-Myc (Salghetti 1999; Gregory and Hann 2000). PP2A and the Pin1 prolyl isomerase regulate Ser 62 dephosphorylation, which is indispensable for ubiquitin-triggered c-Myc decay. When human cells are transformed, c-Myc stabilization may substitute for the place of the minor T antigen SV40, indicating its significance in the process. (Yeh et al. 2004; Stukenberg and Kirschner 2001; Zhou 2000; Liao 2000).
It seems that the transformation driven by c-Myc occurs when there is abnormal expression or when there are genetic alterations affecting the proto-oncogene. In specific cell types, it necessitates simultaneous transfection with other oncogenes or growth factor genes, like c-rasH or transforming growth factor-(tgf) (Amati 1998). c-Myc can cause genetic instability and affects various cell lines in various ways (Felsher and Bishop 1999). Its control over the human telomerase transcriptase gene (hTERT), which is involved in cell immortalization, may be related to its capacity to transform cells (Greenberg et al. 1999). The transcriptional activation of hTERT is mediated by the interaction between c-Myc and other proteins including Sp1 and estrogen (Oh et al. 1999; Kyo 2000). Indirectly, the expression of hTERT can be suppressed by Mad, a protein that competes with c-Myc (Günes 2000; Oh et al. 2000). The specific mechanisms and relationships governing how c-Myc regulates hTERT and its transformation activities requires further study (Liao 2000).
The epigenome confers functional specialization to cells in the body that have the same genome. Mutations in the epigenome are frequently linked to the start of several diseases, including cancer (Fatma et al. 2022). Malignant phenotypes are often conferred by genetic and epigenetic changes in cancer cells. These changes are incorporated into the chemoresistant phenotype in all phases of cancer development (Zhao and Shilatifard 2019). Epigenetic changes are dynamic and reversible in nature. Instead of altering the DNA sequence, epigenetic modifications control the alterations of different biological processes or cellular activities by the imposition of chromatin, nucleosome remodeling, histone, RNA, and protein modifications (Han et al. 2019). In c-Myc dysregulation, numerous epigenetic markers have been identified. The epigenetically altered c-MYC protein is more stable, which contributes to reduced cell death and increased cell proliferation in cancer cells and cancer stem cells (Ba et al. 2018). Epigenetic changes in critical proteins have been associated to chemoresistance in various cancer types and may serve as a biomarker for epigenetic therapy (Matkar et al. 2015). It has been shown that DNA demethylating drugs, histone deacetylase (HDAC) inhibitors, and HAT sensitize chemoresistant cells to therapy. Epidrugs, also known as epigenetic drugs, are a class of pharmacological substances that can epigenetically modify specific proteins, reactivating or suppressing their activity (Miranda Furtado et al. 2019). The epigenome includes reversible DNA and histone protein modifications like as hypermethylation, hypomethylation, acetylation, and deacetylation, which can result in chromatin remodeling and changed transcriptional and translational activity of the genes (Miranda Furtado et al. 2019) (Fatma et al. 2022).
While c-Myc can serve as the primary instigator of cancers such as Burkitt lymphoma when it undergoes translocation beneath the promoter regions of immunoglobulin genes, it is more commonly an ‘immediate-response gene downstream of diverse signaling pathways that becomes activated (Lombardi et al. 1987). It is yet unknown if the metabolic changes brought on by transformation are largely caused by c-Myc overexpression or whether this overexpression is frequently seen as an outcome of the intricate metabolic alterations occurring as cells undergo malignant transformation (Miller et al. 2012). In non-transformed cells, the expression and function of c-Myc are closely monitored by development and growth-inducing signals. Since c-Myc protein levels are minimal its mRNA has a brief half-life, the absence of positive regulatory signals means there is no stimulus for proliferation. Conversely, in tumor cells, c-Myc activity is almost always elevated in tumor cells. This often occurs due to mutations in the gene itself, but more frequently as a result of the activation of c-Myc expression through oncogenic pathway further upstream. The potential of c-Myc to cause apoptosis through many mechanisms balances out its oncogenic characteristics. This paradox likely clarifies why c-Myc is seldom the oncogene responsible for initiating early-stage tumors (Pelengaris et al. 2002).
Post-translational changes may potentially lead to c-Myc overexpression. For instance, mutations within the coding region of c-Myc, specifically in the Thr58 phosphorylation site, are frequently discovered in human lymphomas. This mutation causes ineffective ubiquitination and reduced proteasome-mediated protein turnover, which together increase the transforming activity of c-Myc (Bahram et al. 2000). c-Myc exhibits a multifaceted role in cell behavior, influencing proliferation and apoptosis. Under serum-rich conditions, it targets Cdc25A and ornithine decarboxylase, promoting proliferation, while in serum-limited environments, it induces apoptosis. Direct regulation of H-ferritin, IRP2, and telomerase contributes to proliferation and immortalization. c-Myc’s involvement in Fas ligand-induced apoptosis, alongside its dual function in proliferation and apoptosis, is intricately governed by a context-specific regulatory network (Fuhrmann et al. 1999).
Recent developed inhibitors against c-MYC
Researchers all around are attempting to directly target Myc at laboratory level as well as clinical trials. Several compounds have been found to prevent MYC from heterodimerizing with Max, with 10058-F4 being the most well studied of these (Yin et al. 2003). When 10058-F4 (see Fig. 5) is given to tissue culture, it displaces Myc and -N from chromatin, reverses Myc-dependent activities on RNAPII, and morphs gene expression. In a neuroblastoma transgenic model, administration of 10058-F4 to mice slows the growth of the tumor and lengthens survival. Recently identified as strong chemicals that bind to different portions of this area and create highly localized distortions that inhibit fruitful interaction with Max’s bHLH-ZIP domain (Yap et al. 2013). It has been demonstrated to cause G0/G1 arrest in HL60 cells. The treatment with 10058-F4 inhibits c-Myc’s connection with Max in a dose-dependent manner while having no effect on the former protein’s total level (Jung et al. 2015). An alternative approach involves both the stabilization of Max homodimers and the displacement of Myc from the joined heterodimer (Jiang et al. 2009). Newly discovered types of medications have been found to hinder Myc-related gene activity within cells at approximately 10 µm concentrations. These drugs also reduce the utilization of Myc in chromatin and effectively impede the progression of tumors driven by Myc when tested in live organisms (Struntz et al. 2019). Also available as a therapeutic peptide, OmoMyc.

A Chemical structure of peptide Inhibitor 10,058-F4. PubChem [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004-. PubChem Compound Summary for CID 1271002, 5-[(4-Ethylphenyl)methylene]-2-thioxo-4-thiazolidinone; [cited 2023 July 11]. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/10058-f4., B., Jung, L.A., Gebhardt, A., Koelmel, W., Ade, C.P., Walz, S., Kuper, J., von Eyss, B., Letschert, S., Redel, C., d’Artista, L., Biankin, A., Zender, L., Sauer, M., Wolf, E., Evan, G., Kisker, C., Eilers, M. Structure of Human apo OmoMYC (2017) https://doi.org/10.1038/onc.2016.354
enters cultured cells, ousts endogenous Myc from its proper adhesion spots, and prevents Myc-mediated gene expression (see Fig. 5). After intravenous injection, OmoMyc reduces tumor growth, indicating that it may be able to mitigate Myc’s neoplastic effects in vivo (Beaulieu et al. 2019). OmoMyc falls into the category of Mini proteins or protein domains used as therapeutic agents (Annibali et al. 2014). It directly disrupts the protein–protein interaction (PPI) of MYC and MAX and/or their binding to DNA. OmoMyc stands out as one of the most well-studied compounds in vivo against MYC, confirmed effectively as a gene therapy (Whitfield et al. 2017). Its structure includes the bHLH-Zip domain of Myc with four amino acid modifications that modify its dimerization specificity. This modification allows OmoMyc to not only bind to Myc’s natural partner, Max, but also form heterodimers with Myc and homodimers (Savino et al. 2011). Consequently, OmoMYC acts as a dominant negative regulator of Myc’s transcriptional function, disrupting the interaction between Myc and Max. It can isolate Myc away from DNA and occupy the E-box with transcriptionally inactive dimers, such as OmoMyc/OmoMyc and/or OmoMyc/Max (Soucek et al. 2008; Savino et al. 2011). The therapeutic impact of OmoMyc influences the mutation or tissue of origin, indicating Myc’s vital role in carcinogenesis downstream of the different oncogenic diseases. It has minimal negative effects, implying its safety and possible application in patients. Work is currently being done to convert its use from gene therapy to pharmaceutical applications (Galardi et al. 2016; Soucek et al. 2008; Annibali et al. 2014). Another G4 disruptor is Pyridostatin (PDS), a small synthetic molecule specifically designed to target G4 stabilization and c-Myc structure disruption at the promoter region. Circular dichroism experiments demonstrate that adding PDS to c-Myc G-quadruplex oligonucleotide stretch in a 1:1 ratio attenuates the G4 structure at 260 nm in all spectra (Cuny 2017). Quarfloxin (CX-3543) is a molecular drug that targets and inhibit RNA pol I activity. In neuroblastoma cell lines, quarfloxin cause DNA damage, p53 signaling, cell death, and cell cycle arrest (Hald et al. 2019). Dur to lack of efficacy, quarfloxin was withdrawn from phase II clinical trial(Jin et al. 2023). G2/M arrest is induced in TP53-mutated neuroblastoma cell lines. Quarfloxin is a G-quadruplex stabilizer that inhibits rRNA production by disrupting the connection between putative G-quadruplex structures in rDNA and nucleolin (Li et al. 2016). The inhibition of transcription via the stabilization of G-quadruplexes in promoter regions provides another potential mechanism for MYC gene suppression by quarfloxin (Hald et al. 2019). Quarfloxin finished its phase 1 and 2 trials in patients with advanced solid tumors and lymphomas (NCT00955786) and neuroendocrine/carcinoid tumors (NCT00780663). It has been demonstrated that inhibiting RNA pol I activity causes apoptosis, nucleolar surveillance signaling, p53 pathway activation, senescence, and pro-death autophagy (Li et al. 2016; Bywater et al. 2012). Berberine is a natural alkaloid therapeutic substance, is one of the most actively researched and pursued G quadruplex (G4)-ligands. A noteworthy example and model structure for parallel G-quadruplexes is the primary G-quadruplex generated in the promoter region of the MYC oncogene (MycG4) (Dickerhoff et al. 2021a, b). Berberine has been demonstrated to bind human oncogene telomeric and promoter G-quadruplexes, including MYC. Berberine and its derivatives have been demonstrated to bind MYC G4 and suppress MYC expression. The structural specifics of berberine recognition are unknown (Arora et al. 2008; Ma et al. 2008). One of the many ubiquitin ligases and a target protein can form or strengthen a molecular bond through the use of small-molecule ligands (Sakamoto et al. 2001). These particular compounds, known as PROTACs (proteolysis-targeting chimeras) or degronomids, exhibit a dual role by connecting the target protein to either cereblon or VHL ubiquitin ligase. It’s possible that by conducting chemical screenings, we may uncover simpler structures that can improve the affinity of one of the inherent ubiquitin ligases linked to Myc. Another theory proposes that the activation of transcription takes place within specialized compartments, referred to as phase-separated domains, where enhancers and the transcription apparatus amass (Boija et al. 2018). Instead of focusing on specific PPIs involving Myc, it is conceivable to find drugs that derail Myc-regulated transcription nexus in the situation where Myc proteins assist to phase segregation and this adds to their oncogenic role (Wolf and Eilers 2020). Klien et al. explore the phase-separation capacity of transcription factor (TF) activation domains (ADs) and their significance in gene activation. It has been observed that various ADs form phase-separated condensates with the Mediator coactivator, implying that TFs can interact with Mediator via their ADs’ phase-separating capacity. While the research does not name Myc-regulated transcription directly, it does provide evidence that phase separation with Mediator is involved in gene activation for several TFs. As the phase-separation capacity of ADs appears to play a role in gene activation, it is reasonable to consider targeting Myc-regulated transcription inside phase-separated areas for therapeutic development (Boija et al. 2018). A novel approach is to develop drugs that affect Myc-regulated transcription inside phase-separated domains. Designing small molecules that interfere with or improve Myc’s phase separation with coactivators like the Mediator complex is made possible by an understanding of the role that phase separation plays in gene activation. By altering this process, drugs that specifically affect Myc target genes may be developed, providing a treatment option for maladies like cancer that have dysregulated Myc. Compared to conventional transcription factor techniques, which concentrate on physical contacts and condensate formation in gene activation, targeting phase-separated domains gives precision. To understand Myc-regulated transcription mechanisms in these areas and find possible targets for drugs, more research is necessary (Boija et al. 2018).
By using antisense oligonucleotides or RNAs inducing RNA interference, Myc mRNA silencing attempts were conducted (Yuan Li et al. 2013). Despite the fact that the preliminary results were positive and prompted multiple research projects, including phase I trials, all clinical attempts failed as a result of poor stability of drugs and delivery problems (Devi et al. 2005). There are continuous efforts to enhance the usage and delivery of therapies based on oligonucleotides. The intracellular delivery of oligonucleotides poses a multifaceted challenge, extending beyond mere transportation to tissues and emphasizing the critical need for effective delivery to specific intracellular sites. Concerns arise regarding the functional biodistribution, intricately regulated by endogenous cellular activities. Despite the potential exhibited by ligand-oligonucleotide conjugates, persistent challenges mandate further exploration, encompassing factors such as rapid renal clearance, tissue-specific considerations, and unique liver features that contribute to the success of specific conjugates. Achieving selective delivery to non-hepatic tissues requires a profound understanding of receptor biology, including factors like abundance, turnover, and intracellular trafficking pathways (Sebestyén et al. 2015). The intricate aspects of valency and topology in ligand-oligonucleotide conjugates play a pivotal role in receptor internalization, with the potential for accelerated uptake through cross-linking but posing a risk of disrupting the delicate balance between receptor recycling and degradation. Lipid and polymer-based nanocarriers, crucial for oligonucleotide delivery, confront challenges related to restricted biodistribution and potential toxicity from polycationic components, while emerging unconventional nanocarriers like SNAs and DNA nanostructures offer alternative solutions. Targeted macromolecular scale carriers, utilizing proteins or small non-toxic polymers, amalgamate features from traditional nanocarriers and molecular scale conjugates; however, the incorporation of multiple features raises concerns about potential toxicity and poses challenges in scale-up and production. The emergence of small molecules capable of enhancing oligonucleotide effectiveness opens a novel avenue, yet persistent challenges arise due to the intricate nature of intracellular trafficking, necessitating further research in the chemical biology of this process. Ensuring the delivery of broadly applicable, highly effective, and non-toxic oligonucleotide therapeutics to humans requires a focus on cost-effectiveness, particularly in light of the ongoing pressures on the healthcare system (Juliano 2016).
The translation of antisense nucleotide for Myc mRNA silencing failed in stability and delivery issues because of various reasons like lacking Studies of oligonucleotide metabolism, Pharmacokinetics and pharmacodynamic., completely different uptake of drug In vitro and in vivo, inadequate approaches to study off-target effects and Drug delivery approaches and incomplete Drug–drug interaction studies. The safety of oligonucleotides in humans remains unconfirmed, with a lack of toxicokinetic profiles and unknown species differences. Comprehensive studies on drug–drug interactions in relation to toxicity and mechanisms of toxicity are still lacking. Pharmacokinetic investigations are limited, as are biomarkers for monitoring drug responses. There is a severe limitation in pharmacodynamic studies, and pharmacogenomic approaches are underdeveloped. Phase III trials are largely constrained. Large-scale synthesis of oligonucleotides with GMP-quality is both limited and expensive, and impurity remains a significant concern. The understanding of this novel therapeutic class is limited, reflecting a scarcity of innovative clinical trial approaches. Additionally, standards for combination therapy to ensure both safety and efficacy are not well established in this field (Rayburn and Zhang 2008).
G-quadruplex (G4) formations within the Myc promoter (Simonsson 1998) serve as inhibitory components. The promoter is suppressed and MYC levels are decreased by substances that increase the long-term viability of the G4 structure. Although CX-3543, a G4 stabilizing compound with potential anti-cancer properties, progressed to phase II clinical trials, its mode of action seems to involve mechanisms unrelated to Myc (Drygin et al. 2009). This highlights the difficulty in developing drugs that specifically reduce Myc transcription, and it remains uncertain whether target selectivity can be completely achieved (Wolf and Eilers 2020). TMPyP4 (Tetra (N-methyl-4-pyridyl) porphyrin), a porphyrin derivative, is another that is shown to stabilize G4 at the promoter region. It has a unique effect on a variety of cell cycle regulating genes and proto-oncogenes, including c-MYC. TMPyP4 can suppress the expression of the c-MYC oncogene as well as other c-MYC-regulated genes, implying that c-MYC down-regulation is a potential TMPyP4 target pathway (Grand et al., 2002). A stretch of guanine-rich DNA (the NHE III1) in the c-MYC promoter region can create G-quadruplexes. Human telomeres, with their hexanucleotide repeats (TTAGGG)n, have the capacity to create G-quadruplexes. This is especially intriguing given that the cata subunit of telomerase, hTERT, is transcriptionally controlled by c-MYC. Because TMPyP4 inhibits c-MYC expression and hTERT is controlled by this transcription factor, the effect of TMPyP4 on telomerase activity is explained. TMPyP4 reduces c-MYC RNA and protein expression in MiaPaCa-2 and HeLa S3 cells. (Wang et al. 1998; Grand et al. 2002). Another synthetic small moiety PDS is designed to specifically target and stabilize G4 structures and its stabilization.
Stabilizing the G-quadruplex structure within the MYC promoter region presents a potent strategy for downregulating MYC expression, holding significant promise for treating various cancers. Achieving this involves obtaining high-resolution structural insights into the G-quadruplex and its interactions with ligands, facilitating the rational design of improved drugs. Ligands must exhibit specificity by recruiting DNA bases in a conserved and sequence-specific manner, complemented by the design of complementary hydrogen bonds for enhanced efficacy (Calabrese et al. 2018). Compounds with drug-like properties, such as the quinoline derivative PEQ, which lacks an extensive aromatic moiety, offer suitability for targeting G-quadruplexes in the MYC promoter (Dai et al. 2011; Calabrese et al. 2018). Precise targeting of the MycG4-ligand structure with wild-type binding sites is deemed crucial for achieving specific inhibition of MYC(Dickerhoff et al. 2021a, b).
By preventing Myc function, transcriptional inhibitors with a narrow target specificity exhibit anti-tumor effects. JQ1, for instance, was originally created to prevent the BET subgroup of bromodomain proteins (BRD2, BRD3, and BRD4) from functioning (Filippakopoulos et al. 2010). BET proteins initiate transcription by attaching themselves to histones’ acetylated lysine sites and enlisting coactivators like P-TEFb (see Fig. 6) (Bisgrove et al. 2007). Surprisingly, when treating multiple myeloma cells with JQ1 (Delmore et al. 2011), Myc expression was predominantly affected, although BRD4 and other BET proteins are typically considered global transcription activators (Muhar et al. 2018). The preferential action of JQ1 on Myc transcription could arise from a few factors, including clusters of BET proteins on enhancer regions known as super enhancers in proximity to Myc, or the relatively brief lifespan of Myc’s mRNA and protein (Lovén et al. 2013). Clinical trials for a variety of tumors, including multiple myeloma, glioblastoma, and prostate cancer, are being conducted with a number of unique BET inhibitors (Stathis and Bertoni 2018). Some studies were terminated due to toxicity unrelated to Myc inhibition (Postel-Vinay et al. 2019). BET inhibitors have also been utilized to create compounds that break down BET proteins, but their efficacy as anticancer drugs require further investigation (Raina et al. 2016). Inhibiting transcription-associated cyclin-dependent kinases (CDKs), like BET inhibitors, is under examination as a treatment for cancer (Chipumuro et al. 2014). Potentially therapeutic small compounds aimed at eukaryotic translation initiation factors have been spotted as strong inhibitors of both cap- and IRES-dependent translation of Myc (Wiegering et al. 2015). Myc proteins, including -N, are characterized by their inherent instability and undergo constant degradation through the ubiquitin/proteasome system. There are about 30 different ubiquitin ligases that have been found, and they interact via Myc or -N to affect the stability and functionality of those proteins.

Chemical structures of drug inhibitor against c-Myc. A TMPyP4 https://www.sigmaaldrich.com/IN/en/product/mm/613560#product-documentation, B JQ1 (Drugbank accession no- DB17021) Shi X, Liu C, Liu B, Chen J, Wu X, Gong W: JQ1: a novel potential therapeutic target. Pharmazie. 2018 Sep 1; 73(9):491–493. https://doi.org/10.1691/ph.2018.8480, C Quarfloxin (CX-3543) Drugbank accession no- DB06638
Threonine 58 (T58), present in the highly conserved region of amino acid MBI plays a critical role. When T58 is phosphorylated, it is recognized by the FBXW7 and FBXL3 ubiquitin ligases (Welcker et al. 2004; Huber et al. 2016), whereas the deubiquitinating enzyme USP11 indirectly recognizes dephosphorylated T58. (Herold et al. 2019). In addition to USP11, other deubiquitinating enzymes, including USP28 [84], USP7, USP22, USP36, and USP37, have the capability to stabilize Myc or -N proteins. Recently, there have been successful efforts to specifically inhibit these deubiquitinating enzymes. As a result, it’s very likely that researchers will investigate the use of USP inhibitors in the near future to diminish cellular Myc levels and manage Myc’s activity (Turnbull et al. 2017).
Alisertib, an Aurora-A kinase inhibitor, shows strong therapeutic action against human -N tumors that targets Myc stability (Dauch et al. 2016; DuBois et al. 2018). Clinical findings indicate a dose-limiting toxicity that prevents a precise assessment of treatment efficacy. In more recent studies, it has been demonstrated that the Aurora-A complex plays a vital role in inhibiting -N-mediated transcriptional elongation during the S phase, thereby coordinating -N-driven transcription with DNA replication. This discovery suggests that a viable approach to mitigate the toxicity of high levels of Aurora-A inhibitors is to combine them with drugs that reduce cells’ capacity to cope with replication stress (Büchel et al. 2017). By doing so, the aim is to achieve a therapeutic strategy that combines Aurora-A inhibitors with agents that target replication stress.
Finding that Myc breakdown by ubiquitin plays a dual role in regulating Myc levels and promoting Myc transactivation (Adhikary et al. 2005). VCP ATPase removes ubiquitinated Myc from chromatin (Heidelberger et al. 2018), enabling the transfer of elongation factors bound to Myc to RNAPII (Jaenicke et al. 2016). Inhibitors of HUWE1 ubiquitin ligase and VCP protein disrupt Myc-dependent transcription in colon tumor cells (Peter et al. 2014). Developing therapeutic strategies requires exploring in vivo HUWE1 inhibitors. The comprehensive identification of proteins that interact with Myc enables the discovery of prospective druggable protein–protein confluence that are critical to Myc’s oncologic potential (Baluapuri et al. 2019).
Focusing on disrupting the crucial interaction between Myc and its binding companion Max presents a feasible strategy. A powerful and specific inhibitor called MYCMI-6 inhibits Myc-regulated transcription by exclusively binding itself to the Myc bHLHZip domain. This binding induces apoptosis and has a Kd of 1.6 µM. With an IC50 of less than 0.5 µM, MYCMI-6 shows considerable tumorigenic cell proliferation suppression, specifically guided by Myc. It has been observed that MYCMI-6 exhibits substantial, dose-dependent inhibition of Burkitt’s lymphoma cells (including Mutu, Daudi, and ST486), which are characterized by Myc translocations to immunoglobulin loci. The average GI50 for this inhibition is approximately 0.5 μM. When MCF7 cells are treated with MYCMI-6 for 24 h, there is a significant reduction in Myc: Max PLA signals to only 7%. A titration analysis indicates that the IC50 for inhibiting Myc: Max interaction via PLA is less than 1.5 μM. Furthermore, MYCMI-6 hampers the formation of Myc: Max heterodimers effectively, with an IC50 of 3.8 μM. Additionally, MYCMI-6 is highly efficient in hindering the augmentation of -N-amplified neuroblastoma cells in a free-growth manner, with GI50 values falling below 0.4 μM. (Castell et al. 2018).
There are numerous ways to attack Myc. BRD4, CDK7, and CDK9 inhibitors reduce transcriptional expression of Myc. Myc translation is halted by inhibiting the PI3K/AKT/mTOR pathway. Myc is destabilized following synthesis by inhibitors of USP7, AURKA, and PLK1. OmoMyc and 10,058-F4 combine to break down the Myc-Max dimer complex. The abbreviations BRD4, CDK7, CDK9, PLK1, and mTOR (mammalian target of Rapamycin) are short for bromodomain-containing 4, cyclin-dependent kinase 7, polo-like kinase 1, and cyclin-dependent kinase 9 (Fig. 7).

MYC is both directly and indirectly inhibited. A When aimed at the Myc/Max. or OmoMyc, obstruct DNA binding and hinder the Myc transcriptional pathway. B Inhibiting CDK7 or BRD4, crucial factors in the initiation and elongation of transcription, using compounds like JQ1/dBET1 or THZ1/THZ2 respectively, indirectly regulates Myc expression. Specifically, targeting CDK7 or BRD4 leads to decrease in Myc protein expression. Created with BioRender.com
IDPs (intrinsically disordered proteins) have primary sequences that contain over 70% disorder and are crucial for controlling cell proliferation and death (Sullivan and Weinzierl 2020). Due to this intrinsic disorder, computational methods that explore secondary and tertiary structural conformations offer the most practical approach for studying such proteins. By employing molecular dynamics simulations, utilizing force fields and water models one can analyze c-Myc (Mark and Nilsson 2001). The investigation of the conformations and dynamic behaviors of IDPs has recently attracted attention thanks to computational methods like MD (molecular dynamics) simulations. Since GPUs (graphics processing units) have been developed, MD simulations can now be executed quickly on workstations, allowing for the efficient parallelization of simulations (Stone et al. 2016). The results are compared to experimental secondary structure assignments acquired by NMR, and a specific implicit solvation method is found to be quite consistent (Salomon-Ferrer et al. 2013). This type of experimentation sheds important light on the molecular structure of c-Myc and acts as a good manual for more experimental studies.
IDPs have been analyzed extensively using computational methods and GPU-accelerated simulations. Trends and potential advancements in this area includes the following: A. Enhanced Conformational Space Sampling-GPU acceleration is useful for computational techniques, especially for MD simulations, as it improves the sampling of the large conformational space that is accessible to IDPs like as c-Myc. This makes it possible to depict their dynamic character more accurately (Zhu et al. 2023). B. Longer time simulations to record uncommon events and gain a deeper understanding of the dynamic behavior of IDPs. C. Ensemble-based simulation- For intrinsically disordered proteins (IDPs), ensemble-based simulations comprise the construction of structural ensembles and the investigation of disorder-to-order transitions via molecular simulations. These simulations provide vital information about the dynamics and structural organization of IDPs, which is necessary to comprehend their function. For IDP research, a variety of simulation methods have been developed, including meta dynamics, replica exchange molecular dynamics (REMD) simulations, and Markov State modelling. By facilitating rare event sampling, meta dynamics makes the free energy landscape visible. Replica exchanges at various temperatures are a feature of REMD simulations that improve the accuracy of conformational sampling. Molecular dynamics simulations are utilized in Markov State modelling to build a kinetic model that clarifies conformational dynamics and transitions in IDPs. The combined results of these simulations provide important insights into disorder-to-order transitions and IDP structure ensembles, which greatly aid in the biophysical characterization of these proteins (Fatafta et al. 2021). D. Exploration of the Free Energy Landscape- Simulations accelerated by GPUs help investigate the free energy landscape of IDPs. Finding functionally relevant states and comprehending the transitions between IDP conformations—which are frequently necessary for their biological activity are critical. The newly proposed energy landscape visualization technique (ELViM) is a reaction coordinate-free methodology for exploring IDPs’ frustrated energy landscapes. Using the same effective phase space, ELViM can identify and compare population distributions of distinct conformational ensembles of IDPs. ELViM appears to be an effective tool for analyzing the highly frustrated energy landscape representation of IDPs, where determining proper reaction coordinates is difficult (Oliveira Junior et al. 2021). E. Machine Learning Methodologies-Machine learning approaches combined with GPU-accelerated simulations are becoming increasingly popular for analyzing and interpreting massive amounts of data. These methods can help anticipate IDP structure ensembles and understand how they interact with binding partners (Kawaguchi et al. 2022).
Applications and advantages pertaining to GPU usage include Parallel Processing Efficiency allows for the simultaneous management of several activities while also greatly accelerating computations for simulations involving complex calculations and big datasets. The parallel architecture of GPUs provides a significant speed gain over traditional CPUs, which is especially useful for quickly completing computationally expensive activities such as molecular dynamics simulations or sophisticated physics simulations. High Throughput Capability can handle multiple jobs at the same time, ensuring high throughput. This is useful for performing numerous simulations at the same time or efficiently analyzing large datasets. GPUs offer cost-effective performance per dollar, particularly for computations critical to scientific simulations where computational efficiency is critical. Energy Efficiency Advantage over traditional CPUs due to their design for efficient parallel tasks. GPUs are widely available and used in a variety of computing contexts, ranging from desktop PCs to high-performance computing clusters (Bridges et al. 2017). And while no system is absolute in its working, some challenges faced by researchers include Algorithm parallelization poses a significant challenge for achieving optimal GPU performance, necessitating substantial modifications to algorithms, as not all are easily parallelizable. Adapting existing code or crafting new algorithms for parallelization presents a substantial hurdle. GPUs, with their limited memory compared to CPUs, create challenges for large-scale simulations handling extensive datasets, requiring efficient memory management strategies (Chadwick et al. 2012). GPU programming’s complexity, demanding expertise in specific languages like CUDA or OpenCL, makes the transition from CPU-based to GPU-accelerated code a demanding task, requiring additional training and development efforts. Compatibility issues arising from variations in GPU architectures and capabilities make ensuring simulation code compatibility across different GPU models a challenging aspect. Minimizing data transfer overhead between CPU and GPU is crucial for optimizing simulation performance. While GPUs excel in parallel processing, their limited general-purpose capability may not significantly benefit certain simulations or computations. Dependency on vendor-specific technologies, such as CUDA for NVIDIA GPUs, hampers portability across different GPU architectures and vendors, posing challenges for widespread adoption and compatibility (Taufer et al. 2013; Chadwick et al. 2012).
Synthetic lethality is an idea in cancer research in which the combination of two genetic or molecular events causes cell death, despite the fact that neither event alone is fatal. It entails focusing on a specific genetic or chemical change in cancer cells that is dependent on another change for survival.
Synthetic lethality has been reported between G-quadruplex-targeting drugs, such as Pidnarulex (CX-5461), and the BRCA1/2-mediated homologous recombination (HR) pathway. This suggests that cancer cells with HR pathway abnormalities are more vulnerable to G4-targeting drugs, resulting in cell death, revealing synthetic lethal interactions with HR genes. This is a leading to promising clinical efficacy in treating solid tumors deficient in BRCA2 and PALB2. G4 stabilizers have also been linked to genes involved in additional DNA damage repair pathways, transcription, epigenetic control, and RNA processing deficits. These genetic relationships shed light on patient identification and the development of G4-targeting medication combination treatments (Jin et al. 2023).
Conclusion
The underlying structural strategy of human c-Myc and its role as an oncoprotein in more than 70% of tumors have been covered in this brief overview. Here are some of the main and cutting-edge therapeutic inhibitory strategies to stop Myc from attaching to its functional proteins, Max and Mad, or from either targeting the homodimer produced to prevent binding to the promoter region of DNA. Although Myc has not yet been directly targeted, it is still possible to attack this cancer super-controller with precision and effectiveness by creating novel strategies. Yet, before the emergence of fragment-based nuclear magnetic resonance (NMR) screening, which expanded the outlook on this potential inhibitory molecule, BCL-2 was considered extremely challenging to target. Better treatments will be needed in the future to target Myc-dependent malignancies, whether by direct or indirect Myc targeting. The intricate network of interactions and complexity associated with the c-Myc protein pose significant challenges in developing effective strategies to target it. Nonetheless, several emerging approaches and technologies show promise in addressing the complex c-Myc interactions. These strategies aim to hinder c-Myc function or disrupt its interactions with other molecules involved in oncogenesis. Examples includes, small molecule inhibitors, designed to selectively target specific interactions of c-Myc. For instance, researchers have investigated inhibitors with the potential to hinder the interaction between c-Myc and its partner protein Max, as a potential avenue for therapeutic drugs. Using tiny compounds, peptides, or designed proteins that precisely bind to important interaction sites, one can target PPI and prevent c-Myc from interacting with other proteins. Proteolysis-Targeting Chimeras (PROTACs) are another dual-function molecule capable of binding both c-Myc and an E3 ubiquitin ligase. The ubiquitination and consequent destruction of c-Myc are caused by this interaction, providing a possible method to lower c-Myc protein levels. RNA Interference (RNAi)-based techniques, such as small interfering RNA (siRNA) or short hairpin RNA (shRNA), have the ability to specifically suppress the expression of c-Myc. This approach aims to reduce c-Myc protein levels and its downstream effects. Genome Editing Technologies like CRISPR-Cas9 hold promise in precisely targeting and modifying the c-Myc gene or its interacting partners. This enables the investigation of specific interactions and their functional consequences.
Data availability
Not applicable.
References
Adams, J.M., Harris, A.W., Pinkert, C.A., Corcoran, L.M., Alexander, W.S., Cory, S., Palmiter, R.D., Brinster, R.L.: The C-Myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318(6046), 533–538 (1985). https://doi.org/10.1038/318533a0
Adhikary, S., Marinoni, F., Hock, A., Hulleman, E., Popov, N., Beier, R., Bernard, S., et al.: The Ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell 123(3), 409–421 (2005). https://doi.org/10.1016/j.cell.2005.08.016
Alexandrova, R., Podlipnik, Č: MYC oncogenes as potential anticancer targets. In: Oncogenic viruses, pp. 191–219. Elsevier, Amsterdam (2023). https://doi.org/10.1016/B978-0-12-824156-1.00011-X
Amati, B., Alevizopoulos, K., Vlach, J.: Myc and the cell cycle. Front. Biosci. Landmark 3, 250–68 (1998)
Annibali, D., Whitfield, J.R., Favuzzi, E., Jauset, T., Serrano, E., Cuartas, I., Redondo-Campos, S., et al.: Myc inhibition is effective against Glioma and REVEALS A ROLE for Myc in proficient mitosis. Nat. Commun. 5(1), 4632 (2014). https://doi.org/10.1038/ncomms5632
Arora, A., Balasubramanian, C., Kumar, N., Agrawal, S., Ojha, R.P., Maiti, S.: Binding of berberine to human telomeric quadruplex—spectroscopic, calorimetric and molecular modeling studies. FEBS J. 275(15), 3971–3983 (2008). https://doi.org/10.1111/j.1742-4658.2008.06541.x
Atchley, W.R., Fitch, W.M.: Myc and max: molecular evolution of a family of proto-oncogene products and their dimerization partner. Proc. Natl. Acad. Sci. 92(22), 10217–10221 (1995). https://doi.org/10.1073/pnas.92.22.10217
Atchley, W.R., Hall, B.K.: A model for development and evolution of complex morphological structures. Biol. Rev. 66(2), 101–57 (1991). https://doi.org/10.1111/j.1469-185X.1991.tb01138.x
Ba, M., Long, H., Yan, Z., Wang, S., Yinbing, Wu., Yinuo, Tu., Gong, Y., Cui, S.: BRD4 promotes gastric cancer progression through the transcriptional and epigenetic regulation of C-MYC. J. Cell. Biochem. 119(1), 973–982 (2018). https://doi.org/10.1002/jcb.26264
Bahram, F., von der Lehr, N., Cetinkaya, C., Larsson, L.-G.: C-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 95(6), 2104–2110 (2000). https://doi.org/10.1182/blood.V95.6.2104
Baluapuri, A., Hofstetter, J., Stankovic, N.D., Endres, T., Bhandare, P., Vos, S.M., Adhikari, B., et al.: MYC recruits SPT5 to RNA polymerase II to promote processive transcription elongation. Mol. Cell 74(4), 674-687.e11 (2019). https://doi.org/10.1016/j.molcel.2019.02.031
Battey, J., Moulding, C., Taub, R., Murphy, W., Stewart, T., Potter, H., Lenoir, G., Leder, P.: The human C-Myc oncogene: structural consequences of translocation into the igh locus in Burkitt Lymphoma. Cell 34(3), 779–787 (1983). https://doi.org/10.1016/0092-8674(83)90534-2
Beaulieu, M.-E., Jauset, T., Massó-Vallés, D., Martínez-Martín, S., Rahl, P., Maltais, L., Zacarias-Fluck, M.F., et al.: Intrinsic cell-penetrating activity propels omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med. (2019). https://doi.org/10.1126/scitranslmed.aar5012
Berg, T., Cohen, S.B., Desharnais, J., Sonderegger, C., Maslyar, D.J., Goldberg, J., Boger, D.L., Vogt, P.K.: Small-molecule antagonists of Myc/max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. 99(6), 3830–3835 (2002). https://doi.org/10.1073/pnas.062036999
Bisgrove, D.A., Mahmoudi, T., Henklein, P., Verdin, E.: Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. 104(34), 13690–13695 (2007). https://doi.org/10.1073/pnas.0705053104
Blackwood, E.M., Eisenman, R.N.: Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251(4998), 1211–1217 (1991). https://doi.org/10.1126/science.2006410
Boija, A., Klein, I.A., Sabari, B.R., Dall’Agnese, A., Coffey, E.L., Zamudio, A.V., Li, C.H., et al.: Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175(7), 1842-1855.e16 (2018). https://doi.org/10.1016/j.cell.2018.10.042
Bridges, R.A., Imam, N., Mintz, T.M.: Understanding GPU power. ACM Comput. Surv. 49(3), 1–27 (2017). https://doi.org/10.1145/2962131
Büchel, G., Carstensen, A., Mak, K.-Y., Roeschert, I., Leen, E., Sumara, O., Hofstetter, J., et al.: Association with aurora-A controls N-MYC-dependent promoter escape and pause release of RNA polymerase II during the cell cycle. Cell Rep. 21(12), 3483–3497 (2017). https://doi.org/10.1016/j.celrep.2017.11.090
Bywater, M.J., Poortinga, G., Sanij, E., Hein, N., Peck, A., Cullinane, C., Wall, M., et al.: Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of P53. Cancer Cell 22(1), 51–65 (2012). https://doi.org/10.1016/j.ccr.2012.05.019
Calabrese, D.R., Chen, X., Leon, E.C., Gaikwad, S.M., Phyo, Z., Hewitt, W.M., Alden, S., et al.: Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat. Commun. 9(1), 4229 (2018). https://doi.org/10.1038/s41467-018-06315-w
Carabet, L., Rennie, P., Cherkasov, A.: Therapeutic inhibition of Myc in cancer. Structural bases and computer-aided drug discovery approaches. Int. J. Mol. Sci. 20(1), 120 (2018). https://doi.org/10.3390/ijms20010120
Cartwright, P., McLean, C., Sheppard, A., Rivett, D., Jones, K., Dalton, S.: LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 132(5), 885–896 (2005). https://doi.org/10.1242/dev.01670
Cashman, D.J., Buscaglia, R., Freyer, M.W., Dettler, J., Hurley, L.H., Lewis, E.A.: Molecular modeling and biophysical analysis of the C-MYC NHE-III1 silencer element. J. Mol. Model. 14(2), 93–101 (2008). https://doi.org/10.1007/s00894-007-0254-z
Castell, A., Yan, Q., Fawkner, K., Hydbring, P., Zhang, F., Verschut, V., Franco, M., et al.: A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 8(1), 10064 (2018). https://doi.org/10.1038/s41598-018-28107-4
Chadwick, J., Francois, T., Jonathan, B.: From CPU to GP-GPU. In Proceedings of the 10th International Workshop on Middleware for Grids, Clouds and e-Science, 1–6. New York, NY, USA: ACM. (2012) https://doi.org/10.1145/2405136.2405142.
Chen, B.J., Yan Ling, Wu., Tanaka, Y., Zhang, W.: Small molecules targeting C-Myc oncogene: promising anti-cancer therapeutics. Int. J. Biol. Sci. 10(10), 1084–1096 (2014). https://doi.org/10.7150/ijbs.10190
Chen, H., Liu, H., Qing, G.: Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 3(1), 5 (2018). https://doi.org/10.1038/s41392-018-0008-7
Chipumuro, E., Marco, E., Christensen, C.L., Kwiatkowski, N., Zhang, T., Hatheway, C.M., Abraham, B.J., et al.: CDK7 Inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159(5), 1126–1139 (2014). https://doi.org/10.1016/j.cell.2014.10.024
Cross, D.A.E., Alessi, D.R., Cohen, P., Andjelkovich, M., Hemmings, B.A.: Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378(6559), 785–789 (1995). https://doi.org/10.1038/378785a0
Cuny, Z. G.: Characterization of pyridostatin and its interactions with c-MYC g-quadruplexes. Mississippi State University (2017) https://scholarsjunction.msstate.edu/honorstheses/21?utm_source=scholarsjunction.msstate.edu%2Fhonorstheses%2F21&utm_medium=PDF&utm_campaign=PDFCoverPages.
Dai, J., Carver, M., Hurley, L.H., Yang, D.: Solution structure of a 2:1 quindoline–c-MYC G-quadruplex: insights into G-quadruplex-interactive small molecule drug design. J. Am. Chem. Soc. 133(44), 17673–17680 (2011). https://doi.org/10.1021/ja205646q
Dauch, D., Rudalska, R., Cossa, G., Nault, J.-C., Kang, T.-W., Wuestefeld, T., Hohmeyer, A., et al.: A MYC–aurora kinase A protein complex represents an actionable drug target in P53-altered liver cancer. Nat. Med. 22(7), 744–753 (2016). https://doi.org/10.1038/nm.4107
David, A., Arnaud, N., Fradet, M., Lascaux, H., Ouk-Martin, C., Gachard, N., Zimber-Strobl, U., Feuillard, J., Faumont, N.: C-Myc Dysregulation is a co-transforming event for nuclear factor-ΚB activated B cells. Haematologica 102(5), 883–894 (2017). https://doi.org/10.3324/haematol.2016.156281
Delmore, J.E., Issa, G.C., Lemieux, M.E., Rahl, P.B., Shi, J., Jacobs, H.M., Kastritis, E., et al.: BET bromodomain inhibition as a therapeutic strategy to target C-Myc. Cell 146(6), 904–917 (2011). https://doi.org/10.1016/j.cell.2011.08.017
DePinho, R.A., Legouy, E., Feldman, L.B., Kohl, N.E., Yancopoulos, G.D., Alt, F.W.: Structure and expression of the murine N-Myc gene. Proc. Natl. Acad. Sci. 83(6), 1827–1831 (1986). https://doi.org/10.1073/pnas.83.6.1827
Devi, G.R., Beer, T.M., Corless, C.L., Arora, V., Weller, D.L., Iversen, P.L.: In Vivo bioavailability and pharmacokinetics of a c-MYC antisense phosphorodiamidate morpholino oligomer, AVI-4126, in solid tumors. Clin. Cancer Res. 11(10), 3930–3938 (2005). https://doi.org/10.1158/1078-0432.CCR-04-2091
Dickerhoff, J., Brundridge, N., McLuckey, S.A., Yang, D.: Berberine molecular recognition of the parallel MYC G-quadruplex in solution. J. Med. Chem. 64(21), 16205–16212 (2021a). https://doi.org/10.1021/acs.jmedchem.1c01508
Dickerhoff, J., Dai, J., Yang, D.: Structural recognition of the MYC promoter G-quadruplex by a quinoline derivative: insights into molecular targeting of parallel G-quadruplexes. Nucleic Acids Res. 49(10), 5905–5915 (2021b). https://doi.org/10.1093/nar/gkab330
Drygin, D., Siddiqui-Jain, A., O’Brien, S., Schwaebe, M., Lin, A., Bliesath, J., Ho, C.B., et al.: Anticancer activity of CX-3543: a direct inhibitor of RRNA biogenesis. Can. Res. 69(19), 7653–7661 (2009). https://doi.org/10.1158/0008-5472.CAN-09-1304
DuBois, S.G., Mosse, Y.P., Fox, E., Kudgus, R.A., Reid, J.M., McGovern, R., Groshen, S., et al.: Phase II trial of Alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma. Clin. Cancer Res. 24(24), 6142–6149 (2018). https://doi.org/10.1158/1078-0432.CCR-18-1381
Dudley, J.P., Mertz, J.A., Rajan, L., Lozano, M., Broussard, D.R.: What retroviruses teach us about the involvement of C-Myc in Leukemias and Lymphomas. Leukemia 16(6), 1086–1098 (2002). https://doi.org/10.1038/sj.leu.2402451
Evan, G., Littlewood, T.: A matter of life and cell death. Science 281(5381), 1317–1322 (1998). https://doi.org/10.1126/science.281.5381.1317
Fatafta, H., Samantray, S., Sayyed-Ahmad, A., Coskuner-Weber, O., Strodel, B.: Molecular simulations of IDPs: from ensemble generation to IDP interactions leading to disorder-to-order transitions. Prog. Mol. Biol. Transl. Sci. (2021). https://doi.org/10.1016/bs.pmbts.2021.06.003
Fatma, H., Maurya, S.K., Siddique, H.R.: Epigenetic modifications of C-MYC: role in cancer cell reprogramming, progression and chemoresistance. Semin. Cancer Biol. 83(August), 166–176 (2022). https://doi.org/10.1016/j.semcancer.2020.11.008
Felsher, D.W., Michael Bishop, J.: Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc. Natl. Acad. Sci. 96(7), 3940–3944 (1999). https://doi.org/10.1073/pnas.96.7.3940
Fernandez, P.C., Frank, S.R., Wang, L., Schroeder, M., Liu, S., Greene, J., Cocito, A., Amati, B.: Genomic targets of the human C-Myc protein. Genes Dev. 17(9), 1115–1129 (2003). https://doi.org/10.1101/gad.1067003
Filippakopoulos, P., Qi, J., Picaud, S., Shen, Y., Smith, W.B., Fedorov, O., Morse, E.M., et al.: Selective inhibition of BET bromodomains. Nature 468(7327), 1067–1073 (2010). https://doi.org/10.1038/nature09504
Fuhrmann, G., Rosenberger, G., Grusch, M., Klein, N., Hofmann, J., Krupitza, G.: The MYC dualism in growth and death. Mutat. Res. Rev. Mutat. Res. 437(3), 205–217 (1999). https://doi.org/10.1016/S1383-5742(99)00084-8
Galardi, S., Savino, M., Scagnoli, F., Pellegatta, S., Pisati, F., Zambelli, F., Illi, B., et al.: Resetting cancer stem cell regulatory nodes upon MYC inhibition. EMBO Rep. 17(12), 1872–89 (2016). https://doi.org/10.15252/embr.201541489
Gordan, J.D., Bertout, J.A., Cheng-Jun, Hu., Alan Diehl, J., Celeste Simon, M.: HIF-2α promotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer Cell 11(4), 335–347 (2007). https://doi.org/10.1016/j.ccr.2007.02.006
Grand, C.L., Han, H., Muñoz, R.M., Weitman, S., Von Hoff, D.D., Hurley, L.H., Bearss, D.J.: The cationic porphyrin TMPyP4 down-regulates c-MYC and human telomerase reverse transcriptase expression and inhibits tumor growth in vivo. Mol. Cancer Ther. 1(8), 565–573 (2002)
Greenberg, R.A., O’Hagan, R.C., Deng, H., Xiao, Q., Hann, S.R., Adams, R.R., Lichtsteiner, S., Chin, L., Morin, G.B., DePinho, R.A.: Telomerase reverse transcriptase gene is a direct target of C-Myc but is not functionally equivalent in cellular transformation. Oncogene 18(5), 1219–1226 (1999). https://doi.org/10.1038/sj.onc.1202669
Greenwood, E.: The many faces of MYC. Nat. Rev. Cancer 2(7), 485–485 (2002). https://doi.org/10.1038/nrc851
Gregory, M.A., Hann, S.R.: C-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol. Cell. Biol. 20(7), 2423–2435 (2000). https://doi.org/10.1128/MCB.20.7.2423-2435.2000
Günes, Ç., et al.: Expression of the HTERT gene is regulated at the level of transcriptional initiation and repressed by Mad1. Cancer Res. 60, 2116–2121 (2000)
Hald, Ø.H., Olsen, L., Gallo-Oller, G., Elfman, L.H.M., Løkke, C., Kogner, P., Sveinbjörnsson, B., Flægstad, T., Johnsen, J.I., Einvik, C.: Inhibitors of ribosome biogenesis repress the growth of MYCN-amplified neuroblastoma. Oncogene 38(15), 2800–2813 (2019). https://doi.org/10.1038/s41388-018-0611-7
Han, M., Jia, L., Lv, W., Wang, L., Cui, W.: Epigenetic enzyme mutations: role in tumorigenesis and molecular inhibitors. Front. Oncol. (2019). https://doi.org/10.3389/fonc.2019.00194
Heidelberger, J.B., Voigt, A., Borisova, M.E., Petrosino, G., Ruf, S., Wagner, S.A., Beli, P.: Proteomic profiling of VCP substrates links VCP to K6-linked ubiquitylation and C-Myc function. EMBO Rep. (2018). https://doi.org/10.15252/embr.201744754
Henriksson, M., Bakardjiev, A., Klein, G., Lüscher, B.: Phosphorylation sites mapping in the N-terminal domain of c-Myc modulate its transforming potential. Oncogene 8, 3199–3209 (1993)
Hermeking, H., Rago, C., Schuhmacher, M., Li, Q., Barrett, J.F., Obaya, A.J., O’Connell, B.C., et al.: Identification of CDK4 as a target of c-MYC. Proc. Natl. Acad. Sci. 97(5), 2229–2234 (2000). https://doi.org/10.1073/pnas.050586197
Herold, S., Kalb, J., Büchel, G., Ade, C.P., Baluapuri, A., Jiajia, Xu., Koster, J., et al.: Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nature 567(7749), 545–549 (2019). https://doi.org/10.1038/s41586-019-1030-9
Huber, A.-L., Papp, S.J., Chan, A.B., Henriksson, E., Jordan, S.D., Kriebs, A., Nguyen, M., et al.: CRY2 and FBXL3 cooperatively degrade C-MYC. Mol. Cell 64(4), 774–789 (2016). https://doi.org/10.1016/j.molcel.2016.10.012
Jaenicke, L.A., von Eyss, B., Carstensen, A., Wolf, E., Wenshan, Xu., Greifenberg, A.K., Geyer, M., Eilers, M., Popov, N.: Ubiquitin-dependent turnover of MYC antagonizes MYC/PAF1C complex accumulation to drive transcriptional elongation. Mol. Cell 61(1), 54–67 (2016). https://doi.org/10.1016/j.molcel.2015.11.007
Jiang, H., Bower, K.E., Beuscher, A.E., Zhou, B., Bobkov, A.A., Olson, A.J., Vogt, P.K.: Stabilizers of the max homodimer identified in virtual ligand screening inhibit Myc function. Mol. Pharmacol. 76(3), 491–502 (2009). https://doi.org/10.1124/mol.109.054858
Jin, M., Hurley, L.H., Hong, Xu.: A synthetic lethal approach to drug targeting of G-quadruplexes based on CX-5461. Bioorg. Med. Chem. Lett. 91, 129384 (2023). https://doi.org/10.1016/j.bmcl.2023.129384
Juliano, R.L.: The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 44(14), 6518–6548 (2016). https://doi.org/10.1093/nar/gkw236
Jung, K.Y., Wang, H., Teriete, P., Yap, J.L., Chen, L., Lanning, M.E., Angela, Hu., et al.: Perturbation of the C-Myc-max protein-protein interaction via synthetic α-helix mimetics. J. Med. Chem. 58(7), 3002–3024 (2015). https://doi.org/10.1021/jm501440q
Kalkat, M., Resetca, D., Lourenco, C., Chan, P.-K., Wei, Y., Shiah, Y.-J., Vitkin, N., et al.: MYC protein interactome profiling reveals functionally distinct regions that cooperate to drive tumorigenesis. Mol. Cell 72(5), 836-848.e7 (2018). https://doi.org/10.1016/j.molcel.2018.09.031
Kato, G.J., Barrett, J., Villa-Garcia, M., Dang, C.V.: An amino-terminal c-Myc domain required for neoplastic transformation activates transcription. Mol. Cell. Biol. 10(11), 5914–5920 (1990). https://doi.org/10.1128/mcb.10.11.5914-5920.1990
Kawaguchi, H., Yuichi, N., Shogo O.: IDPS signature classification with a reject option and the incorporation of expert knowledge. In 2022 21st IEEE International Conference on Machine Learning and Applications (ICMLA), 623–28. IEEE. (2022). https://doi.org/10.1109/ICMLA55696.2022.00096.
Kawauchi, D., Robinson, G., Uziel, T., Gibson, P., Rehg, J., Gao, C., Finkelstein, D., et al.: A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 21(2), 168–180 (2012). https://doi.org/10.1016/j.ccr.2011.12.023
Kit, O.I., Vodolazhsky, D.I., Kutilin, D.S., Enin, Y.S., Gevorkyan, Y.A., Zolotukhin, P.V., Boumber, Y., Kharin, L.V., Panina, S.B.: A proteomics analysis reveals 9 up-regulated proteins associated with altered cell signaling in colon cancer patients. Protein. J. 36(6), 513–522 (2017). https://doi.org/10.1007/s10930-017-9746-6
Kohl, N.E., Kanda, N., Schreck, R.R., Bruns, G., Latt, S.A., Gilbert, F., Alt, F.W.: Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 35(2), 359–367 (1983). https://doi.org/10.1016/0092-8674(83)90169-1
Kumar, D., Sharma, N., Giri, R.: Therapeutic interventions of cancers using intrinsically disordered proteins as drug targets: C-Myc as model system. Cancer Inform. 16, 117693511769940 (2017). https://doi.org/10.1177/1176935117699408
Kyo, S.: Sp1 cooperates with C-Myc to activate transcription of the human telomerase reverse transcriptase gene (HTERT). Nucleic Acids Res. 28(3), 669–677 (2000). https://doi.org/10.1093/nar/28.3.669
Lancho, O., Herranz, D.: The MYC enhancer-ome: long-range transcriptional regulation of MYC in cancer. Trends in Cancer 4(12), 810–822 (2018). https://doi.org/10.1016/j.trecan.2018.10.003
Land, H., Parada, L.F., Weinberg, R.A.: Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304(5927), 596–602 (1983). https://doi.org/10.1038/304596a0
Li, Xu., Li, L., Li, Y., Zhao, J., Fan, S., Wang, L.: CX-5461 Induces autophagy and inhibits tumor growth via mammalian target of rapamycin-related signaling pathways in osteosarcoma. Onco. Targets. Ther. 9(September), 5985–5997 (2016). https://doi.org/10.2147/OTT.S104513
Li, Y., Sun, X.-X., Qian, D.Z., Dai, M.-S.: Molecular crosstalk between MYC and HIF in cancer. Front. Cell Develop. Biol. (2020). https://doi.org/10.3389/fcell.2020.590576
Li, Y., Zhang, B., Zhang, H., Zhu, X., Feng, D., Zhang, D., Zhuo, B., Li, L., Zheng, J.: Oncolytic adenovirus armed with ShRNA targeting MYCN gene inhibits neuroblastoma cell proliferation and in vivo xenograft tumor growth. J. Cancer Res. Clin. Oncol. 139(6), 933–941 (2013). https://doi.org/10.1007/s00432-013-1406-4
Liao, D.J., Dickson, R.B.: C-Myc in breast cancer. Endocr. Relat. Cancer 7(3), 143–164 (2000)
Lombardi, L., Newcomb, E.W., Dalla-Favera, R.: Pathogenesis of Burkitt Lymphoma: expression of an activated c-Myc oncogene causes the tumorigenic conversion of EBV-infected human B Lymphoblasts. Cell 49(2), 161–170 (1987). https://doi.org/10.1016/0092-8674(87)90556-3
Lorentzen, A., Lewinsky, R.H., Bornholdt, J., Vogel, L.K., Mitchelmore, C.: Expression profile of the N-Myc downstream regulated gene 2 (NDRG2) in human cancers with focus on breast cancer. BMC Cancer 11(1), 14 (2011). https://doi.org/10.1186/1471-2407-11-14
Louis, S.F., Vermolen, B.J., Garini, Y., Young, I.T., Guffei, A., Lichtensztejn, Z., Kuttler, F., et al.: C-Myc induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. Proc. Natl. Acad. Sci. 102(27), 9613–9618 (2005). https://doi.org/10.1073/pnas.0407512102
Lovén, J., Hoke, H.A., Lin, C.Y., Lau, A., Orlando, D.A., Vakoc, C.R., Bradner, J.E., Lee, T.I., Young, R.A.: Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153(2), 320–334 (2013). https://doi.org/10.1016/j.cell.2013.03.036
Ma, Y., Tian-Miao, Ou., Hou, J.-Q., Yu-Jing, Lu., Tan, J.-H., Lian-Quan, Gu., Huang, Z.-S.: 9-N-Substituted Berberine derivatives: stabilization of G-quadruplex DNA and down-regulation of oncogene c-Myc. Bioorg. Med. Chem. 16(16), 7582–7591 (2008). https://doi.org/10.1016/j.bmc.2008.07.029
Mark, P., Nilsson, L.: Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 105(43), 9954–9960 (2001). https://doi.org/10.1021/jp003020w
Matkar, S., Sharma, P., Gao, S., Gurung, B., Katona, B.W., Liao, J., Muhammad, A.B., et al.: An epigenetic pathway regulates sensitivity of breast cancer cells to HER2 inhibition via FOXO/c-Myc axis. Cancer Cell 28(4), 472–485 (2015). https://doi.org/10.1016/j.ccell.2015.09.005
Miller, D.M., Thomas, S.D., Islam, A., Muench, D., Sedoris, K.: C-Myc and cancer metabolism. Clin. Cancer Res. 18(20), 5546–5553 (2012). https://doi.org/10.1158/1078-0432.CCR-12-0977
Furtado, M., Libardi, C., Luciano, M.C.D.S., Da Silva, R., Santos, G.P., Furtado, M.O., Moraes, C.P.: Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics 14(12), 1164–76 (2019). https://doi.org/10.1080/15592294.2019.1640546
Muhar, M., Ebert, A., Neumann, T., Umkehrer, C., Jude, J., Wieshofer, C., Rescheneder, P., et al.: SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360(6390), 800–805 (2018). https://doi.org/10.1126/science.aao2793
Murphy, D.J., Junttila, M.R., Pouyet, L., Karnezis, A., Shchors, K., Bui, D.A., Brown-Swigart, L., Johnson, L., Evan, G.I.: Distinct thresholds Govern Myc’s biological output in vivo. Cancer Cell 14(6), 447–457 (2008). https://doi.org/10.1016/j.ccr.2008.10.018
Nair, S.K., Burley, S.K.: X-Ray structures of Myc-max and mad-max recognizing DNA. Cell 112(2), 193–205 (2003). https://doi.org/10.1016/S0092-8674(02)01284-9
Nau, M.M., Brooks, B.J., Battey, J., Sausville, E., Gazdar, A.F., Kirsch, I.R., Wesley McBride, O., Bertness, V., Hollis, G.F., Minna, J.D.: L-Myc, a new Myc-related gene amplified and expressed in human small cell lung cancer. Nature 318(6041), 69–73 (1985). https://doi.org/10.1038/318069a0
Oh, S., Song, Y.-H., Kim, U.-J., Yim, J., Kim, T.K.: In Vivo and in vitro analyses of Myc for differential promoter activities of the human telomerase (HTERT) gene in normal and tumor cells. Biochem. Biophys. Res. Commun. 263(2), 361–365 (1999). https://doi.org/10.1006/bbrc.1999.1366
Oh, S., Song, Y.-H., Yim, J., Kim, T.K.: Identification of mad as a repressor of the human telomerase (HTERT) gene. Oncogene 19(11), 1485–1490 (2000). https://doi.org/10.1038/sj.onc.1203439
Junior, O., Antonio, B., Lin, X., Kulkarni, P., Onuchic, J.N., Roy, S., Leite, V.B.P.: Exploring energy landscapes of intrinsically disordered proteins: insights into functional mechanisms. J. Chem. Theory Comput. 17(5), 3178–3187 (2021). https://doi.org/10.1021/acs.jctc.1c00027
Palm, W., de Lange, T.: How Shelterin protects mammalian telomeres. Annu. Rev. Genet. 42(1), 301–334 (2008). https://doi.org/10.1146/annurev.genet.41.110306.130350
Pelengaris, S., Khan, M., Evan, G.: C-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2(10), 764–776 (2002). https://doi.org/10.1038/nrc904
Peter, S., Bultinck, J., Myant, K., Jaenicke, L.A., Walz, S., Müller, J., Gmachl, M., et al.: Tumor cell-specific inhibition of MYC function using small molecule inhibitors of the HUWE 1 ubiquitin ligase. EMBO Mol. Med. 6(12), 1525–41 (2014). https://doi.org/10.15252/emmm.201403927
Ponzielli, R., Katz, S., Barsyte-Lovejoy, D., Penn, L.Z.: Cancer therapeutics: targeting the dark side of Myc. Eur. J. Cancer 41(16), 2485–2501 (2005). https://doi.org/10.1016/j.ejca.2005.08.017
Postel-Vinay, S., Herbschleb, K., Massard, C., Woodcock, V., Soria, J.-C., Walter, A.O., Ewerton, F., et al.: First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur. J. Cancer 109(March), 103–110 (2019). https://doi.org/10.1016/j.ejca.2018.12.020
Prendergast, G., Ziff, E.: A new bind for Myc. Trends Genet. 8(3), 91–96 (1992). https://doi.org/10.1016/0168-9525(92)90196-B
Raina, K., Jing, Lu., Qian, Y., Altieri, M., Gordon, D., Ann, M.K., Rossi, J.W., et al.: PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. 113(26), 7124–7129 (2016). https://doi.org/10.1073/pnas.1521738113
Rayburn, E., Zhang, R.: Antisense, RNAi, and gene silencing strategies for therapy: mission possible or impossible? Drug Discov. Today 13(11–12), 513–21 (2008). https://doi.org/10.1016/j.drudis.2008.03.014
Sakamoto, K.M., Kim, K.B., Kumagai, A., Mercurio, F., Crews, C.M., Deshaies, R.J.: Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. 98(15), 8554–8559 (2001). https://doi.org/10.1073/pnas.141230798
Sakamuro, D., Prendergast, G.C.: New Myc-interacting proteins: a second Myc network emerges. Oncogene 18(19), 2942–2954 (1999). https://doi.org/10.1038/sj.onc.1202725
Salghetti, S.E.: Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 18(3), 717–726 (1999). https://doi.org/10.1093/emboj/18.3.717
Salomon-Ferrer, R., Götz, A.W., Poole, D., Le Grand, S., Walker, R.C.: Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. explicit solvent particle Mesh Ewald. J. Chem. Theory Comput. 9(9), 3878–3888 (2013). https://doi.org/10.1021/ct400314y
Sammak, S., Hamdani, N., Gorrec, F., Allen, M.D., Freund, S.M.V., Bycroft, M., Zinzalla, G.: Crystal structures and nuclear magnetic resonance studies of the Apo form of the C-MYC:MAX BHLHZip complex reveal a helical basic region in the absence of DNA. Biochemistry 58(29), 3144–3154 (2019). https://doi.org/10.1021/acs.biochem.9b00296
Savino, M., Annibali, D., Carucci, N., Favuzzi, E., Cole, M.D., Evan, G.I., Soucek, L., Nasi, S.: The action mechanism of the Myc inhibitor termed omomyc may give clues on how to target Myc for cancer therapy. PLoS ONE 6(7), e22284 (2011). https://doi.org/10.1371/journal.pone.0022284
Sears, R., Leone, G., DeGregori, J., Nevins, J.R.: Ras enhances Myc protein stability. Mol. Cell 3, 169–179 (1999)
Sebestyén, M.G., Wong, S.C., Trubetskoy, V., Lewis, D.L., Wooddell, C.I.: Targeted in vivo delivery of SiRNA and an endosome-releasing agent to hepatocytes. In: RNA interference: challenges and therapeutic opportunities. Springer, New York (2015). https://doi.org/10.1007/978-1-4939-1538-5_10
Sheiness, D., Michael Bishop, J.: DNA and RNA from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian Myelocytomatosis Virus. J. Virol. 31(2), 514–521 (1979). https://doi.org/10.1128/jvi.31.2.514-521.1979
Siegel, R.L., Miller, K.D., Fuchs, H.E., Jemal, A.: Cancer statistics 2022. CA: Cancer J. Clin. 72(1), 7–33 (2022). https://doi.org/10.3322/caac.21708
Simonsson, T.: DNA tetraplex formation in the control region of C-Myc. Nucleic Acids Res. 26(5), 1167–1172 (1998). https://doi.org/10.1093/nar/26.5.1167
Singh, A., Sharma, S., Kumar, P., Garg, N.: Cellular experiments to study the inhibition of C-Myc/MAX heterodimerization. In: Integrated methods in protein biochemistry: Part A, pp. 193–205. Elsevier, Amsterdam (2022). https://doi.org/10.1016/bs.mie.2022.07.009
Soucek, L., Evan, G.: Myc—is this the oncogene from hell? Cancer Cell 1(5), 406–408 (2002). https://doi.org/10.1016/S1535-6108(02)00077-6
Soucek, L., Whitfield, J., Martins, C.P., Finch, A.J., Murphy, D.J., Sodir, N.M., Karnezis, A.N., Swigart, L.B., Nasi, S., Evan, G.I.: Modelling Myc inhibition as a cancer therapy. Nature 455(7213), 679–683 (2008). https://doi.org/10.1038/nature07260
Spencer, Charlotte A., Groudine, Mark: Control of C-Myc regulation in normal and neoplastic cells, pp. 1–48. Elsevier, Amsterdam (1991). https://doi.org/10.1016/S0065-230X(08)60476-5
Staller, P., Peukert, K., Kiermaier, A., Seoane, J., Lukas, J., Karsunky, H., Möröy, T., et al.: Repression of P15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 3(4), 392–399 (2001). https://doi.org/10.1038/35070076
Stathis, A., Bertoni, F.: BET proteins as targets for anticancer treatment. Cancer Discov. 8(1), 24–36 (2018). https://doi.org/10.1158/2159-8290.CD-17-0605
Stone, J., de Lange, T., Ramsay, G., Edward Jakobovits, J., Bishop, M., Varmus, H., Lee, W.: Definition of regions in human C- Myc that are involved in transformation and nuclear localization. Mol. Cell. Biol. 7(5), 1697–1709 (1987). https://doi.org/10.1128/mcb.7.5.1697-1709.1987
Stone, J. E., Michael J. H., James C. P., Joseph R. P., Zaida L.-S., Klaus S.: Evaluation of emerging energy-efficient heterogeneous computing platforms for biomolecular and cellular simulation workloads. In 2016 IEEE International Parallel and Distributed Processing Symposium Workshops (IPDPSW), 89–100. IEEE. (2016). https://doi.org/10.1109/IPDPSW.2016.130.
Struntz, N.B., Chen, A., Deutzmann, A., Wilson, R.M., Stefan, E., Evans, H.L., Ramirez, M.A., et al.: Stabilization of the max homodimer with a small molecule attenuates Myc-driven transcription. Cell Chem. Biol. 26(5), 711-723.e14 (2019). https://doi.org/10.1016/j.chembiol.2019.02.009
Stukenberg, P.T., Kirschner, M.W.: Pin1 acts catalytically to promote a conformational change in Cdc25. Mol. Cell 7(5), 1071–1083 (2001). https://doi.org/10.1016/S1097-2765(01)00245-3
Sullivan, S.S., Weinzierl, R.O.J.: Optimization of molecular dynamics simulations of C-MYC1-88—an intrinsically disordered system. Life 10(7), 109 (2020). https://doi.org/10.3390/life10070109
Taufer, M., Ganesan, N., Patel, S.: GPU-enabled macromolecular simulation: challenges and opportunities. Comput. Sci. Eng. 15(1), 56–65 (2013). https://doi.org/10.1109/MCSE.2012.42
Turnbull, A.P., Ioannidis, S., Krajewski, W.W., Pinto-Fernandez, A., Heride, C., Martin, A.C.L., Tonkin, L.M., et al.: Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 550(7677), 481–486 (2017). https://doi.org/10.1038/nature24451
Vickers, N.J.: Animal communication: When I’m Calling You, Will You Answer Too? Curr. Biol. 27(14), R713–R715 (2017). https://doi.org/10.1016/j.cub.2017.05.064
Wang, J., Xie, L.Y., Allan, S., Beach, D., Hannon, G.J.: Myc activates telomerase. Genes Dev. 12(12), 1769–1774 (1998). https://doi.org/10.1101/gad.12.12.1769
Welcker, M., Orian, A., Jin, J., Grim, J.A., Wade Harper, J., Eisenman, R.N., Clurman, B.E.: The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. 101(24), 9085–9090 (2004). https://doi.org/10.1073/pnas.0402770101
Whitfield, J.R., Beaulieu, M.-E., Soucek, L.: Strategies to inhibit Myc and their clinical applicability. Front. Cell Develop. Biol. (2017). https://doi.org/10.3389/fcell.2017.00010
Wiegering, A., Uthe, F.W., Jamieson, T., Ruoss, Y., Hüttenrauch, M., Küspert, M., Pfann, C., et al.: Targeting translation initiation bypasses signaling crosstalk mechanisms that maintain high MYC levels in colorectal cancer. Cancer Discov. 5(7), 768–781 (2015). https://doi.org/10.1158/2159-8290.CD-14-1040
Wolf, E., Eilers, M.: Targeting MYC proteins for tumor therapy. Ann. Rev. Cancer Biol. 4(1), 61–75 (2020). https://doi.org/10.1146/annurev-cancerbio-030518-055826
World Health Organization.: Cancer Control: A Global Snapshot in 2015. (2016). https://www.who.int/publications-detail-redirect/cancer-control-a-global-snapshot-in-2015.
World Health Organization.: Cancer. World Health Organization. (2022). https://www.who.int/news-room/fact-sheets/detail/cancer.
Yap, J.L., Wang, H., Angela, Hu., Chauhan, J., Jung, K.-Y., Gharavi, R.B., Prochownik, E.V., Fletcher, S.: Pharmacophore identification of C-Myc inhibitor 10074–G5. Bioorg. Med. Chem. Lett. 23(1), 370–374 (2013). https://doi.org/10.1016/j.bmcl.2012.10.013
Yeh, E., Cunningham, M., Arnold, H., Chasse, D., Monteith, T., Ivaldi, G., Hahn, W.C., et al.: A signalling pathway controlling C-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 6(4), 308–318 (2004). https://doi.org/10.1038/ncb1110
Yin, X., Giap, C., Lazo, J.S., Prochownik, E.V.: Low molecular weight inhibitors of Myc-max interaction and function. Oncogene 22(40), 6151–6159 (2003). https://doi.org/10.1038/sj.onc.1206641
Zhang, Q., West-Osterfield, K., Spears, E., Li, Z., Panaccione, A., Hann, S.: MB0 and MBI are independent and distinct transactivation domains in MYC that are essential for transformation. Genes 8(5), 134 (2017). https://doi.org/10.3390/genes8050134
Zhao, Z., Shilatifard, A.: Epigenetic modifications of histones in cancer. Genome Biol. 20(1), 245 (2019). https://doi.org/10.1186/s13059-019-1870-5
Zhou, X.Z.: Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and Tau proteins. Mol. Cell 6, 873–883 (2000)
Zhu, J.-J., Zhang, N.-J., Wei, T., Chen, H.-F.: Enhancing conformational sampling for intrinsically disordered and ordered proteins by Variational Autoencoder. Int. J. Mol. Sci. 24(8), 6896 (2023). https://doi.org/10.3390/ijms24086896
Zimmerman, K.A., Yancopoulos, G.D., Collum, R.G., Smith, R.K., Kohl, N.E., Denis, K.A., Nau, M.M., et al.: Differential expression of Myc family genes during murine development. Nature 319(6056), 780–783 (1986). https://doi.org/10.1038/319780a0
Acknowledgements
I would like to thank Manipal University Jaipur for granting me with Ramdas Pai Scholarship for PhD scholars.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no competing interest amongst the authors.
Ethical approval
There is an ethical approval to submit and publish the Manuscript.
Consent to participate
Authors gave their consent to participate in any kind of survey, questionnaire, etc.
Consent to publish
Authors gave their consent to publish the manuscript.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bamrah, G.K., Kumari, N. & Srivastava, S. Recent advances in anti-tumor therapeutic approaches for the universally active transcriptional factor c-MYC. Proc.Indian Natl. Sci. Acad. 90, 576–593 (2024). https://doi.org/10.1007/s43538-024-00244-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43538-024-00244-7