
Abstract
Cancer remains one of the leading causes of death in the world. Despite the considerable success of conventional treatment strategies, the incidence and mortality rates are still high, making developing new effective anticancer therapies an urgent priority. Ginsenoside Rg5 (Rg5) is a minor ginsenoside constituent obtained exclusively from ginseng species and is known for its broad spectrum of pharmacological activities. This article aimed to comprehensively review the anticancer properties of Rg5, focusing on action mechanisms, structure–activity relationship (SAR), and pharmacokinetics attributes. The in vitro and in vivo activities of Rg5 have been proven against several cancer types, such as breast, liver, lung, bone, and gastrointestinal (GI) cancers. The modulation of multiple signaling pathways critical for cancer growth and survival mediates these activities. Nevertheless, human clinical studies of Rg5 have not been addressed before, and there is still considerable ambiguity regarding its pharmacokinetics properties. In addition, a significant shortage in the structure–activity relationship (SAR) of Rg5 has been identified. Therefore, future efforts should focus on further optimization by performing extensive SAR studies to uncover the structural features essential for the potent anticancer activity of Rg5. Thus, this review highlights the value of Rg5 as a potential anticancer drug candidate and identifies the research areas requiring more investigation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
As the topmost cause of death in the twenty-first century, cancer continues to be a severe global health challenge [1]. The data for the year 2020 alone showed 10 million deaths and 19.3 million new cases worldwide [1, 2]. The Global Cancer Observatory (GLOBOCAN) estimated that by 2040, there will be 30.2 million cases around the globe, and cancer-related deaths will reach 16.3 million [3]. Cancer is characterized by abnormal cells’ rapid and uncontrolled growth resulting from successive gene mutations [4]. The occurrence of these mutations has been attributed to several factors, such as exposure to (i) genotoxic chemicals, (ii) radiation, and (iii) infectious agents such as bacteria and viruses [5, 6]. So far, more than 200 types of cancer have been identified, varying widely in their pathological features alongside their response to treatment [7]. For several decades, surgery, chemotherapy, and radiotherapy have served as the standard strategies for treating different types of cancer. Despite their effectiveness, these strategies have several limitations, such as variable efficacy due to the heterogeneous nature of the disease, toxicity, increasing incidence of therapeutic failures, low selectivity, high cost, and, most notably, the emergence of drug resistance [8]. Therefore, developing cost-effective anticancer agents with improved therapeutic properties is of utmost priority.
Over the last 50 years, a significant number of anticancer drugs have been isolated from natural sources [9]. They usually influence multiple singling pathways linked to the promotion of cancer survival and growth via either modulation of the macromolecular targets’ activity or regulation of their expression [10]. Due to their diverse and complex chemical natures, which cover more expansive chemical space, safety, and low toxicity, anticancer agents of natural origin continue to attract the attention of researchers around the globe [11]. In this context, over 60% of the recently approved anticancer drugs originate from natural sources. These drugs belong to several structural classes of natural products, such as terpenes, alkaloids, essential oils, polyphenols, and polysaccharides [12, 13].
As a large class of natural products, terpenes constitute a rich source of structurally diverse bioactive compounds. Their biosynthesis involves polymerizing five-carbon building blocks called isoprenoid units [14]. They may occur as simple hydrocarbons or complex structures with additional functional groups [15]. Terpenoids can conveniently be classified into six classes, namely monoterpenes, sesquiterpenes, diterpenes, triterpenes, tetraterpenes, and polyterpenes [16]. Several studies have reported that terpenoids are an invaluable source of compounds with a vast range of pharmacological activities such as antimicrobial [17], antiviral [18], anti-inflammatory [19], and anticancer effects [20].
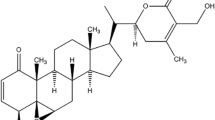
Panax ginseng is one of the world’s most prominent and widely used herbs for medicinal purposes [21]. In this regard, the therapeutic potentials of ginseng have been documented in different types of cancers, including breast cancer, lung cancer, and liver cancer [22]. The active ingredients in this herb are triterpene glycosides, commonly known as ginsenosides, which exclusively occur in Panax species [23]. Until recently, more than 200 ginsenosides have been isolated and identified, of which ginsenosides Rb1, Rb2, Rc, Rd, Re, Rf, and Rg1 are considered major constituents (> 90% of the total ginsenoside contents) [21]. On the other hand, some ginsenosides, such as ginsenoside Rg5 (Rg5), exist in minute concentrations (Fig. 1). This minor ginsenoside is obtained during the steam heating process via deglycosylation of ginsenoside Rb1 and dehydration of C-20 of ginsenoside Rg3 [24]. Rg5 displays a broad spectrum of pharmacological activity, including anticancer [25], antidiabetic [26, 27], anti-inflammatory [28] anti-osteoarthritic [29], sedative and hypnotic [30], and cardioprotective effects [31]. Rg5 exhibited superior therapeutic properties to the major ginsenosides [24, 32]. Nevertheless, difficulties pertinent to its extraction and isolation and the suboptimal pharmacokinetics profile have limited the further development of this promising drug candidate [25].

Chemical structure of ginsenoside Rg5. The structure made by ChemDraw (https://revvitysignals.com)
Accumulating evidence from preclinical studies indicates the potential of Rg5 as an anticancer agent against different types of cancer. The anticancer activity of Rg5 is mediated through the modulation of multiple singling pathways considered essential for cancer cells’ growth and multiplication. In this context, the present review outlines the in vitro and in vivo anticancer activity of Rg5, focusing mainly on mechanisms of action, structure–activity relationship (SAR), and pharmacokinetics attributes. The barriers and opportunities for its further development are also discussed. In this regard, this review would be helpful to establish the clinical development of Rg5 as a promising nature-derived anticancer drug candidate.
Review methodology
Searched databases
1. PubMed
https://pubmed.ncbi.nlm.nih.gov.
2. National science and technology library
3. Web of knowledge
http://apps.webofknowledge.com.
Inclusion/exclusion criteria:
-
Articles were included if reporting in vitro or in vivo human/animal studies using well-defined experimental dosages of Rg5.
-
Exclusion criteria include the studies that did not use Rg5 as a single ingredient and instead combined it with other bioactive chemical entity(s).
Sources, phytochemistry
Ginseng is a herbaceous perennial plant, a member of the Araliaceae family and the genus Panax. Therapeutically, the three most commonly used species of ginseng herbs are Panax ginseng (Korean ginseng), P. quinquefolius (American ginseng), and P. notoginseng (Chinese notoginseng) [33]. The freshly harvested ginseng is called “raw ginseng” while that prepared by steam heating and drying of fresh ginseng (Fig. 2) is commonly known as “red ginseng.” In addition, simple drying of raw ginseng furnishes the so-called “white ginseng” [34, 35]. At room temperature, the raw ginseng is chemically unstable and hence processed into red ginseng, which exhibits a higher biological efficacy when compared to white ginseng. Due to the different processing methods used to prepare red and white ginsengs, implying variation in chemical composition, they are separately regulated in the Korean, Japanese, and Chinese compendia [34].

Red ginseng manufacturing process
Since the first work by Shin et al. on the bioactivities of ginseng extracts, thousands of articles have been published on the traditional usage, chemical profile, and biological activities [36]. Such effects of ginseng extracts have been linked to the presence of certain phytoconstituents known as ginsenosides [37,38,39,40,41,42,43,44]. Ginsenosides are diverse saponins composed of triterpenoid structures directly linked to a sugar moiety. They can conveniently be classified into the dammarane and oleanane types based on the triterpene skeleton [45, 46]. Based on sugar moieties at C-3 and C-6, dammarane-type ginsenosides can further be classified into (i) protopanaxadiol-type group including Rb1, Rb2, Rc, and Rd; (ii) protopanaxatriol-type group including Re, Rf, and Rg1 (Fig. 3). Additionally, the chirality of the C-20 (denoted by a red asterisk in Fig. 3) in both groups could allow the occurrence of two stereoisomeric ginsenoside groups 20(R) and 20(S). Likewise, oleanane-type ginsenosides (Fig. 3) have been divided into the oleanolic acid and ocotillol groups [46, 47].

Chemical structures of four ginsenosides types. The structures made by ChemDraw (https://revvitysignals.com)
Ginsenosides can also be divided based on their polarity into (i) polar ginsenosides and (ii) less-polar ginsenosides, which exist in low quantity in the plant and are therefore called “rare saponins” [48]. Members of the latter group, such as Rg3, Rg5, Rg6, Rk1, and Rh2, display enhanced pharmacological efficacy relative to their polar counterparts, and this has been attributed to their better pharmacokinetics properties [48,49,50,51,52].
In addition to the naturally occurring ginsenosides that exist in the raw ginseng (“white ginseng”), the processing method of red ginseng production generates new ginsenosides (Fig. 4) such as Rh1, Rg2, Rg3, and Rg5 [47]. These ginsenosides are relatively less polar when compared with the naturally existing ones, such as Rb1, Rc, Rd, Re, and Rg1, from which they are produced [53]. The specifications of red ginseng imply that it must contain 2.5–34 mg/g combinations of ginsenosides Rb1, Rg1, and Rg3 [47].

Chemical structures of ginsenosides generated during the red ginseng manufacturing process. The structures made by ChemDraw (https://revvitysignals.com)
As mentioned above, ginsenosides are a unique class of saponins characterized by a triterpene (e.g., dammarane-type) core linked to one or more sugar molecules. Accordingly, the biosynthetic pathway of ginsenosides proceeds through three main steps: (i) formation of the ginsenosides core structure (Fig. 5); (ii) synthesis of sugar donor part; and (iii) core structure modification step [54]. Ginsenosides are biosynthesized from oxidosqualene.

Biosynthetic pathways for the major four ginsenosides structures. The name of the four main core structures of ginsenosides is marked in green. Solid arrows indicate the proven biosynthetic pathways; dashed arrows are for unproven pathways, and double arrows indicate the multiple biosynthetic steps. The enzymes that catalyze each reaction step are shown in red. The structures made by ChemDraw (https://revvitysignals.com)
Rg5 is a minor ginsenoside generated during the red ginseng manufacturing process, and it displays diverse and superior pharmacological activities relative to the major ginsenosides [24, 32, 55, 56]. It was first isolated in 1966 from red ginseng with an overall yield of less than 0.025% [57]. Since then, different methods have been developed to increase its yield [58,59,60], though harsh extraction conditions (higher temperature or pressure with extended heating time), among other challenges, hindered extensive evaluation of its biodynamic activities [57].
Chemically, Rg5 belongs to protopanaxadiol-type ginsenosides and is obtained through sugar elimination at the C20 position of ginsenoside Rb1 to furnish ginsenoside Rg3. Dehydration of the latter with subsequent generation of double bonds between C20 and C22 affords Rg5 [24, 47]. It occurs as a white crystalline powder with a melting point of 188‒190 °C, readily dissolves in water (logP = 3.5) at room temperature, and its aqueous solubility could reach up to 503.46 µg/mL [25]. Figure 6 illustrates the essential spectral features of Rg5.

Spectral characteristics of ginsenoside Rg5 [120]
Structure-anticancer activity relationship
A thorough review of the literature has identified a significant shortage in the structure–activity relationship of Rg5, as only very few reports addressed the anticancer activity of Rg5 in comparison to its natural or synthetic analogs. However, the literature is very rich in the structure–activity relationship of ginsenosides in general [32, 61,62,63]. To this end, the chemical structures of ginsenosides have been associated with different biological activities. For instance, the anticancer activities of ginseng saponins vary depending on the number of sugar moieties, the number and the position of hydroxyl functionalities, and stereochemical features [64]. Ginsenosides with four or more sugar molecules had no significant anticancer activity, which could be attributed to their low lipophilicity [65]. The polar hydroxy groups in a sugar moiety reduce the ginsenoside’s hydrophobic character, rendering it less likely to cross the cell membrane. Thus, the cellular uptake capability is decreased, and effective concentration is not achieved [66]. The finding that ginsenosides with higher lipophilicity (fewer or no sugars) possess higher anticancer activity indicates the inverse correlation between anticancer activity and the number of sugar residues [67]. Liu and Fan reported that the deglycosylation of ginsenoside Rb1 and subsequent dehydration of carbon at position 20 of the resultant ginsenoside Rg3 yielded Rg5. Then, they evaluated the antiproliferative activities of Rb1, 20(R)-Rg3, 20(S)-Rg3, and Rg5 on various human cancer cell lines using the MTT assay. The most remarkable cytotoxicity in the various cancer cells was associated with Rg5, which has two fewer sugar moieties than Rb1 [25]. Rg5 exhibits higher lipophilicity when compared to Rb1, and this could rationalize why it was found to be more cytotoxic. Rg5 differs from Rg3 only by the presence of a hydroxyl group at C-20 in Rg3 and, thus, the number of sugar moieties cannot explain the more significant cytotoxicity of Rg5 when compared to both enantiomers of Rg3.
Nonetheless, literature data revealed that differences in the number and position of hydroxyl groups influence the anticancer activity of different ginsenosides [61]. Dehydration at C-20 of Rg3 appears to be essential for the anticancer activity of Rg5. In this context, Rg5 demonstrated the highest activity against all human cancer cell lines examined [25]. In addition, Rg5 promotes breast cancer cell apoptosis with higher potency than Rg3 [50].
Bioavailability and pharmacokinetics of ginsenoside Rg5
Over the last 50 years, ginsenosides have been recognized to exert countless pharmacological effects; nevertheless, their low oral bioavailability (< 20%) hindered further development [68, 69]. The reason behind this low bioavailability is multifactorial. It has been linked to their (i) relatively high molecular weights (> 600), (ii) low water-solubility, (iii) biodegradation by the intestinal microflora and/or gastric acid conditions, and (iv) extensive hepatic metabolism [70]. Therefore, several studies aimed at enhancing their bioavailability and consequently their efficacy have been carried out [71, 72]. PPD-type ginsenosides have demonstrated a low rate of absorption and elimination (Tmax was 0.83–6 h and t1/2 was 1.48–39.4 h) relative to PPT-type ginsenosides (Tmax was 0.5–0.92 h and t1/2 0.2–5.01 h [70]. This variation has been attributed to the probable influence of the C-6 hydroxyl group present in PPT-type ginsenosides [73]. Further, PPD-type ginsenosides exhibited higher plasma protein binding relative to their counterparts, a property that could explain their slow elimination rate [70].
Concerning biotransformation, several studies have focused on the role of the gut microflora in transforming ginsenosides before their absorption. As illustrated in Fig. 7, ginsenoside Rg5 is metabolized by removing one sugar moiety to afford ginsenoside Rh3. Deglycosylation of the latter resulted in the hydrophobic metabolite, the aglycone moiety, and PPD [74]. On the other hand, little is known about the metabolism of ginsenosides in the liver, which should be an essential area of future research.

Proposed metabolism of ginsenoside Rg5 by the gut microflora. The structures made by ChemDraw (https://revvitysignals.com)
As already mentioned, Rg5 is PPD-type ginsenoside and, therefore, is expected to display a pharmacokinetics profile similar to that of other structural class members. However, there is still considerable ambiguity regarding its pharmacokinetics properties, and the research in this area is minimal. A single report by Kim et al. has provided some data concerning the pharmacokinetics properties of Rg5. Unfortunately, this report has been limited in several ways: (i) small sample size, (ii) short experimental period, and (iii) restricted to the evaluation of the red ginseng extract rather than the individual ginsenosides. To this end, future studies on the pharmacokinetics profile of Rg5 are highly recommended [75].
Preclinical anticancer studies
Many compounds are selected as therapeutic drug candidates for various types of cancer, one of which is Rg5. Research on the anticancer activity of Rg5 has been carried out in vitro on various cancer cells, as shown in Table 1.
Breast cancer is the most malignant cancer in women. The morbidity from this disease is one in 20 globally and one in eight in high-income countries [82]. The most common breast cancers are invasive lobular and ductal carcinoma or a mixture. Rg5 suppresses the signaling of phosphorylated phosphorylated phospatidyl inositol-kinase (PI3K), group serine-threonine enzyme (Akt), mammalian target of rapamycin (mTOR), and Bad but increases Bax and cleaved caspase-3. These are mechanisms for Rg5 to inhibit breast cancer. Kim and Kim [50] conducted an in vitro study using MCF-7 and MDA-MB-453 cells treated with Rg5. MCF-7 cells were further inhibited by 200 µM Rg5 for 72 h. This inhibition is due to Rg5-induced cell cycle arrest in the G0/G1 phase and polyadenosine diphosphate ribose polymerase (PARP) cleavage. Decreased gene expression of cyclin D1, cyclin E2, and CDK4 and increased expression of p15 geneINK4B, p53, and p21WAF1/CIP1 also trigger inhibition of MCF-7 cell growth. In vitro studies have been carried out using BT-474 and T-47D breast cancer cells. Through adenosine monophosphate (AMP) kinase inhibition and inactivation of p70S6K and S6, the viability of these cells was reduced by 73.3% for BT-474 and 76.5% for T-47D at Rg5 concentrations of 10 mg/ml [76]. The mRNA translation mechanism causing this phenomenon was also proposed.
Another in vitro study states that Rg5 can inhibit the growth of MCF-7 breast cancer cells. Rg5 has an IC50 of 78.39 ± 4.63 µM in MCF-7 cancer cells, the lowest value when compared to several other cancer cells [25]. The inhibition of MCF-7 growth was due to the presence of mitochondria-mediated apoptosis and autophagy mechanisms. Rg5 increased cleavage of caspase-3, -8, -9, PARP, cytochrome c, and Bax. The expression of Bcl-2, PI3K/Akt, mTOR, and Bad decreased due to Rg5 treatment. This compound inhibits p62 but promotes LC3B, LC3B-II, Atg5, Atg12, and Beclin expressions.
The GIT is a complex digestive system that includes several vital organs, such as the mouth, esophagus, stomach, and intestines. Malignant cancers of the GIT include esophageal, colorectal, and stomach cancer [83]. Several studies revealed the anticancer activity of Rg5 as a potential candidate for treating gastrointestinal cancer.
Liu and Fan [25] conducted an in vitro study regarding the effects of Rg5 on several cancer cells, including colorectal (CACO-2) and gastric (SGC-7901) cancer cells [25]. Rg5 showed considerable concentration-dependent toxicity in CACO-2 and SGC-7901 cells. However, SGC-7901 was more stunted than CACO-2 after treatment with Rg5. The IC50 values for CACO-2 and SGC-7901 after being treated with Rg5 for 48 h were 101.46 ± 4.75 µM and 89.09 ± 6.47 µM, respectively.
Liu and Fan added Rg5 treatment to gastric cancer cells, namely SGC-7901 and BGC-823 [84]. As a result, Rg5 inhibited both cells depending on the dose and duration of treatment. A dose of 200 µM Rg5 with a treatment duration of 48 h gave a very high inhibitory effect. The reduction in cancer cell growth is thought to be due to apoptosis resulting from increased expression levels of p21, cyclin B1, phosphor-cdc25c, and phosphor-cdc2, as well as decreased expression levels of cdc2. Rg5 also induces autophagy by increasing the expression of LC3B-II, Atg5, Atg12, and Beclin-1 and decreasing the expression of p26. Reactive oxygen species (ROS) and adenosine monophosphate-activated protein kinase (MAPK) signaling pathways were also activated by Rg5 treatment as evidenced by the increased phosphorylation level of p38, jun N-terminal kinase (JNK), and extracellular signal-regulated kinase (ERK).
Eye cancer, especially retinoblastoma, is common in children. The ratio reaches 1:20,000 births, and almost half of the cases worldwide are hereditary [85]. Therefore, research on the discovery of novel anticancer agents and treatment of retinoblastoma is urgently needed. Cui et al. [77, 79] have shown that Rg5 could reduce the proliferation of WERI-RB-1 and Y79 cells. This inhibition, due to the induction of apoptosis, was dependent on the dose and duration of the treatment. In addition, the mechanism of apoptosis was linked to a decrease in Bcl2 expression at the mRNA level. Bcl 2 is a protein that has a vital role in the process of apoptosis in various cancers. Akt phosphorylation activity was also disrupted and decreased by treating cancer cells with Rg5.
Esophageal cancer can be divided into adenocarcinoma and squamous cell carcinoma types. These two types of cancer are the most common deadly cancers in Africa and Southeast Asia [86]. Rg5 induces apoptosis in Eca-109 esophageal cancer cells as revealed by Zhang et al. [78, 80]. They have shown that concentrations ranging from 2 to 32 µM could inhibit Eca-109 proliferation in a dose-dependent manner. The inhibition of Eca-109 proliferation was due to increased caspase-3, -8, and -9 signaling pathways. In addition, apoptosis was also induced by increased PARP cleavage and decreased Bcl-2/Bax ratio, expression of B-cell lymphoma 2, p-Akt, and PI3K/Akt signaling [78].
Liver cancer is a disease that occurs due to chronic cirrhosis, which causes the third most deaths worldwide [87]. Chen et al. [79] confirmed the anticancer activity, specific to liver cancer, of Rg5. The susceptibility of MHCC-97H cells was evident due to reduced cell numbers after Rg5 treatment, especially at a 50 µg/ml concentration. The IC50 value for Rg5 was 4.94 µg/ml. The mechanism that occurs in the anti-liver cancer activity of Rg5 was also linked to the induction of apoptosis. Apoptosis is caused by releasing cytochrome c and Smac into the cytoplasm from mitochondria, caspase-3 and -9 activation. Thus, the Bcl-xL, Bcl2, and c-IAP2 MHCC-97H levels decreased upon administration of Rg5 in cancer cells.
Osteosarcoma is a type of bone cancer that generally begins in the mesenchymal stem cells. Osteosarcoma is often located in the metaphysis of the bones around the knee (distal femur or proximal tibia) [88]. Based on in vitro studies using MG-63, HOS, and U2OS cells, Liu et al. (2021) stated that Rg5 could potentially inhibit bone cancer cells [89]. The study showed that treatment with Rg5 for 24 h and concentrations ranging from 0 to 1280 nM led to more inhibition of the growth of MG-63 cells than other cells. The minimum inhibitory concentration was recorded as 80 nM. This compound inhibits MG-63 cell growth through caspase-3 activity related to the LC3 autophagy pathway. In addition, Rg5 inhibits the PI3K/Akt/mTORC1 pathway by downregulating phosphorylated PI3K and upregulating Raptor.
The lungs are vital organs that are exposed to extrinsic factors that cause cancer, such as smoking and pollution. Lung cancer accounted for 12% of cancer cases globally in 2018 [90]. The effect of Rg5 on lung cancer has been studied in vitro using A549 cells [81]. The compound suppressed cell proliferation associated with inhibition of E-cadherin expression and increased expression of CD44 and CD133. Treatment with Rg5 also caused a reduction in the TGF-β1 signaling pathway and nuclear factor kappa-light-chain-enhancer of activated B cells/extracellular signal-regulated kinase (NF-κB/ERK) activation.
In vivo research on the anticancer activity of Rg5 is also being carried out to determine the efficacy of this compound. The commonly used animal models are nude mice with xenografts or direct injection of cancer cells into the body’s system (Table 2).
Dong et al. [91, 93] studied the in vivo anticancer effect of ginsenoside Rg5 by employing a mouse model of a xenograft subcutaneous tumor of human MCF-7 breast cancer. The in vitro IC50 values for Rg5 were 26.55 ± 0.84 µM and 19.75 ± 2.58 µM for 24 and 48 h treatment, respectively. The signaling pathway for this remains unclear, but Rg5 effectively reduced the tumor volume that develops in animal models. Rg5 has also been shown to reduce breast tumor volume in the BALB/c nude mouse model in vivo [92]. In this study, high doses (20 mg/kg) of Rg5 could reduce tumor volume by up to 71.4 ± 9.4%, an amount almost the same as the positive control used (docetaxel) of 72.0 ± 9.1%, but with minimal side effects on the body. The compound’s intrinsic mitochondrial and extrinsic death receptor activation caused caspase-dependent apoptosis. Induction of apoptosis and autophagy also occurred due to inhibition of the PI3K/Akt signaling pathways because PI3K and Akt phosphorylation decreased drastically. In addition, cleavage of caspase-3, -8, -9, Bax, cytochrome c, PARP, and Ras levels increased after Rg5 treatment. This compound also suppressed Bcl-2 expression. The autophagy corresponded to an increase of LC3B-II, Atg5, Atg7, Atg12, and a decrease of p26. A high dose of Rg5 further decreased the PI3K/Akt phosphorylation level.
Liu and Fan [86] also proved that Rg5 can inhibit the growth of gastric cancer cells in vivo using a mouse model injected with BGC-823 subcutaneously on the left side [84]. The 40 mg/kg (injected intraperitoneally) Rg5 treatment significantly suppressed the tumor volume formed. The reduced tumor volume was due to increased expressions of pro-caspase-3, pro-PARP, p-ERK/ERK, LC3-II, p-JNK/JNK, cyclin b1, and p38/p38, but there was inhibition in p62 and cdc levels expression.
In vivo research on the effect of Rg5 on lung cancer has been carried out by Feng et al. [93, 95]. In experimental animals injected with A549/T, various doses of Rg5 showed significant activity in tumor volume reduction. However, this study did not elucidate the mechanism or signaling pathway associated with the observed anticancer effect. Apart from lung cancer, Rg5 can also inhibit the growth of ovarian cancer in the nude mice model in a dose-dependent manner [94]. At a 20 mg/kg dose of Rg5, tumor volume reduction of up to 83% was observed. This was suggested to be due to decreased expression of the FGF-8b gene after treatment with Rg5 for 30 days.
In general, the mechanism of Rg5 as an anticancer drug in vitro and in vivo is summarized in Fig. 8. The in vitro data appeared to be supported by the in vivo research results.

Overview of mechanisms of action of ginsenoside Rg5 as an anticancer agent. p26 polyclonal-26 antibody; PI3K phosphatidyl inositol-3-kinase; Ras Ras oncogenes; p53 polyclonal-53 antibody; NF-κB nuclear factor kappa-B; Akt group of serine-threonine enzyme; Erk extracelullar signal-regulated kinase; Bad proapoptotic BH3-only protein; AMPK AMP-activated protein kinase; mTOR mammalian or mechanistic target of rapamycin; p70S6k p70 ribosomal s6 kinase; S6 serine-threonine protein-6; Cyclin B1 regulatory protein in mitosis of cell cycle from G2 to M phase; Cdc: cell division cycle; CDK 4: cyclin dependent kinase-4; Cyclin E2 regulatory protein in mitosis of cell cycle, i.e. S-phase; p21 polyclonal-21 antibody; p15 polyclonal-15 antibody; Cyclin D1 regulator of cell cycle progression, oncogenic driver; Bcl-xL B-cell lymphoma extra-large; Bcl-2 B-cell lymphoma-2; Bax Bcl-2 associated X-protein; Bak Bcl-2 homologous antagonist/killer; Ca2+ calcium cation; Cyt C cytochrome C; PARP poly-adenosine diphospate-ribose polymerase; ROS reactive oxygen species; Smad 2/3 suppressor of mothers against decapentaplegic 2/3. The figure made by Biorender (https://www.biorender.com)
Mechanism of antitumor action of Ginsenoside Rg5
In the 2010s, the mechanism of anticancer activity of Rg5 was elucidated by examining the expression of proteins involved in signaling pathways associated with cell proliferation. The primary mechanism related to the effect was the PI3K/AKT, NF‑κB, and ataxia telangiectasia mutation (ATM)/mouse double minute 2 (MDM2) pathways [95]. It is involved in cancer migration by affecting its degradation by lysosomes. Rg5 inhibits the phosphorylation of NF‑κB and erythropoietin-producing hepatocellular receptor A2 (EphA2). It suppressed the expression of EphA2 due to lysosomal degradation that led to inhibition of cancer cell migration [96].
Regarding the anticancer effect of Rg5, there has been intense research recently, primarily using bioinformatics and network pharmacology to identify essential target proteins. The informatics analysis elicited forty-four estimated target proteins for hepatocellular carcinoma [79]. Docking simulation estimated Estrogen Receptor 2 (ESR2) and Heat Shock Protein (HSP) 90AA1 were the candidates for target proteins—suppression of ESR2-induced apoptosis in gastric carcinoma [97]. However, details of the drug-protein interaction and inhibition of the biological activity need further research.
Annexin A2 was also identified as the target protein of Rg5 [98]. Annexin A2 activates NF-κB via interaction with the NF-κB p50 subunit. In this connection, NF-κB activation promotes tumorigenesis and cancer cell growth. The inhibition of the activity of NF-κB-induced apoptosis due to down-regulation of anti-apoptosis protein expressions such as inhibitors of apoptosis proteins (IAPs) in HepG2 cells. The mechanism of the anticancer effect of Rg5 was investigated using osteosarcoma cells (MG-63, HOS, and U2OS cells). It inhibited the phosphorylation of PI3K, Akt, and mTORC1, which induce apoptosis through active caspase-3 [24, 92]. Rg5 also suppresses human esophageal cancer cells by inhibiting the PI3K/Akt pathway [78]. Annexin A2 accelerated the PI3K pathway [29]. There is the possibility that the anticancer activity in osteosarcoma cells is mediated through the PI3K pathway as well as inhibition of Annexin A2 as the target protein. The PI3K is also studied as the target protein of Rg5. The growth of breast cancer cells (MCF-7 cells) was shown to be suppressed by treatment with Rg5. The compound also inhibits the phosphorylation of PI3K, Akt, mTOR, and Bad. The docking simulation study predicted a strong binding between ginsenoside Rg5 and PI3K [25]. However, data on the actual binding and in vitro inhibition should be collected to substantiate the docking study.
A series of critical reports on the mechanism of antitumor activity of Rg5 have been published in recent years. The reports identified some target proteins, suggesting an effect mediated through not one but multiple target proteins. The combined effect is thought to regulate the cancer signaling pathway and lead to inhibition of cell proliferation via apoptosis.
Barriers and opportunities for conducting human clinical studies
Over the last five decades, research on Rg5 was limited to human cell culture and/or experimental animal studies. Generally, studies based on clinical trials involving ginsenosides have been minimal, even though ginsenosides-containing products have long been recognized to have efficient anticancer properties when used alone or in combination with other medications [99]. Furthermore, the clinical trials had substantial flaws, such as short study periods, irrational experimental design, and limited sample size [46, 75]. In addition, the low bioavailability of natural ginsenosides, in general, has been identified as the major limitation to stepping further into human clinical trials [100]. To address this issue, three fundamental approaches should be followed (i) preparation of large amounts of pure Rg5; (ii) promotion of Rg5 stability in the GIT, and (iii) design of specific drug delivery systems based on a systematic analysis of Rg5 physicochemical properties [99]. Therefore, further investigations to explain the pharmacokinetics profile and toxicity in animal models and humans are in high demand. These investigations could help boost the potential of Rg5 as a promising natural drug candidate for contribution to global health promotion.
Toxicity, side effects, and safety
The ability of Rg5 to cause apoptosis underlies its effect as an anticancer agent. However, apart from being an inducer of apoptosis, Rg5 also has an anti-apoptotic effect on nerve cells [101], liver cells [59], cartilage, and chondrocyte matrix in cases of rheumatism [29], cisplatin-induced kidney damage [102] and cardiomyocyte [103]. Cell apoptosis, inflammation, and oxidative stress damage are pathogenic mechanisms underlying chemotherapy-induced toxicity to immune response and vital organs such as renal, gastrointestinal mucosal, neuron, hematopoiesis, heart, immune response, liver, and hair loss [104]. Rg5 has an antioxidative effect on HT22 cells induced by thermal stress. In HT22 cells exposed to heat stressors, the production of oxidative stress indicators, namely nitric oxide (NO), is attenuated through the regulation of antioxidant enzymes such as heme oxygenase-1/nuclear factor erythroid 2-related factor 2 (HO-1/Nrf2) and glutathione [105]. These enzymes are crucial in redox homeostasis [105]. Thus, side effects or toxic effects of Rg5 may occur because of the drug’s action on regulating multiple signaling pathways and targets.
A toxicity study on Rg5 in vitro showed that it was non-toxic to the Murine Hippocampal Neuronal Cell Line HT22 cells at concentrations 20 and 40 µg/mL [105] and up to 100 µg/ml to 3T3-L1 cells [27]. However, at > 60 µg/mL, Rg5 displayed a toxic effect on HTT2 cells, and at a dose of 100 µg/ml, the gene expression of HTT2 was up and down-regulated [106], in which at non-toxic levels, Rg5 exhibits neuroinflammation, apoptosis, and immune response effects. The potential toxic effect of Rg5, which is indicated by the LC50 value of Rg5 on MCF-7 cells for 24 h, was 98 ± 3.76 µM and 38.61 ± 7.72 µM on exposure for 48 h. Meanwhile, the LC50 on L929 cells for 24 h of exposure was 65.02 ± 18.17 µM [91]. These data indicate that the potential toxic effect of Rg5 as indicated by the LC50 value, is classified as highly toxic [107]. This data also presented differences in the sensitivity of target cells to Rg5.
Safety and effectiveness studies on Rg5 had been conducted in vivo on tumor-transplanted mouse models for breast cancer. When injected intraperitoneally at a dose of 20 mg/kg for 30 consecutive days, Rg5 showed no significant changes in all aspects of clinical and pathological parameters like the white blood cell, lymphocyte, and granulocyte contents in peripheral blood, two transaminases (ALT 257 and AST), the blood urea nitrogen (BUN), and creatinine level in serum. Pathological patterns of the lung, spleen, kidney, and liver showed no significant differences from those of normal mice. Meanwhile, at the same dose of 20 mg/kg, Rg5 inhibited the growth of breast cancer cells through apoptosis and autophagy due to the inhibition of the 2 PI3K/Akt pathway [92]. Variations in the toxicity effects in vivo and in vitro may be connected to Rg5’s kinetic profile, which has a low oral bioavailability (< 20%) [68, 69] due to its extensive hepatic metabolism and poor oral absorption [70]. It was concluded that Rg5 at this dose is safe to treat breast cancer. The safety of Rg5 was also demonstrated at twice as high as the dose used for breast cancer. At a dose that showed a reduction in the size of gastric tumors, namely 40 mg/kg intraperitoneally for 30 days, Rg5 did not affect the mice’s weight gain or histopathological analysis parameters.
Even though Rg5, up to a dose of 40 mg/kg body weight, is safe with no harm to vital organs and the immune response, there is a possibility that it may have side effects. Rg5 can potentially reduce the activity of the acetylcholinesterase enzyme [108], which breaks down acetylcholine into choline and acetate. Acetylcholine is a neurotransmitter of the parasympathetic nervous system in the peripheral and central nervous systems. Peripheral parasympathetic nerves control the performance of the heart, lungs, gastrointestinal, and mucus expenditure. Inhibition of acetylcholinesterase may lead to hypotension and bradycardia due to increased acetylcholine activity. The hypotensive effect of Rg5 is also supported by the action of Rg5 on vascular cells in which NO/cGMP pathway and angiogenic signaling were inhibited by Rg5, leading to relaxation of the aortic blood vessels [109, 110], and anti-platelet aggregation [111] as well as inhibition of angiotensin-converting enzyme (ACE). The potential inhibitory effect of Rg5 on ACE was found at level IC50 = 0.124 µM [112].
Another side effect of Rg5 is related to its ability to inhibit the performance of gamma-aminobutyric aminergic (GABAA) and serotonin receptors. Oral administration of Rg5 into rats for 7 consecutive days at a dose of 45 mg/kg and into mice at doses of 30 and 60 mg/kg significantly promoted the sleep quality index and lessened the locomotor activity. The sedative and hypnotic effects of Rg5 were elicited by acting on GABA and the serotonin nervous system [30]. The doses affected (GABAergic) and serotonin were within the range of those used for treating gastric tumors. Thus, the sedative and hypnotic effects are likely to be side effects accompanying the use of Rg5 as an antitumor drug.
Safety effect of Ginsenoside Rg5 under pathophysiological conditions
The potential damage to these two organs requires the dosage of some drugs readjusted to avoid toxic effects. Besides that, pathophysiological changes in the body will cause changes in the function of the liver and kidneys, resulting in unwanted effects of Rg5.
Administration of Rg5 at 40 mg/kg for 30 days in breast cancer mice did not affect the liver morphology in male mice. However, the liver function in glucose and lipid metabolism of diabetic mice was influenced by Rg5 at a dosage higher than 40 mg/kg mg/kg. At 90 mg/kg body weight/day for 8 weeks, Rg5 reduced hepatic insulin resistance, leading to improved glucose intolerance and lipid accumulation [113]. Hepatic clearance can be reduced in the presence of fatty liver of non-insulin-dependent diabetes mellitus (NIDDM). In addition, uncontrolled diabetes causes an overall increase in cytochrome P450 in the rat [114]. Rg5 decreases the subtype of cytochrome P450, namely CYP450 2 E1 [59]. The ability of Rg5 to suppress cytochromes CYP450 2 E1 and its ability to inhibit other parameters related to liver damage indicates the potential of Rg5 as a hepatoprotective agent. The protective effect was also demonstrated in mice that were given Rg5, followed by acetaminophen. The CYP450 2 E1 in these mice also decreased.
Cytochrome P450 subfamily 2E1 (CYP450 2E1) was involved in the oxidation of acetaminophen, which produces N-acetyl-p-benzoquinone imine (NAPQI) as a reactive metabolite [115]. When the hepatic glutathione supply is depleted, NAPQI reacts with hepatic cellular membrane molecules, causing hepatocyte damage and death. The widespread damage of hepatocytes leads to acute liver necrosis [116,117,118].
Glomerular filtration is elevated in diabetic patients [114]. Furthermore, Diabetes mellitus was reported to be a risk factor for pancreatic, liver, colon, breast, and endometrial cancer [119]. The safety of Rg5 to renal function in diabetic conditions was demonstrated by improving oxidative stress in the renal injury of mice. Rg5 at an oral dose of 90 mg/kg body weight/day for 8 weeks reduced inflammatory responses by inhibiting oxidative stress and NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation. Studies in vitro also displayed that Rg5 had renoprotective effects on cisplatin-induced cytotoxicity in LLC-PK1 cells at concentrations of 50,100, and 250 µg/mL. Rg5 is safe for the liver and kidneys; however, the dose of Rg5 that protects the kidneys of mice is higher than the dose that causes sedation in mice. Therefore, it is speculated that sedation will occur as a side effect when RG5 is used to protect these organs.
Conclusions and future prospects
A literature survey has identified a significant shortage in the SAR of Rg5 as only very few reports addressed the anticancer activity of Rg5 in comparison to its natural or synthetic analogs. Consequently, future efforts should focus on performing critical and profound SAR studies to unveil the structural features essential for the potent anticancer activity of Rg5. This will help further optimization. Rg5 demonstrated low oral bioavailability (< 20%), restricting its clinical use. This low bioavailability has been attributed to its relatively high molecular weights (> 600) and the extensive degradation by intestinal microflora. Thus, further studies on enhancing its bioavailability are crucial. Further, little is known about the effect of Rg5 on liver metabolizing enzymes and/or identification of its actual active metabolites. There is still considerable ambiguity regarding the pharmacokinetics properties of Rg5, and the research in this area is minimal. Furthermore, its therapeutic effects have not been clinically evaluated, which could be considered a significant challenge for developing Rg5 in medical use. This review documented the various therapeutic attributes of Rg5 in the field of cancer and thus provides a valuable platform for developing Rg5 as a potential anticancer drug candidate.
Data availability
Not applicable.
Abbreviations
- Akt:
-
Group of serine-threonine enzyme
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- AMPK:
-
AMP-activated protein kinase
- AMP:
-
Adenosine monophosphate
- ATM:
-
Ataxia telangiectasia mutation
- Bad:
-
Proapoptotic BH3-only protein
- Bak:
-
Bcl-2 homologous antagonist/killer
- Bax:
-
Bcl-2 associated X-protein
- Bcl-2:
-
B-cell lymphoma-2
- Bcl-xL:
-
B-cell lymphoma extra large
- BUN:
-
Blood urea nitrogen
- Ca2+ :
-
Calcium cation
- Cdc:
-
Cell division cycle
- CDK 4:
-
Cyclin-dependent kinase-4
- Cyclin B1:
-
Regulatory protein in mitosis of the cell cycle from G2 to M phase
- Cyclin D1:
-
A regulator of cell cycle progression, oncogenic driver
- Cyclin E2:
-
Regulatory protein in mitosis of the cell cycle, i.e. S-phase
- Cyp450:
-
Cytochrome P450
- Cyt C:
-
Cytochrome C
- EphA2:
-
Erythropoietin-producing hepatocellular receptor A2
- Erk:
-
Extracellular signal-regulated kinase
- ESR2:
-
Estrogen receptor 2
- GABA:
-
Gamma-aminobutyric acid
- Ginsenoside Rg5:
-
Rg5
- GIT:
-
Gastrointestinal tract
- HSP:
-
Heat shock protein
- IAPs:
-
Inhibitors of apoptosis proteins
- IR:
-
Infrared
- MDM2:
-
Mouse double minute 2
- MS:
-
Mass spectrometry
- mTOR:
-
Mammalian target of rapamycin
- NAPQI:
-
N-acetyl-p-benzoquinone imine
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NIDDM:
-
Non-insulin-dependent diabetes mellitus
- NLRP3:
-
NOD-like receptor family pyrin domain-containing 3
- NMR:
-
Nuclear magnetic resonance
- PARP:
-
Poly-adenosine diphosphate-ribose polymerase
- p21:
-
Polyclonal-21 antibody
- p15:
-
Polyclonal-15 antibody
- p26:
-
Polyclonal-26 antibody
- p70S6k:
-
P70 ribosomal s6 kinase
- p53:
-
Polyclonal-53 antibody
- PI3K:
-
Phosphatidyl inositol-3-kinase
- PARP:
-
Poly-adenosine diphosphate-ribose polymerase
- PPD:
-
Protopanaxadiol
- PPT:
-
Protopanaxatriol
- Ras:
-
Ras oncogenes
- ROS:
-
Reactive oxygen species
- SAR:
-
Structure–activity relationship
- S6:
-
Serine-threonine protein-6
- Smad 2/3:
-
Suppressor of mothers against decapentaplegic 2/3
- TGF-β:
-
Transforming growth factor-beta:
- T max :
-
Peak time
- V d :
-
Volume of distribution
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Sobral PJM, Vicente ATS, Salvador JAR. Recent advances in oridonin derivatives with anticancer activity. Front Chem. 2023;11:1066280.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Hassanpour SH, Dehghani M. Review of cancer from perspective of molecular. J Cancer Res Pract. 2017;4(4):127–9.
Antwi SO, Eckert EC, Sabaque CV, Leof ER, Hawthorne KM, Bamlet WR, Chaffee KG, Oberg AL, Petersen GM. Exposure to environmental chemicals and heavy metals, and risk of pancreatic cancer. Cancer Causes Control. 2015;26(11):1583–91.
Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118(12):3030–44.
Gm C. The cell: a molecular approach. Sunderland: Sinauer Associates; 2000.
Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther. 2016;38(7):1551–66.
Naeem A, Hu P, Yang M, Zhang J, Liu Y, Zhu W, Zheng Q. Natural products as anticancer agents: current status and future perspectives. Molecules. 2022;27(23):8367.
Sun LR, Zhou W, Zhang HM, Guo QS, Yang W, Li BJ, Sun ZH, Gao SH, Cui RJ. Modulation of multiple signaling pathways of the plant-derived natural products in cancer. Front Oncol. 2019;9:1153.
Atanasov AG, Zotchev SB, Dirsch VM, International Natural Product Sciences T, Supuran CT. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov. 2021;20(3):200–16.
Shen J, Li J, Yu P, Du G. Research status and hotspots of anticancer natural products based on the patent literature and scientific articles. Front Pharmacol. 2022;13: 903239.
Priya S, Satheeshkumar PK. Natural products from plants. In: Functional and Preservative Properties of Phytochemicals. Academic Press; 2020. p. 145–163.
Klos P, Chlubek D. Plant-derived terpenoids: a promising tool in the fight against melanoma. Cancers (Basel). 2022;14(3):502.
Shagufta P. Introductory chapter: terpenes and terpenoids. In: Shagufta P, Areej A-T, editors. Terpenes and terpenoids. Rijeka: IntechOpen; 2018.
Swamy MK, Akhtar MS. Natural bio-active compounds. Singapore: Springer; 2019.
Mahizan NA, Yang SK, Moo CL, Song AA, Chong CM, Chong CW, Abushelaibi A, Lim SE, Lai KS. Terpene derivatives as a potential agent against antimicrobial resistance (AMR) pathogens. Molecules. 2019;24(14):2631.
Darshani P, Sen Sarma S, Srivastava AK, Baishya R, Kumar D. Anti-viral triterpenes: a review. Phytochem Rev. 2022;21(6):1761–842.
Del Prado-Audelo ML, Cortes H, Caballero-Floran IH, Gonzalez-Torres M, Escutia-Guadarrama L, Bernal-Chavez SA, Giraldo-Gomez DM, Magana JJ, Leyva-Gomez G. Therapeutic applications of terpenes on inflammatory diseases. Front Pharmacol. 2021;12: 704197.
Kamran S, Sinniah A, Abdulghani MAM, Alshawsh MA. Therapeutic potential of certain terpenoids as anticancer agents: a scoping review. Cancers (Basel). 2022;14(5):1100.
Ratan ZA, Haidere MF, Hong YH, Park SH, Lee JO, Lee J, Cho JY. Pharmacological potential of ginseng and its major component ginsenosides. J Ginseng Res. 2021;45(2):199–210.
Zhao L, Zhao H, Zhao Y, Sui M, Liu J, Li P, Liu N, Zhang K. Role of ginseng, quercetin, and tea in enhancing chemotherapeutic efficacy of colorectal cancer. Front Med (Lausanne). 2022;9: 939424.
Vayghan HJ, Ghadimi SS, Nourazarian AR. Preventive and therapeutic roles of ginseng - focus on colon cancer. Asian Pac J Cancer Prev. 2014;15(2):585–8.
Liu MY, Liu F, Gao YL, Yin JN, Yan WQ, Liu JG, Li HJ. Pharmacological activities of ginsenoside Rg5 (Review). Exp Ther Med. 2021;22(2):840.
Liu Y, Fan D. The preparation of ginsenoside Rg5, its antitumor activity against breast cancer cells and its targeting of PI3K. Nutrients. 2020;12(1):246.
Ponnuraj SP, Siraj F, Kang S, Noh HY, Min JW, Kim YJ, Yang DC. Amelioration of insulin resistance by Rk1 + Rg5 complex under endoplasmic reticulum stress conditions. Pharmacognosy Res. 2014;6(4):292–6.
Simu SY, Ahn S, Castro-Aceituno V, Yang D-C. Ginsenoside Rg5: Rk1 exerts an anti-obesity effect on 3T3-L1 cell line by the downregulation of PPARγ and CEBPα. Iran J Biotechnol. 2017;15(4):252.
Zhu Y, Zhu C, Yang H, Deng J, Fan D. Protective effect of ginsenoside Rg5 against kidney injury via inhibition of NLRP3 inflammasome activation and the MAPK signaling pathway in high-fat diet/streptozotocin-induced diabetic mice. Pharmacol Res. 2020;155: 104746.
Zhang P. Ginsenoside-Rg5 treatment inhibits apoptosis of chondrocytes and degradation of cartilage matrix in a rat model of osteoarthritis. Oncol Rep. 2017;37(3):1497–502.
Shao J, Zheng X, Qu L, Zhang H, Yuan H, Hui J, Mi Y, Ma P, Fan D. Ginsenoside Rg5/Rk1 ameliorated sleep via regulating the GABAergic/serotoninergic signaling pathway in a rodent model. Food Funct. 2020;11(2):1245–57.
Cho YL, Hur SM, Kim JY, Kim JH, Lee DK, Choe J, Won MH, Ha KS, Jeoung D, Han S, Ryoo S, Lee H, Min JK, Kwon YG, Kim DH, Kim YM. Specific activation of insulin-like growth factor-1 receptor by ginsenoside Rg5 promotes angiogenesis and vasorelaxation. J Biol Chem. 2015;290(1):467–77.
Qi LW, Wang CZ, Yuan CS. Ginsenosides from American ginseng: chemical and pharmacological diversity. Phytochemistry. 2011;72(8):689–99.
Kim HJ, Kim P, Shin CY. A comprehensive review of the therapeutic and pharmacological effects of ginseng and ginsenosides in central nervous system. J Ginseng Res. 2013;37(1):8–29.
Lee SM, Bae BS, Park HW, Ahn NG, Cho BG, Cho YL, Kwak YS. Characterization of Korean Red Ginseng (Panax ginseng Meyer): history, preparation method, and chemical composition. J Ginseng Res. 2015;39(4):384–91.
Park SK, Hyun SH, In G, Park CK, Kwak YS, Jang YJ, Kim B, Kim JH, Han CK. The antioxidant activities of Korean Red Ginseng (Panax ginseng) and ginsenosides: a systemic review through in vivo and clinical trials. J Ginseng Res. 2021;45(1):41–7.
Shin BK, Kwon SW, Park JH. Chemical diversity of ginseng saponins from Panax ginseng. J Ginseng Res. 2015;39(4):287–98.
Kim SK, Park JH. Trends in ginseng research in 2010. J Ginseng Res. 2011;35(4):389–98.
Lee DH, Cho HJ, Kim HH, Rhee MH, Ryu JH, Park HJ. Inhibitory effects of total saponin from Korean red ginseng via vasodilator-stimulated phosphoprotein-Ser(157) phosphorylation on thrombin-induced platelet aggregation. J Ginseng Res. 2013;37(2):176–86.
Siddiqi MH, Siddiqi MZ, Ahn S, Kang S, Kim YJ, Sathishkumar N, Yang DU, Yang DC. Ginseng saponins and the treatment of osteoporosis: mini literature review. J Ginseng Res. 2013;37(3):261–8.
Kang KS, Ham J, Kim YJ, Park JH, Cho EJ, Yamabe N. Heat-processed Panax ginseng and diabetic renal damage: active components and action mechanism. J Ginseng Res. 2013;37(4):379–88.
Lee S, Kim MG, Ko SK, Kim HK, Leem KH, Kim YJ. Protective effect of ginsenoside Re on acute gastric mucosal lesion induced by compound 48/80. J Ginseng Res. 2014;38(2):89–96.
Lee CH, Kim JH. A review on the medicinal potentials of ginseng and ginsenosides on cardiovascular diseases. J Ginseng Res. 2014;38(3):161–6.
Hyun SH, Bhilare KD, In G, Park CK, Kim JH. Effects of Panax ginseng and ginsenosides on oxidative stress and cardiovascular diseases: pharmacological and therapeutic roles. J Ginseng Res. 2022;46(1):33–8.
Zhao A, Liu N, Yao M, Zhang Y, Yao Z, Feng Y, Liu J, Zhou G. A review of neuroprotective effects and mechanisms of Ginsenosides from Panax Ginseng in treating ischemic stroke. Front Pharmacol. 2022;13: 946752.
Ru W, Wang D, Xu Y, He X, Sun YE, Qian L, Zhou X, Qin Y. Chemical constituents and bioactivities of Panax ginseng (C. A. Mey). Drug Discov Ther. 2015;9(1):23–32.
Fan W, Huang Y, Zheng H, Li S, Li Z, Yuan L, Cheng X, He C, Sun J. Ginsenosides for the treatment of metabolic syndrome and cardiovascular diseases: pharmacology and mechanisms. Biomed Pharmacother. 2020;132: 110915.
So SH, Lee JW, Kim YS, Hyun SH, Han CK. Red ginseng monograph. J Ginseng Res. 2018;42(4):549–61.
Zhang F, Tang S, Zhao L, Yang X, Yao Y, Hou Z, Xue P. Stem-leaves of Panax as a rich and sustainable source of less-polar ginsenosides: comparison of ginsenosides from Panax ginseng, American ginseng and Panax notoginseng prepared by heating and acid treatment. J Ginseng Res. 2021;45(1):163–75.
Le TH, Lee SY, Lee GJ, Nguyen NK, Park JH, Nguyen MD. Effects of steaming on saponin compositions and antiproliferative activity of Vietnamese ginseng. J Ginseng Res. 2015;39(3):274–8.
Kim SJ, Kim AK. Anti-breast cancer activity of fine black ginseng (Panax ginseng Meyer) and ginsenoside Rg5. J Ginseng Res. 2015;39(2):125–34.
Lee MR, Yun BS, Sung CK. Comparative study of white and steamed black Panax ginseng, P. quinquefolium, and P. notoginseng on cholinesterase inhibitory and antioxidative activity. J Ginseng Res. 2012;36(1):93–101.
Duana Z, Deng J, Dong Y, Zhu C, Lia W, Fana D. Anticancer effects of ginsenoside Rk3 on non-small cell lung cancer cells: in vitro and in vivo. Food Funct. 2017;8:3723–36.
Potenza MA, Montagnani M, Santacroce L, Charitos IA, Bottalico L. Ancient herbal therapy: a brief history of Panax ginseng. J Ginseng Res. 2022;47(3):359–65.
Hou M, Wang R, Zhao S, Wang Z. Ginsenosides in Panax genus and their biosynthesis. Acta Pharm Sin B. 2021;11(7):1813–34.
Ramesh J, Thilakan RC, Gopalakrishnan RM, Vijayapoopathi S, Dorschel A, Venugopal B. Ginsenoside Rg5 sensitizes paclitaxel-resistant human cervical-adeno-carcinoma cells to paclitaxel-and enhances the anticancer effect of paclitaxel. Genes (Basel). 2022;13(7):1142.
Bai L, Gao J, Wei F, Zhao J, Wang D, Wei J. Therapeutic potential of ginsenosides as an adjuvant treatment for diabetes. Front Pharmacol. 2018;9:423.
Guo DD, Cheng LQ, Zhang YW, Zheng HC, Ma HY, Li L. An improved method for the preparation of Ginsenoside Rg5 from ginseng fibrous root powder. Heliyon. 2019;5(10): e02694.
Jo SK, Kim IS, Yoon KS, Yoon HH, Yoo HH. Preparation of ginsenosides Rg3, Rk1, and Rg5-selectively enriched ginsengs by a simple steaming process. Eur Food Res Technol. 2014;240(1):251–6.
Wang Z, Hu JN, Yan MH, Xing JJ, Liu WC, Li W. Caspase-mediated anti-apoptotic effect of ginsenoside Rg5, a main rare ginsenoside, on acetaminophen-induced hepatotoxicity in mice. J Agric Food Chem. 2017;65(42):9226–36.
Kang KS, Yamabe N, Kim HY, Yokozawa T. Effect of sun ginseng methanol extract on lipopolysaccharide-induced liver injury in rats. Phytomedicine. 2007;14(12):840–5.
Nag SA, Qin J-J, Wang W, Wang M-H, Wang H, Zhang R. Ginsenosides as anticancer agents: in vitro and in vivo activities, structure–activity relationships, and molecular mechanisms of action. Front Pharmacol. 2012;3:25.
Feng R, Liu J, Wang Z, Zhang J, Cates C, Rousselle T, Meng Q, Li J. The structure-activity relationship of ginsenosides on hypoxia-reoxygenation induced apoptosis of cardiomyocytes. Biochem Biophys Res Commun. 2017;494(3–4):556–68.
Quan K, Liu Q, Wan J-Y, Zhao Y-J, Guo R-Z, Alolga RN, Li P, Qi L-W. Rapid preparation of rare ginsenosides by acid transformation and their structure-activity relationships against cancer cells. Sci Rep. 2015;5(1):8598.
Qi L-W, Wang C-Z, Yuan C-S. American ginseng: potential structure–function relationship in cancer chemoprevention. Biochem Pharmacol. 2010;80(7):947–54.
Li W, Liu Y, Zhang J-W, Ai C-Z, Xiang N, Liu H-X, Yang L. Anti-androgen-independent prostate cancer effects of ginsenoside metabolites in vitro: mechanism and possible structure-activity relationship investigation. Arch Pharmacal Res. 2009;32:49–57.
Fan W, Fan L, Wang Z, Mei Y, Liu L, Li L, Yang L, Wang Z. Rare ginsenosides: a unique perspective of ginseng research. J Adv Res. 2024. https://doi.org/10.1016/j.jare.2024.01.003.
Wang CZ, Yuan CS. Potential role of ginseng in the treatment of colorectal cancer. Am J Chin Med. 2008;36(6):1019–28.
Leung KW, Wong AS-T. Pharmacology of ginsenosides: a literature review. Chin Med. 2010;5(1):1–7.
Yao W, Guan Y. Ginsenosides in cancer: a focus on the regulation of cell metabolism. Biomed Pharmacother. 2022;156: 113756.
Pan W, Xue B, Yang C, Miao L, Zhou L, Chen Q, Cai Q, Liu Y, Liu D, He H. Biopharmaceutical characters and bioavailability improving strategies of ginsenosides. Fitoterapia. 2018;129:272–82.
Cai H, Wen X, Wen L, Tirelli N, Zhang X, Zhang Y, Su H, Yang F, Chen G. Enhanced local bioavailability of single or compound drugs delivery to the inner ear through application of PLGA nanoparticles via round window administration. Int J Nanomed. 2014;9:5591.
Li T, Shu Y-J, Cheng J-Y, Liang R-C, Dian S-N, Lv X-X, Yang M-Q, Huang S-L, Chen G, Yang F. Pharmacokinetics and efficiency of brain targeting of ginsenosides Rg1 and Rb1 given as Nao-Qing microemulsion. Drug Dev Ind Pharm. 2015;41(2):224–31.
Kong L-T, Wang Q, Xiao B-X, Liao Y-H, He X-X, Ye L-H, Liu X-M, Chang Q. Different pharmacokinetics of the two structurally similar dammarane sapogenins, protopanaxatriol and protopanaxadiol, in rats. Fitoterapia. 2013;86:48–53.
Kim D-H. Gut microbiota-mediated pharmacokinetics of ginseng saponins. J Ginseng Res. 2018;42(3):255–63.
Kim HJ, Oh TK, Kim YH, Lee J, Moon JM, Park YS, Sung CM. Pharmacokinetics of Ginsenoside Rb1, Rg3, Rk1, Rg5, F2, and compound K from Red Ginseng Extract in Healthy Korean Volunteers. Evid-Based Complement Altern Med. 2022. https://doi.org/10.1155/2022/8427519.
Zou Y, Liu P. Ginsenoside-Rg5 inhibits proliferation of the breast carcinoma cells through promotion of the proteins involved in AMP kinase pathway. Int J Clin Exp Med. 2016;9(9):17664–9.
Cui Y, Su Y, Deng L, Wang W. Ginsenoside-Rg5 inhibits retinoblastoma proliferation and induces apoptosis through suppressing BCL2 expression. Chemotherapy. 2019;63(5):293–300.
Zhang D, Wang A, Feng J, Zhang Q, Liu L, Ren H. Ginsenoside Rg5 induces apoptosis in human esophageal cancer cells through the phosphoinositide-3 kinase/protein kinase B signaling pathway. Mol Med Rep. 2019;19(5):4019–26.
Chen C, Lv Q, Li Y, Jin Y-H. The anti-tumor effect and underlying apoptotic mechanism of ginsenoside Rk1 and Rg5 in human liver cancer cells. Molecules. 2021;26(13):3926.
Liu M-Y, Liu F, Gao Y-L, Yin J-N, Yan W-Q, Liu J-G, Li H-J. Pharmacological activities of ginsenoside Rg5. Exp Ther Med. 2021;22(2):1–9.
Kim H, Choi P, Kim T, Kim Y, Song BG, Park Y-T, Choi S-J, Yoon CH, Lim W-C, Ko H. Ginsenosides Rk1 and Rg5 inhibit transforming growth factor-β1-induced epithelial-mesenchymal transition and suppress migration, invasion, anoikis resistance, and development of stem-like features in lung cancer. J Ginseng Res. 2021;45(1):134–48.
Britt KL, Cuzick J, Phillips K-A. Key steps for effective breast cancer prevention. Nat Rev Cancer. 2020;20(8):417–36.
Wang D, DuBois RN. Role of prostanoids in gastrointestinal cancer. J Clin Investig. 2018;128(7):2732–42.
Liu Y, Fan D. Ginsenoside Rg5 induces G2/M phase arrest, apoptosis and autophagy via regulating ROS-mediated MAPK pathways against human gastric cancer. Biochem Pharmacol. 2019;168:285–304.
Kivelä T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. BMJ Publishing Group Ltd.; 2009. p. 1129–31.
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, Zhang G, Wang X, Dong Z, Chen F. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5(1):8.
Kim Y-J, Zhang D, Yang D-C. Biosynthesis and biotechnological production of ginsenosides. Biotechnol Adv. 2015;33(6):717–35.
Eaton BR, Schwarz R, Vatner R, Yeh B, Claude L, Indelicato DJ, Laack N. Osteosarcoma. Pediatr Blood Cancer. 2021;68: e28352.
Liu M-Y, Liu F, Li Y-J, Yin J-N, Gao Y-L, Wang X-Y, Yang C, Liu J-G, Li H-J. Ginsenoside Rg5 inhibits human osteosarcoma cell proliferation and induces cell apoptosis through PI3K/Akt/mTORC1-related LC3 autophagy pathway. Oxid Med Cell Longev. 2021;2021:1–12.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. 2018;68(6):394–424.
Dong Y, Fu R, Yang J, Ma P, Liang L, Mi Y, Fan D. Folic acid-modified ginsenoside Rg5-loaded bovine serum albumin nanoparticles for targeted cancer therapy in vitro and in vivo. Int J Nanomed. 2019;14:6971–88.
Liu Y, Fan D. Ginsenoside Rg5 induces apoptosis and autophagy via the inhibition of the PI3K/Akt pathway against breast cancer in a mouse model. Food Funct. 2018;9(11):5513–27.
Feng S-L, Luo H-B, Cai L, Zhang J, Wang D, Chen Y-J, Zhan H-X, Jiang Z-H, Xie Y. Ginsenoside Rg5 overcomes chemotherapeutic multidrug resistance mediated by ABCB1 transporter: in vitro and in vivo study. J Ginseng Res. 2020;44(2):247–57.
Li H, Li H. Ginsenoside-Rg5 inhibits growth and metastasis of ovarian carcinoma via suppressing expression of fibroblast growth factor-8b (FGF8b). J King Saud Univ-Sci. 2020;32(1):1162–7.
Chen Y, Chen G, Li J, Huang YY, Li Y, Lin J, Chen LZ, Lu JP, Wang YQ, Wang CX, Pan LK, Xia XF, Yi X, Chen CB, Zheng XW, Guo ZQ, Pan JJ. Association of tumor protein p53 and ataxia-telangiectasia mutated comutation with response to immune checkpoint inhibitors and mortality in patients with non-small cell lung cancer. JAMA Netw Open. 2019;2(9): e1911895.
Song L, Yang F, Wang Z, Yang L, Zhou Y. Ginsenoside Rg5 inhibits cancer cell migration by inhibiting the nuclear factor-κB and erythropoietin-producing hepatocellular receptor A2 signaling pathways. Oncol Lett. 2021;21(6):1–8.
Zhou X, Jiao D, Dou M, Zhang W, Hua H, Chen J, Li Z, Li L, Han X. Association of APC gene promoter methylation and the risk of gastric cancer: a meta-analysis and bioinformatics study. Medicine (Baltimore). 2020;99(16): e19828.
Wang YS, Li H, Li Y, Zhu H, Jin YH. Identification of natural compounds targeting Annexin A2 with an anti-cancer effect. Protein Cell. 2018;9(6):568–79.
Yu SE, Mwesige B, Yi Y-S, Yoo BC. Ginsenosides: the need to move forward from bench to clinical trials. J Ginseng Res. 2019;43(3):361–7.
Kim H, Lee JH, Kim JE, Kim YS, Ryu CH, Lee HJ, Kim HM, Jeon H, Won H-J, Lee J-Y. Micro-/nano-sized delivery systems of ginsenosides for improved systemic bioavailability. J Ginseng Res. 2018;42(3):361–9.
Wu J, Jeong HK, Bulin SE, Kwon SW, Park JH, Bezprozvanny I. Ginsenosides protect striatal neurons in a cellular model of Huntington’s disease. J Neurosci Res. 2009;87(8):1904–12.
Park JY, Choi P, Kim T, Ko H, Kim H-K, Kang KS, Ham J. Protective effects of processed ginseng and its active ginsenosides on cisplatin-induced nephrotoxicity: in vitro and in vivo studies. J Agric Food Chem. 2015;63(25):5964–9.
Yang Y-L, Li J, Liu K, Zhang L, Liu Q, Liu B, Qi L-W. Ginsenoside Rg5 increases cardiomyocyte resistance to ischemic injury through regulation of mitochondrial hexokinase-II and dynamin-related protein 1. Cell Death Dis. 2017;8(2):e2625–e2625.
Wan Y, Wang J, Xu J-F, Tang F, Chen L, Tan Y-Z, Rao C-L, Ao H, Peng C. Panax ginseng and its ginsenosides: potential candidates for the prevention and treatment of chemotherapy-induced side effects. J Ginseng Res. 2021;45(6):617–30.
Choi S-Y, Kim K-J, Song J-H, Lee B-Y. Ginsenoside Rg5 prevents apoptosis by modulating heme-oxygenase-1/nuclear factor E2-related factor 2 signaling and alters the expression of cognitive impairment-associated genes in thermal stress-exposed HT22 cells. J Ginseng Res. 2018;42(2):225–8.
Panossian A, Abdelfatah S, Efferth T. Network pharmacology of Red Ginseng (Part I): effects of ginsenoside Rg5 at physiological and sub-physiological concentrations. Pharmaceuticals. 2021;14(10):999.
Clarkson C, Maharaj VJ, Crouch NR, Grace OM, Pillay P, Matsabisa MG, Bhagwandin N, Smith PJ, Folb PI. In vitro antiplasmodial activity of medicinal plants native to or naturalised in South Africa. J Ethnopharmacol. 2004;92(2–3):177–91.
Chu S, Gu J, Feng L, Liu J, Zhang M, Jia X, Liu M, Yao D. Ginsenoside Rg5 improves cognitive dysfunction and beta-amyloid deposition in STZ-induced memory impaired rats via attenuating neuroinflammatory responses. Int Immunopharmacol. 2014;19(2):317–26.
Cho Y-L, Hur S-M, Kim J-Y, Kim J-H, Lee D-K, Choe J, Won M-H, Ha K-S, Jeoung D, Han S. Specific activation of insulin-like growth factor-1 receptor by ginsenoside Rg5 promotes angiogenesis and vasorelaxation. J Biol Chem. 2015;290(1):467–77.
Kim E-J, Jung I-H, Van Le TK, Jeong J-J, Kim N-J, Kim D-H. Ginsenosides Rg5 and Rh3 protect scopolamine-induced memory deficits in mice. J Ethnopharmacol. 2013;146(1):294–9.
Lee JG, Lee YY, Kim SY, Pyo JS, Yun-Choi HS, Park JH. Platelet antiaggregating activity of ginsenosides isolated from processed ginseng. Die Pharm-An Int J Pharm Sci. 2009;64(9):602–4.
Liu J, Liu Y, Lin H, Zhou B, Yu H, Li L, Wang C, Li X, Li P, Liu J. The effect of ginsenoside Rg5, isolated from black ginseng, on heart failure in zebrafish based on untargeted metabolomics. J Funct Foods. 2021;76: 104325.
Wei Y, Yang H, Zhu C, Deng J, Fan D. Ginsenoside Rg5 relieves type 2 diabetes by improving hepatic insulin resistance in db/db mice. J Funct Foods. 2020;71: 104014.
Gwilt PR, Nahhas RR, Tracewell WG. The effects of diabetes mellitus on pharmacokinetics and pharmacodynamics in humans. Clin Pharmacokinet. 1991;20:477–90.
Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis. 2007;11:525–48.
Rumack BH. Acetaminophen hepatotoxicity: the first 35 years. J Toxicol Clin Toxicol. 2002;40(1):3–20.
Nelson SD. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. In: Seminars in liver disease. Thieme Medical Publishers, Inc.; 1990. p. 267–78.
Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 1995;333(17):1118–27.
Shahid RK, Ahmed S, Le D, Yadav S. Diabetes and cancer: risk, challenges, management and outcomes. Cancers. 2021;13(22):5735.
Kim SI, Park JH, Ryu J-H, Park JD, Lee YH, Park J-H, Kim T-H, Kim JM, Baek N-I. Ginsenoside Rg 5, a genuine dammarane glycoside from Korean red ginseng. Arch Pharmacal Res. 1996;19:551–3.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Conceptualization and design were performed by J.S.-R.; investigation, data curation, and writing were performed by T.E., A.M.M., E.A. M. M., I.B., M.R., K.Y., M.A.M., S.F.A.; validation, review, and editing were performed by D.B., S.H., J.S.-R. All the authors contributed equally, read, and approved the final manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors wish to confirm that there are no known conflicts of interest associated with this publication, and there has been no significant financial support for this work that could have influenced its outcome.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Elsaman, T., Muddathir, A.M., Mohieldin, E.A.M. et al. Ginsenoside Rg5 as an anticancer drug: a comprehensive review on mechanisms, structure–activity relationship, and prospects for clinical advancement. Pharmacol. Rep 76, 287–306 (2024). https://doi.org/10.1007/s43440-024-00586-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43440-024-00586-5