
Abstract
Excessive administration of the antipyretic and analgesic drug acetaminophen can cause acute liver injury. The mechanism is that a large amount of N-acetyl-p-benzoquinone-imine is generated through the metabolism of cytochrome P450 (CYP450), which leads to cell necrosis, oxidative stress, and inflammation. The purpose of this study was to investigate the protective effect of psoralen on acetaminophen-induced acute liver injury, and to clarify the isoform CYP2E1 of human cytochrome P450 in the efficacy of it. The results showed that psoralen ameliorated hepatocyte necrosis by decreasing serum aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, oxidative stress index malondialdehyde and inflammatory cytokines, such as interleukins (Il6 and Il1β) and tumor necrosis factor (Tnfα) in mice. Molecular docking suggested that psoralen could bind to active sites of CYP2E1, CYP1A2 and CYP3A11. A single dose of psoralen could significantly inhibit the enzymatic activity of CYP2E1 in rats, while administration of psoralen for 5 days had no significant effect on CYP2E1. In conclusion, psoralen can ameliorate acetaminophen-induced acute liver injury, specifically by reducing hepatocyte necrosis, inflammation and oxidative stress. Psoralen could be the substrate of CYP2E1 which can competitively bind to its active site, thereby competing with acetaminophen, thus inhibiting the enzymatic activity of CYP2E1 to have an impact on rates of drug metabolism in the liver.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Acetaminophen (1) is a common antipyretic and analgesic drug, but overdose of it can cause acute liver injury (Ruepp et al. 2002; Lee 2017; Bunchorntavakul and Reddy 2018). Eighty five to ninety percent of acetaminophen undergoes phase II conjugation to sulfated and glucuronidated metabolites, which are excreted into bile, and the remaining 5–9% is metabolized by the phase I metabolic cytochrome P450 (CYP450) enzymatic system, mainly the isoform CYP2E1, producing the extremely toxic intermediate to liver tissue N-acetyl-p-benzoquinone-imine (2), which reacts at physiological pH with glutathione to be inactivated, forming an acerminophen-glutathione conjugate, which is rapidly detoxified (Bunchorntavakul and Reddy 2013, 2018). However, excessive acetaminophen will generate a large amount of N-acetyl-p-benzoquinone-imine, resulting in a subsequent covalent bounding to proteins and other biomolecules, mitochondrial oxidative stress, and hepatocyte necrosis, ultimately causing acute liver injury (Jaeschke et al. 2018). Scheme 1 illustrates the acetaminophen metabolism in the liver (Larson 2007).

Acetaminophen (1) metabolism in the liver. Pathways shown in blue and purple lead to nontoxic metabolites; the pathways in red lead to N-acetyl-p-benzoquinone-imine (2), which is toxic if not conjugated to glutathione
At present, acetaminophen-induced acute liver injury is mainly treated by liver transplantation or the antioxidant N-acetylcysteine. However, liver transplantation is expensive, immune rejection is prone to occur, and treatment with the antioxidant N-acetylcysteine is only used for the early stage of the disease (Hu et al. 2020). Both methods have limitations, so it is of great significance to find a drug that can effectively treat acetaminophen-induced acute liver injury.
Psoralea corylifolia L., Fabaceae, is a commonly used traditional Chinese medicine in clinical therapies. It has multiple pharmacological effects, such as anti-cancer, anti-osteoporosis and anti-psoralenriasis (Zhang et al. 2016). Psoralen (3), a furocoumarin, is one of the active components of P. corylifolia. It has been reported that furanocoumarins have complex interactions with CYP450 enzymes. Furanocoumarins are usually metabolized by CYP enzyme systems, and in turn inhibit the activity of them (Uesawa and Mohri 2010). Psoralen has curative effect on liver diseases such as hepatic steatosis, liver fibrosis, and liver cancer (Ren et al. 2020). Studies have shown that P. corylifolia can improve non-alcoholic steatosis of L02 cells induced by palmitic acid (Zhou et al. 2017). Psoralen can inhibit the malignant proliferation of SMMC7721 hepatoma cells (Wang et al. 2019). However, there are few studies on the protective effects of psoralen against acute liver injury. Therefore, this study is aimed to investigate the ameliorative effect of psoralen on acetaminophen-induced acute liver injury and to clarify the role of CYP2E1 in its efficacy.

Materials and Methods
Animals
Eight-week-old C57BL/6J male mice (16–19 g) were purchased from Shanghai Slac Laboratory Animal Co., Ltd., Shanghai, China (Production License No. SCXK 2018-0003). Eight-week-old Wistar male rats (180–200 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China (Production License No. SCXK 2016-0006). The animals were acclimated for 1 week before treatments. The animals were kept in the barrier system, and the laboratory temperature was 24 ± 2 °C relative humidity 60 ~ 80%; light cycle: 12 h day/night. Adequate amounts of rodent chow and purified water were provided daily. All animal care and handling protocols were approved by the Animal Ethics Committee of China Pharmaceutical University. All experiments conformed with the recommendations of the Guidelines for Ethical Conduct in the Care and Use of Animals.
Experimental Design
The mice were randomly divided into six groups (n = 8 per group) as follows: control, model, diallyl sulfide, psoralen-low, psoralen-medium and psoralen-high. The drug administration group was given psoralen 25, 50 and 100 mg/kg (0.5% carboxymethyl cellulose sodium suspension) daily, the positive control group was given 200 mg/kg diallyl sulfide daily, and the solvent control group and the model group were given 0.5% carboxymethyl cellulose sodium by intragastric administration for 5 consecutive days. On day 4, all groups were given 300 mg/kg acetaminophen (administration volume 0.4 ml/20 g, i.p.) 1 h after administration, while the solvent control group was given the same volume of normal saline intraperitoneally. The animals were fasted without water for 6 h the night before the 5th administration and sacrificed 2 h after the last administration of psoralen or diallyl sulfide the next morning (25 h after the administration of acetaminophen).
The rats were randomly divided into three groups (n = 8 per group) as follows: control, psoralen-1, and psoralen-5. The control group and psoralen-1 group were given 0.5% carboxymethyl cellulose sodium intragastric administration for 5 consecutive days, while the psoralen-5 group was given 70 mg/kg psoralen intragastric administration for 5 consecutive days, and the animals in each group were fasted for 10 h at night on the 5th day. Blood was collected at 0 h in the morning on the 6th day. Then, the control group and psoralen-5 group were given 0.5% carboxymethyl cellulose sodium and mixed probe substrate solution simultaneously, while the psoralen-1 group was given 70 mg/kg psoralen and mixed probe substrate simultaneously. Blood was collected from the mouse orbit at 0.083, 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h after the mixed probe solution was given.
Reagents and Preparation
Acetaminophen was purchased from Shanghai Aladdin Reagent Co., Ltd., China; diallyl sulfide, phenacetin, and chlorzoxazone were purchased from Sigma Company, USA; psoralen (purity > 98%, Lot # JZ18071201) was purchased from Nanjing Jingzhu Biotechnology Co., Ltd., China. Midazolam was purchased from Jiangsu Nhwa Pharmaceutical Co., Ltd, China.
Acetaminophen was dissolved in normal saline at 37 °C. Owing to its poor solubility, it can be prepared the night before modeling; diallyl sulfide was dissolved in distilled water (8 mg/ml); 0.5% carboxymethyl cellulose sodium was chosen as the vehicle for psoralen, which was prepared freshly in 1, 2, 4 and 14 mg/ml. Mixed probe substrates phenacetin, chlorzoxazone and midazolam were dissolved in 40 ml 0.5% carboxymethyl cellulose sodium.
Measurement of Enzymatic Parameters
Blood samples were collected by removing mouse eyeballs before sacrifice and centrifuged at 3500 × g for 15 min. The separated serum was collected to measure serum alanine aminotransferase (Neusoft Wittmann Biotechnology Co., Ltd., China), aspartate aminotransferase (Neusoft Wittmann Biotechnology Co., Ltd., China) and lactate dehydrogenase (Nanjing Jiancheng Bioengineering Institute, China).
Malondialdehyde Determination
Liver tissues were collected and homogenized in normal saline and centrifuged at 3500 × g for 10 min. Protein concentrations were measured with a BCA protein assay kit (Beyotime Institute of Biotechnology, China). A malondialdehyde detection kit was purchased from Nanjing Jiancheng Bioengineering Institute, China. Reagents were mixed according to the manufacturer’s instructions, and readings were taken by a Varioskan LUX Multimode Microplate Reader (Thermo Fisher Scientific, USA).
Histopathological Analysis
For histological assessments, liver tissues were fixed in 10% buffered formalin for 24 h, embedded in paraffin, and cut into 5 μm sections before being stained with hematoxylin and eosin (H&E) for assessment by BX53 microscopy (Olympus Corporation, JPN).
Assessment of Inflammatory Factors and Cyp2e1
RNA was isolated from mouse tissues using Trizol reagent (Nanjing Vazyme Biotechnology Co., Ltd., China) and was reverse transcribed into cDNA with the HiScript®Q RT SuperMix for qPCR (+ gDNA wiper) RT Reagent Kit (Nanjing Vazyme Biotechnology Co., Ltd., China). qPCR amplification was performed using AceQ®qPCR SYBR®Green Master Mix (Nanjing Vazyme Biotechnology Co., Ltd., China). The cycling conditions were 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Primer sequences are shown below.
Box: Primer sequences of inflammatory factors and Cyp2e1
Gapdh | F: CTTTGGCATTGTGGAAGGGCTC |
R: GCAGGGATGATGTTCTGGGCAG | |
Tnfα | F: GGTGCCTATGTCTCAGCCTCTT |
R: GCCATAGAACTGATGAGAGGGAG | |
Il6 | F: TACCACTTCACAAGTCGGAGGC |
R: CTGCAAGTGCATCATCGTTGTTC | |
Il1β | F: TGGACCTTCCAGGATGAGGACA |
R: GTTCATCTCGGAGCCTGTAGTG | |
Cyp2e1 | F: AGGCTGTCAAGGAGGTGCTACT |
R: AAAACCTCCGCACGTCCTTCCA |
Western Blot Analysis
Preserved livers were homogenized at 4 °C and centrifuged at 12,000 × g for 10 min to obtain the supernatant. Protein concentrations in the extracts were determined using a BCA Protein Assay Kit. The protein samples were separated by sodium dodecyl sulfate‒polyacrylamide gel electrophoresis and transferred onto PVDF membranes (Millipore, USA). The membranes were blocked with 5% bovine serum albumin for 60 min, and incubated overnight at 4 °C with CYP2E1 antibody (Abcam, UK) diluted 1:1000 with 5% bovine serum albumin and secondary antibodies diluted at 1:10000 with TBST at room temperature for 60 min. The bands were visualized by Tanon image scanner (Shanghai Tanon Technology Co., Ltd., China). Image J software was used to quantify signal intensities. The signals were normalized to those of GAPDH (Thermo Fisher Scientific, USA), which was used as the internal control.
Molecular Docking
The binding mode of psoralen to mouse CYP2E1, CYP1A2 and CYP3A11 was performed by homology modeling and molecular docking. The primary protein sequences of CYP2E1 (Q05421), CYP1A2 (P00186) and CYP3A11 (Q64459) in mice were available from the UniProt database (Karim et al. 2015) (https://www.uniprot.org/). Swiss-model software (Arnold et al. 2006) was used to conduct a template search for mouse CYP2E1, CYP1A2 and CYP3A11 first-level sequences, templates with close enzyme species, high homology, and high sequence coverage were screened out, and homology modeling was conducted. The modeling structure was evaluated by PROCHECK (Laskowski et al. 1996) and Verify 3D (Eisenberg et al. 1997) software (https://saves.mbi.ucla.edu/) to evaluate the rationality of the protein structure. The psoralen 3D structure was downloaded in mol2 format via the TCMSP database (Ru et al. 2014) (https://tcmspw.com/). The docking experiment was performed by Autodock 4.0 (Scripps Research, USA) (Morris et al. 2009) with the Lamarckian genetic algorithm (Fuhrmann et al. 2010). The binding site coordinates of CYP2E1, CYP1A2 and CYP3A11 were (−16.954, 36.683, −28.371), (39.523, 29.911, 4.918) and (19.341, −19.041, 9.627), respectively. The default parameters were used unless otherwise specified. Ligand‒protein complexes with the lowest free energy of combination were considered the most favorable structures (Fuhrmann et al. 2010). Visualization and further analysis of the ligand‒protein interactions were carried out using Discovery Studio 4.5 (BIOVIA, USA) (Guedes et al. 2014) and PyMOL Molecular Graphics System (Version 2.0 Schrödinger, LLC). Open Bable software (Version 2.4.1) was used for file format conversion in this study.
UPLC‒MS/MS of CYP Activity
A 10 μg/ml standard mixture mother liquor was prepared by adding 100 μl of 1’-hydroxymidazolam (product of midazolam, 100 μg/ml), 10 μl of other substrates and products (chlorzoxazone substrate of CYP2E1), 6-hydroxyclozoxa (product of chlorzoxazone), midazolam (substrate of CYP1A2), phenacetin (substrate of CYP3A11) and paracetamol (product of phenacetin), 1 mg/ml) and 730 μl of methanol. The linear solution was prepared according to Table S1. Fifty microliters of rat blank plasma and 300 μl acetonitrile were mixed and centrifuged at 12,000 × g for 10 min to obtain the supernatant for UPLC‒MS/MS detection.
A Shimadzu ultra-performance liquid chromatographic system (UPLC, Shimadzu Corporation, Tokyo, Japan) was used for separation and a QTrap 5500 Triple Quadrupole Linear Ion Traps mass spectrometer (AB Sciex, Foster City, CA) was used for detection. Chromatographic separation was achieved on an Agilent ZORBAX Eclipse XDB-C18 column (2.1 × 100 mm, 1.8 μm particle size). A gradient elution lasting 7 min was obtained; the mobile phase contained aqueous 0.1% formic acid as phase A and methanol as phase B. The gradient used was as follows: (0–1 min) 5% methanol, (1–5 min) 5%-80% methanol, (5–6 min) 80% methanol, (6.0–6.1 min) back to 5% methanol and remained for 1 min. The flow rate was 0.35 ml/min and the injection volume was 2 μl.
Statistical Analysis
The experimental data were presented as mean ± standard error (SEM) and were analyzed by one-way ANOVA using GraphPad Prism 7. p < 0.05 was considered significant.
Results and Discussion
Mouse Body Weight and Liver Coefficient
As shown in Fig. 1, after intraperitoneal injection of acetaminophen or saline on day 4, the body weight of mice in each group significantly decreased due to prior fasting. Then, the body weights of the mice in all groups except the model group increased on day 5. Compared with that of the control group, the liver coefficient of the model group was increased. After administration of the positive control drug diallyl sulfide, a low or medium dose of psoralen, the liver coefficient was moderately decreased. However, the liver coefficient of the psoralen-H group was slightly increased compared with that of the model group.

Effects of psoralen (3) for 5 days on body weight and liver coefficient in mice with acetaminophen-induced acute liver injury. control: 0.5% carboxymethyl cellulose sodium; model: 0.5% carboxymethyl cellulose sodium; diallyl sulfide: positive drug, 200 mg/kg diallyl sulfide; psoralen-L: low dose of psoralen, 25 mg/kg; psoralen-M: medium dose of psoralen, 50 mg/kg; psoralen-H: high dose of psoralen, 100 mg/kg. (A) Body weight. (B) Liver coefficient. Data are expressed as mean ± SEM, n = 8
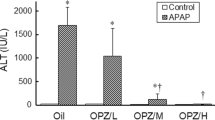
Biochemical and Oxidative Stress Indices
Serum ALT, AST and LDH in the model group were significantly increased by 193, 33 and sixfold, respectively, in comparison to the control group (Fig. 2). Treatment with the positive drug diallyl sulfide significantly decreased serum ALT, AST and LDH. The three doses of psoralen all significantly decreased serum ALT, AST and LDH. However, it was shown that the efficacy of medium and high doses of psoralen rather than a low dose was almost equivalent to that of the positive control drug diallyl sulfide. Compared with the control group, the oxidative stress index MDA in the model group was significantly increased by sixfold. Positive drug efficiently decreased MDA, as well as three doses of psoralen. Compared with the positive control group, the psoralen-M and psoralen-L groups showed better efficacy without apparent dose dependence.

Effects of psoralen (3) on serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), and hepatic malondialdehyde (MDA) in mice with acetaminophen-induced acute liver injury. (A) ALT; (B) AST; (C) LDH; (D) MDA. Data were expressed as mean ± SEM, n = 8. ##p < 0.01 compared with control group, *p < 0.05, **p < 0.01 compared with the model group
Liver Histopathological Investigation
To evaluate the ameliorative effect of psoralen on acetaminophen-induced liver injury, we examined the histopathological changes in the liver. As shown in Fig. 3, the structure of hepatocytes was intact, and the hepatic cords were arranged neatly in the control group, while in the model group, extensive inflammatory cell infiltration and hepatocyte necrosis were observed. After treatment with the positive control drug diallyl sulfide, inflammation and necrosis were significantly reduced. It was found that a low dose of psoralen ameliorated acetaminophen-induced hepatocyte necrosis but was not able to reduce inflammatory cell infiltration, while medium doses and high doses had similar effects as diallyl sulfide by efficiently ameliorating both necrosis and inflammation.

Histological images of mouse livers stained with hematoxylin and eosin. (A) control; (B) model; (C) diallyl sulfide; (D) psoralen-L; (E) psoralen-M; (F) psoralen-H. HE stained, ×100 magnification
Assessment of Inflammatory Factors
As shown in Fig. 4, acetaminophen-induced hepatocyte necrosis was mainly caused by inflammation, so the inflammatory factors Tnfα, Il6 and Il1β were analyzed. The model group showed higher expression of Tnfα, Il6 and Il1β than the control group. Treatment with diallyl sulfide significantly downregulated these inflammatory factors. Compared with the model group, the low dose of psoralen decreased Il6 and Il1β but did not display any significant effect on Tnfα. However, medium and high doses of psoralen dramatically decreased all these inflammatory factors, which seemed to have better effects than the positive control drug diallyl sulfide.

Effects of psoralen (3) on inflammatory factor gene levels in mice with acetaminophen-induced acute liver injury. The samples tested in this experiment were extracted from mouse liver tissues: (A) Tnfα; (B) Il6; (C) Il1β. Data were expressed as mean ± SEM, n = 8. ##p < 0.01 compared with the control group, **p < 0.01 compared with the model group
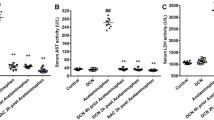
Analysis of CYP2E1 Expression
Acetaminophen-induced liver injury is closely correlated with CYP2E1. As illustrated in Fig. 5, CYP2E1 was significantly decreased in the model group at both the mRNA and protein levels compared with the control group. Diallyl sulfide significantly upregulated both the mRNA and protein expression of CYP2E1. Compared with the model group, treatment with different doses of psoralen increased CYP2E1 at the mRNA and protein levels. However, the expression of CYP2E1 in these groups was lower than that in the diallyl sulfide group. Interestingly, a high dose of psoralen inhibited CYP2E1 at the protein level compared with low and medium doses.

Effects of psoralen (3) on CYP2E1 expression levels in mice with acetaminophen-induced acute liver injury. The samples tested in this experiment were extracted from mouse liver tissues: (A) Cyp2e1 mRNA expression; (B) CYP2E1 protein bands; (C). CYP2E1 protein expression. Data were expressed as mean ± SEM, n = 3/8. #p < 0.05; ##p < 0.01 compared with the control group, *p < 0.05, **p < 0.01 compared with the model group
Molecular Docking
Homology models were established by using three isoforms of human CYP450, namely using human CYP2E1 (PDB: 3E4E), CYP1A2 (PDB: 4I8V) and CYP3A11 (PDB: 5G5J) protein structures as templates. PROCHECK and Verify 3D software were used to evaluate the quality of these models. The results demonstrated that their structures constructed in this study were reasonable (Fig. S3), and psoralen was used for molecular docking.
The amino acid residues and binding energies of psoralen interacting with CYP2E1, CYP1A2 and CYP3A11 are summarized in Table 1. The theoretical binding mode of psoralen with CYP2E1 is shown in Fig. 6A. The carbonyl oxygen of psoralen could form a hydrogen bond with the hydroxyl group of VAL-364 with a bond length of 2.0 Å (Fig. 6B). In addition, van der Waals forces were formed between the psoralen molecule and THR304, THR307, GLN358, ILE361, ASN362, LEU397, and PHE430 of CYP2E1. The predicted binding energy of psoralen was Ef = −6.17 kcal/mol (−25.82 kJ/mol).

Molecular docking results of psoralen (3) with CYP2E1. Three-dimensional and two-dimensional docking diagrams were drawn by PyMOL and Discovery Studio software, respectively: (A) three-dimensional docking diagrams of psoralen with CYP2E1; CYP2E1 was represented by a cartoon model, amino acid residues were represented by a purple stick model and psoralen was represented by a gray stick model, the green dotted line represented hydrogen bonds, the pink dotted line represented pi-pi stacking, bond length unit was Å. (B) Two-dimensional docking diagrams of psoralen with CYP2E1. The color depth within the ring was proportional to the strength of the interaction

Effects of single-dose psoralen (3) on the activity of CYP2E1. The enzymatic activity of CYP2E1 in the liver was evaluated by detecting the changes in metabolites in the blood. control: 0.5% carboxymethyl cellulose sodium + mixed substrate solution; psoralen-1: single dose administration of 70 mg/kg psoralen + mixed substrate solution. (A) Concentration changes of chlorzoxazone. (B) Concentration changes of 6-hydroxychlorzoxazone. (C) Cmax metabolic ratios of chlorzoxazone and its production 6-hydroxychlorzoxazone metabolized by CYP2E1; (D) Area under curve metabolic ratios of chlorzoxazone and its production HCZZ metabolized by CYP2E1. Data were expressed as mean ± SEM, n = 8. #p < 0.05, ##p < 0.01 compared with the control group
The theoretical binding mode of psoralen with CYP1A2 is shown in Fig. S1. Psoralen could form π-π stacking with the benzene ring of phenylalanine of CYP1A2 at distances of 4.5, 4.5, 4.9, and 5.6 Å. Van der Waals forces were formed between the psoralen molecule and THR117, PHE255, PHE259, VAL260, ASN310, ASP311, and GLY314 of CYP1A2. The predicted binding energy of psoralen was Ef = −6.20 kcal/mol (−25.94 kJ/mol).
The theoretical binding mode of psoralen with CYP3A11 is shown in Fig. S2. The carbonyl oxygen of psoralen formed a hydrogen bond with the residual TRP-126 with a bond length of 1.7 Å (Fig. S2B). In addition, van der Waals forces were formed between the psoralen molecule and ARG105, SER119, PHE137, PHE303, ARG441, ASN442, and GLY445 of CYP3A11. The predicted binding energy of psoralen was Ef = −5.41 kcal/mol (−22.64 kJ/mol).
Effect on Phase I Metabolic Enzymes
Chlorzoxazone, a muscle-relaxing drug, can be metabolized to 6-hydroxychlorzoxazone by CYP2E1 (Witt et al. 2016). Phenacetin, is widely used as an analgesic, and can be metabolized to paracetamol by CYP1A2 (Hinson 1983), and midazolam, used for anesthesia in surgical and dental procedures, can be metabolized to 1’- or 4’- hydroxy metabolites (1’-hydroxymidazolam) by CYP3A11 (Gandhi et al. 2012). Compared with the control group, the accumulation of the prototype chlorzoxazone in the psoralen-1 group increased, and the production of the metabolite 6-hydroxyclozoxa decreased at the same time. The Cmax and area under curve metabolic ratios of 6-hydroxyclozoxa/chlorzoxazone all decreased significantly in the psoralen-1 group compared with the control group, indicating that single-dose psoralen can significantly inhibit the activity of CYP2E1 (Fig. 7). Similarly, single-dose psoralen also significantly inhibited the activity of CYP1A2 (Fig. S5), while inducing the activity of CYP3A11 (Fig. S7).
As shown in Fig. 8, the Cmax and area under curve metabolic ratios of 6-hydroxyclozoxa /chlorzoxazone showed no apparent difference between the psoralen-5 group and the control group, indicating that 5-day-dose psoralen had no significant effect on the activity of CYP2E1. The same effect was observed on CYP3A11 (Fig. S8). However, 5-day-dose psoralen significantly increased the Cmax and area under curve metabolic ratios of paracetamol/phenacetin, indicating that it can induce the activity of CYP1A2 (Fig. S6).

Effects of five-day-dose psoralen (3) on the activity of CYP2E1. The enzymatic activity of CYP2E1 in the liver was evaluated by detecting the changes in metabolites in the blood. control: 0.5% carboxymethyl cellulose sodium + mixed substrate solution; psoralen-5: 5-day administration of 70 mg/kg psoralen + mixed substrate solution. (A) Concentration changes of chlorzoxazone. (B) Concentration changes of 6-hydroxychlorzoxazone. (C) Cmax metabolic ratios of chlorzoxazone and its production 6-hydroxychlorzoxazone metabolized by CYP2E1; (D) Area under curve metabolic ratios of chlorzoxazone and its production HCZZ metabolized by CYP2E1. Data were expressed as mean ± SEM, n = 8. ##p < 0.01 compared with the control group
The acetaminophen-induced acute liver injury model is a classic drug-induced liver injury model. The pathogenesis of this model is that when too much acetaminophen is ingested, a large amount of it will be metabolized by the CYP450 enzyme (mainly CYP2E1, CYP1A2, and CYP3A11 in mice) to produce toxic free radicals.This eventually leads to mitochondrial dysfunction, lipid peroxidation, and cell necrosis (Gao et al. 2020).
In our study, male mice were chosen because female mice express a lower level of SH3 domain-binding protein that preferentially associates with Btk through the ERα/p53/miR34a pathway. SH3 domain-binding protein that preferentially associates with Btk is an outer membrane docking protein on mitochondria that can bind with P-JNK to induce oxidative stress in mitochondria, so female mice have a stronger ability to antagonize liver injury than male mice (Win et al. 2019). Histopathological results showed extensive hepatocyte necrosis, missing cellular structure and blurred intercellular space in the acetaminophen model group. In addition, the blood biochemical indices ALT, AST and LDH, the oxidative stress index MDA and the gene expression of inflammatory factors were significantly elevated in the model group. After treatment with psoralen, hepatocyte necrosis was improved, and ALT, AST, LDH, MDA and inflammatory factors Il1β, Il6 and Tnfα were significantly decreased, indicating that psoralen can ameliorate acetaminophen-induced acute liver injury.
Western blot results showed that CYP2E1 expression in the model group was significantly downregulated compared with that in the control group. This result might be the consequence of a toxic dose of acetaminophen causing degradation of CYP450 through the lysosomal pathway. After treatment with psoralen, CYP2E1 expression was increased in both the low-dose and medium-dose psoralen groups compared with the model group. This might be because psoralen can antagonize liver injury and thus weaken the degradation of CYP450 by acetaminophen. Interestingly, CYP2E1 was decreased in the high-dose psoralen group compared with the low-dose and medium-dose psoralen groups. This might be because psoralen itself has an inhibitory effect on CYP2E1 protein expression at high doses. It has been reported that psoralen itself is associated with the inhibition of CYP2E1 expression (Wang et al. 2012). Our previous study also showed that five-day treatment of mice with 100 mg/kg psoralen can significantly inhibit hepatic CYP2E1 expression (Fig. S4).
The above results showed that psoralen has a pharmacological effect on acetaminophen-induced acute liver injury, but as acetaminophen can affect the expression of CYP2E1, the role of CYP2E1 in the efficacy of psoralen is still unclear. Compared with the expression level of upstream proteins and genes of metabolic enzymes, the effect of drugs on metabolic enzyme activity could play a more important role for the drug effects (Fan et al. 2014; Lin et al. 2019). Therefore, we first evaluated whether psoralen can bind to active sites of these major metabolic enzymes in phase I such as CYP2E1, CYP1A2 and CYP3A11 by homology modeling and molecular docking.
The interaction of psoralen with CYP2E1, CYP1A2 and CYP3A11 was studied by homologous modeling and molecular docking. Mouse CYP2E1, CYP1A2 and CYP3A11 protein structures were established using Swiss model homology modeling, and the quality of all structures was assessed by PROCHECK and Verify 3D software. The results showed that the modeling structures were reasonable, and psoralen can bind to the active sites of CYP2E1, CYP1A2 and CYP3A11. The predicted binding energies of psoralen with CYP2E1, CYP1A2 and CYP3A11 were Ef = −6.17, 6.20, and −5.41 kcal/mol, respectively, indicating that the interactions between psoralen and CYP2E1, CYP1A2 and CYP3A11 were spontaneous reactions. Psoralen may be the substrate of these enzymes due to strong interactions between them.
Then, the role of enzyme activity changes in the efficacy of psoralen was investigated by measuring the effects of psoralen on CYP2E1, CYP1A2 and CYP3A11 by using the cocktail method. A single-dose psoralen group was set up in this experiment; that is, psoralen and the probe substrate were simultaneously given to rats to determine whether psoralen itself is the substrate of one or several metabolic enzymes. In addition, five-day-dose psoralen was also set up, that is, the probe substrate was given 24 h after the last administration of psoralen to exclude the influence of psoralen itself. The objective was to determine the effect of continuous administration of psoralen on metabolic enzyme activity. The probe substrates of three metabolic enzymes and their dosages were determined according to previous studies (Tang et al. 2008; Gao et al. 2013; Xu et al. 2014; Lu et al. 2018).
The measured enzyme activity results showed that single-dose psoralen administration could significantly inhibit the enzymatic activity of CYP2E1 and CYP1A2, and had a significant induction effect on CYP3A11. Psoralen administration for 5 days significantly induced the enzymatic activity of CYP1A2, but had no significant effect on CYP2E1 and CYP3A11. Single-dose psoralen significantly inhibited the activity of CYP2E1, indicating that psoralen may be the substrate for competitive binding of CYP2E1. When psoralen occupies the active site of CYP2E1, it results in a reduction in toxicity by preventing the formation of NAPQ1. Psoralen measured in this study mainly inhibited metabolic enzyme activity during a single administration, but induced enzyme activity after 5 days of continuous administration. This suggests that multiple metabolic enzymes may be involved in the metabolism of psoralen. Therefore, special attention should be given to the interaction between drugs to avoid the occurrence of adverse reactions when psoralen is used in combination with other drugs in the future, especially drugs that are known to be metabolized by these enzymes.
Conclusion
In summary, psoralen can significantly reduce the elevation of serum alanine aminotransferase, aspartate aminotransferase, lactate dehydrogenase, oxidative stress index of malondialdehyde, and major pro-inflammatory factors induced by acetaminophen, and recover hepatocyte necrosis. The main reason may be that psoralen is a substrate of CYP2E1, which can competitively bind to the active site with acetaminophen, thus inhibiting the activity of CYP2E1 to avoid the formation of toxic metabolites.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article or its supplementary materials.
References
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. https://doi.org/10.1093/bioinformatics/bti770
Bunchorntavakul C, Reddy KR (2013) Acetaminophen-related hepatotoxicity. Clin Liver Dis 17:587–607. https://doi.org/10.1016/j.cld.2013.07.005
Bunchorntavakul C, Reddy KR (2018) Acetaminophen (APAP or N-acetyl-p-aminophenol) and acute liver failure. Clin Liver Dis 22:325–346. https://doi.org/10.1016/j.cld.2018.01.007
Eisenberg D, Luthy R, Bowie JU (1997) VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol 277:396–404. https://doi.org/10.1016/s0076-6879(97)77022-8
Fan X, Jiang Y, Wang Y, Tan H, Zeng H, Wang Y, Chen P, Qu A, Gonzalez FJ, Huang M, Bi H (2014) Wuzhi tablet (Schisandra sphenanthera extract) protects against acetaminophen-induced hepatotoxicity by inhibition of CYP-mediated bioactivation and regulation of NRF2-ARE and p53/p21 pathways. Drug Metab Dispos 42:1982–1990. https://doi.org/10.1124/dmd.114.059535
Fuhrmann J, Rurainski A, Lenhof HP, Neumann N (2010) A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J Comput Chem 31:1911–1918. https://doi.org/10.1002/jcc.21478
Gandhi AS, Guo T, Shah P, Moorthy B, Chow DL, Hu M, Ghose R (2012) CYP3A-dependent drug metabolism is reduced in bacterial inflammation in mice. Br J Pharmacol 166:2176–2187. https://doi.org/10.1111/j.1476-5381.2012.01933.x
Gao J, Shi Z, Zhu S, Li GQ, Yan R, Yao M (2013) Influences of processed rhubarbs on the activities of four CYP isozymes and the metabolism of saxagliptin in rats based on probe cocktail and pharmacokinetics approaches. J Ethnopharmacol 145:566–572. https://doi.org/10.1016/j.jep.2012.11.030
Gao RY, Wang M, Liu Q, Feng D, Wen Y, Xia Y, Colgan SP, Eltzschig HK, Ju C (2020) Hypoxia-inducible factor-2alpha reprograms liver macrophages to protect against acute liver injury through the production of interleukin-6. Hepatology 71:2105–2117. https://doi.org/10.1002/hep.30954
Guedes IA, de Magalhaes CS, Dardenne LE (2014) Receptor-ligand molecular docking. Biophys Rev 6:75–87. https://doi.org/10.1007/s12551-013-0130-2
Hinson JA (1983) Reactive metabolites of phenacetin and acetaminophen: a review. Environ Health Persp 49:71–79. https://doi.org/10.1289/ehp.834971
Hu C, Zhao L, Wu Z, Li L (2020) Transplantation of mesenchymal stem cells and their derivatives effectively promotes liver regeneration to attenuate acetaminophen-induced liver injury. Stem Cell Res Ther 11:88. https://doi.org/10.1186/s13287-020-01596-9
Jaeschke H, Duan L, Akakpo JY, Farhood A, Ramachandran A (2018) The role of apoptosis in acetaminophen hepatotoxicity. Food Chem Toxicol 118:709–718. https://doi.org/10.1016/j.fct.2018.06.025
Karim R, Aziz MM, Shatabda S, Rahman MS, Mia MA, Zaman F, Rakin S (2015) CoMOGrad and PHOG: from computer vision to fast and accurate protein tertiary structure retrieval. Sci Rep 5:13275. https://doi.org/10.1038/srep13275
Larson AM (2007) Acetaminophen hepatotoxicity. Clin Liver Dis 11:525–548. https://doi.org/10.1016/j.cld.2007.06.006
Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8:477–486. https://doi.org/10.1007/BF00228148
Lee WM (2017) Acetaminophen (APAP) hepatotoxicity-isn’t it time for acetaminophen to go away?". J Hepatol 67:1324–1331. https://doi.org/10.1016/j.jhep.2017.07.005
Lin L, Guan H, Li R, Liao X, Zhao F, Wang M, Li J, Xu G, He X, Zhang J, Li Y, Wang Y, Zhou M, Liao S (2019) Auriculatone sulfate effectively protects mice against acetaminophen-induced liver injury. Molecules 24:3642. https://doi.org/10.3390/molecules24203642
Lu YY, Du ZY, Li Y, Wang JL, Zhao MB, Jiang Y, Guo XY, Tu PF (2018) Effects of Baoyuan decoction, a traditional Chinese medicine formula, on the activities and mRNA expression of seven CYP isozymes in rats. J Ethnopharmacol 225:327–335. https://doi.org/10.1016/j.jep.2018.07.023
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. https://doi.org/10.1002/jcc.21256
Ren Y, Song X, Tan L, Guo C, Wang M, Liu H, Cao Z, Li Y, Peng C (2020) A review of the pharmacological properties of psoralen. Front Pharmacol 11:571535. https://doi.org/10.3389/fphar.2020.571535
Ru J, Li P, Wang J, Zhou W, Li B, Huang C, Li P, Guo Z, Tao W, Yang Y, Xu X, Li Y, Wang Y, Yang L (2014) TCMSP: a database of systems pharmacology for drug discovery from herbal medicines. J Cheminform 6:13. https://doi.org/10.1186/1758-2946-6-13
Ruepp SU, Tonge RP, Shaw J, Wallis N, Pognan F (2002) Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol Sci 65:135–150. https://doi.org/10.1093/toxsci/65.1.135
Tang H, Min G, Ge B, Li Y, Liu X, Jiang S (2008) Evaluation of protective effects of Chi-Zhi-Huang decoction on phase I drug metabolism of liver injured rats by cocktail probe drugs. J Ethnopharmacol 117:420–426. https://doi.org/10.1016/j.jep.2008.02.020
Uesawa Y, Mohri K (2010) Quantitative structure-activity relationship (QSAR) analysis of the inhibitory effects of furanocoumarin derivatives on cytochrome P450 3A activities. Pharmazie 65:41–46. https://doi.org/10.1691/ph.2010.9651
Wang X, Lou YJ, Wang MX, Shi YW, Xu HX, Kong LD (2012) Furocoumarins affect hepatic cytochrome P450 and renal organic ion transporters in mice. Toxicol Lett 209:67–77. https://doi.org/10.1016/j.toxlet.2011.11.030
Wang X, Peng P, Pan Z, Fang Z, Lu W, Liu X (2019) Psoralenralen inhibits malignant proliferation and induces apoptosis through triggering endoplasmic reticulum stress in human SMMC7721 hepatoma cells. Biol Res 52:34. https://doi.org/10.1186/s40659-019-0241-8
Win S, Min RW, Chen CQ, Zhang J, Chen Y, Li M, Suzuki A, Abdelmalek MF, Wang Y, Aghajan M, Aung FW, Diehl AM, Davis RJ, Than TA, Kaplowitz N (2019) Expression of mitochondrial membrane-linked SAB determines severity of sex-dependent acute liver injury. J Clin Invest 129:5278–5293. https://doi.org/10.1172/JCI128289
Witt L, Suzuki Y, Hohmann N, Mikus G, Haefeli WE, Burhenne J (2016) Ultrasensitive quantification of the CYP2E1 probe chlorzoxazone and its main metabolite 6-hydroxychlorzoxazone in human plasma using ultra performance liquid chromatography coupled to tandem mass spectrometry after chlorzoxazone microdosing. J Chromatogr B Analyt Technol Biomed Life Sci 1027:207–213. https://doi.org/10.1016/j.jchromb.2016.05.049
Xu RA, Xu ZS, Ge RS (2014) Effects of hydroxysafflor yellow A on the activity and mRNA expression of four CYP isozymes in rats. J Ethnopharmacol 151:1141–1146. https://doi.org/10.2147/DMSO.S246381
Zhang X, Zhao W, Wang Y, Lu J, Chen X (2016) The chemical constituents and bioactivities of Psoralea corylifolia Linn.: a review. Am J Chin Med 44:35–60. https://doi.org/10.1142/S0192415X16500038
Zhou L, Tang J, Xiong X, Dong H, Huang J, Zhou S, Zhang L, Qin H, Yan S (2017) Psoralea corylifolia L. attenuates nonalcoholic steatohepatitis in juvenile mouse. Front Pharmacol 8:876. https://doi.org/10.3389/fphar.2017.00876
Funding
This study was supported by the National Natural Science Foundation of China (82074114 to ZJ, 81973562 to LZ), the Natural Science Foundation of Jiangsu Province (BK20210430 to QY), the China Postdoctoral Science Foundation (2020M681786 to QY), the Innovation Team Projects in Universities of Guangdong Province (No. 2018KCXTD016 to LZ), and the "Double First-Class" University project (CPU2018GY33 to ZJ).
Author information
Authors and Affiliations
Contributions
YZ: designed the research, conducted the animal experiments and analyzed the data. QL: conducted the molecular docking, analyzed and drafted the manuscript. SL: methodology of molecular docking. YC and XH: methodology of analysis of enzyme activities. MD and ZD: conducted the animal experiments. LZ: Supervision. QY, ZJ: revised the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Ethical Approval
All animal care and handling procedures were approved by the Animal Ethics Committee of China Pharmaceutical University under experimental protocol nº 2021-09-032. All experiments were performed according to the recommendations of the Guidelines for Ethical Conduct in the Care and Use of Animals.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Y., Lu, Q., Liu, S. et al. Psoralen Ameliorates Acetaminophen-Induced Acute Liver Injury by Inhibiting the Enzymatic Activity of CYP2E1. Rev. Bras. Farmacogn. 33, 1060–1071 (2023). https://doi.org/10.1007/s43450-023-00439-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43450-023-00439-x