
Abstract
l-threonine aldolases catalyze the conversion of glycine and aldehydes to synthesize β-hydroxy-α-amino acids with unsatisfactory enzyme activity. Here, we expressed the l-threonine aldolase from Pseudomonas putida KT2440 (l-PpTA) in Escherichia coli BL21 (DE3) and improved the activity and thermostability by protein engineering. Five amino acid residues (Ser10, His89, Asp93, Arg177, and Arg321) located in the substrate-binding pocket were selected and for mutation. Eight mutants (D93A, D93G, D93M, D93F, D93S, D93Q, D93Y and D93H) with increased enzyme activity were identified and their kcat/KM values showed about 1–7-fold higher than wild-type. Among all the variants, D93H showed the highest catalytic efficiency with 2925 and 4515 s−1 mM−1 of kcat/KM values toward l-threonine and l-allo-threonine, respectively. In addition, circular dichroism spectrum exhibited that the melting temperature of D93H (54.2 °C) was 5 °C higher than wild-type (49.2 °C). Molecular dynamics simulations illustrated that the D93H variant shortens the distance between the imidazole group of H93 and the hydroxyl group of substrate, which facilitated the proton extraction and promote the enzymatic reaction. This work affords a candidate for the synthesis of β-hydroxy-α-amino acids with improved catalytic efficiency and thermostability and provides structural insights into the l-TA family by protein engineering.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
β-Hydroxy-α-amino acids have two chiral carbon atoms and are significant precursor of effective components of many drugs, which are widely used in pharmaceutical and fine chemical industries [1,2,3]. For example, 4-hydroxy-l-threonine is an important precursor of the PLP-dependent enzyme inhibitor rhizobitoxine as well as involved in the synthesis of vitamin B6 [4], l-threo-4-methylsulfonylphenylserine is a key intermediate in the synthesis of florfenicol and thiamphenicol [5], and l-threo-3,4-dihydroxyphenylserine is a special remedy for the treatment of Parkinsonism [6].
Numerous efforts have been made for the production of β-hydroxy-α-amino acids using chemical methods such as Sharpless epoxidation [7] that can be used to prepare 2,3-epoxy alcohols from primary or secondary allyl alcohols, Sharpless aminohydroxylation [8] that can convert alkenes to corresponding o-amino alcohols under the action of alkylimino osmium compounds, and dynamic kinetic resolution [9] that can use chiral reagents to interact with racemates to generate diastereomers. Although each of these methods are elegant, limitations still remain, especially insufficient stereoselectivity, harsh reaction conditions or the overuse of organic solvents. Therefore, it is advisable to develop an effective enzymatic process, which can complement the shortage of chemical methods.
Threonine aldolases (TAs) (EC 4.1.2.5) can catalyze aldol reaction with glycine and various aldehydes to synthesize β-hydroxy-α-amino acids as well as its reversible reaction [10, 11]. TAs have been widely found in plants [12], bacteria [13] and fungi [4]. Based on the α-carbon stereoselectivity of natural substrate threonine, TAs can be classified into l-TAs and d-TAs [7]. In addition, according to the β-carbon stereoselectivity of natural substrate l-threonine, l-TAs were further divided into three groups: (I) l-TAs only catalyze l-threonine; (II) l-allo-TAs simply act on l-allo-threonine; (III) low-specificity l-TAs can catalyze both l-threonine and l-allo-threonine [1, 14]. These three l-TAs have highly similar primary structure, while display obvious differences in stereospecificity. Currently, only a few d-TAs have been characterized [15, 16].l-TAs can synthesize β-hydroxy-α-amino acids in single step under mild conditions. Zhao et al. [6] expressed l-TA from gut microbiota of black bear and improve the diastereoselectivity of l-threo-3,4-dihydroxyphenylserine by rational design. Wang et al. [1] synthesized l-threo-4-methylsulfonylphenylserine using recombinant l-TA from Actinocorallia herbida. Blesl et al. [17] produced alpha-quaternary alpha-amino acids by expressing l-TA from Aeromonas jandaei and d-TA from Pseudomonas sp. Therefore, l-TAs as a promising biocatalyst have been attracted extensive attention. Recently, substantial attempts have been devoted for engineering TAs to enhance its features and functions. For example, Chen et al. [18] developed a substrate-binding-guided mutagenesis and stepwise visual screening method to improve or invert Cβ-stereoselectivity of TA from Pseudomonas sp. Zheng et al. [10] explored a combinatorial active-site saturation test/iterative saturation mutagenesis (CAST/ISM) strategy to enhance the diastereoselective of l-TA from Bacillus nealsonii. Liu et al. [2] performed an error-prone PCR and combinatorial mutation approach to increase the yield and stereoselectivity of l-TA from Pseudomonas sp. in the synthesis of 4-(methylsulfonyl)phenylserine. All reported methods represent the methodology to modify the properties of l-TA, while fundamentally improve its characteristics requires more efforts.
Previously, we have characterized a low-specificity l-TA from Pseudomonas putida for catalyzing β-hydroxy-α-amino acids [19]. To further improve the catalytic activity and thermostability, several key amino acid residues located to the binding pocket were selected to conduct NNK saturation mutation, kinetic parameters determination, and circular dichroism (CD) spectrum assay. Moreover, the molecular insights into the structural changes caused by mutations were provided by molecular docking and dynamic simulation. This work affords a candidate with improved catalytic efficiency and thermostability to the l-TA family and provides insights for the β-hydroxy-α-amino acids synthesis by engineering threonine aldolases.
Materials and methods
Chemical and materials
The Escherichia coli BL21 (DE3) and pET-28a were stored in our laboratory as host and expression vector, respectively. The cofactor pyridoxal 5′-phosphate (PLP), NADH and yeast alcohol dehydrogenase (ADH) were bought from the Sigma-Aldrich (Shanghai, China). The ClonExpress MultiS One Step Cloning Kit and Plasmid Isolation Kit was purchased from Vazyme (Nanjing, China). The PrimeSTAR and BamHI, XhoI restriction enzymes were bought from Takara (Dalian, China). All other chemicals were of the analytical grade and commercially available.
Construction of recombinant strains
The gene latE (GenBank: AE015451.2) coding l-TA from Pseudomonas putida KT2440 (l-PpTA) was synthesized by Sangon Biotech Co., Ltd. (Shanghai, China) on pET-28a plasmid to construct the recombinant plasmids pET-KT2440, which was subsequently transformed into E. coli BL21 (DE3). The recombinant strain E. coli BL21/pET28a-l-PpTA was obtained after DNA sequencing.
The saturation mutagenesis strains of five amino acid residues (Ser10, His89, Asp93, Arg177 and Arg321) were constructed using homologous recombination technique [19] with pET-KT2440 as template. The SM libraries of l-PpTA were constructed to encode 20 canonical amino acids using the degenerate codons NNK. The short fragments contain the target mutation sites were amplified using the primers S-F and S-R. And the long fragments include the plasmid pET28a were amplified using the primers L-F and L-R. Then, the short fragments and long fragments were connected using the ClonExpress MultiS One Step Cloning Kit. After that, the recombinant products were transformed into the E. coli BL21 (DE3). Eight recombinant strains E. coli BL21/pET-D93A, E. coli BL21/pET-D93G, E. coli BL21/pET-D93M, E. coli BL21/pET-D93F, E. coli BL21/pET-D93S, E. coli BL21/pET-D93Q, E. coli BL21/pET-D93Y, and E. coli BL21/pET-D93H were identified by DNA sequencing. The primers used in this study are listed in Table 1.
Protein expression and purification
The recombinant strains were cultured at 37 °C in LB medium added with 50 μg mL−1 kanamycin. When OD600 value of the recombinant strains reached 0.6–0.8, 0.1 mM IPTG was added to induce protein expression at 25 °C for 12–14 h. The cells were harvested by centrifugation and suspended in buffer (20 mM Tris–HCl and 150 mM NaCl, pH 8.0) and lysed by an ultrasonic oscillator (Sonic Materials Co., Piscataway, USA). The supernatant was collected by centrifugation at 12,000 × g, 4 °C for 30 min.
The gathered supernatant was loaded on a HisTrap HP affinity column equilibrated with buffer (20 mM Tris–HCl and 150 mM NaCl, pH 8.0), and then it was eluted with buffer (20 mM Tris–HCl, 150 mM NaCl, 1 M imidazole, pH 8.0). Subsequently the fractions were applied to a Resource Q column equilibrated with buffer (20 mM Tris–HCl, pH 8.0). Finally, the fractions were loaded on a Superdex 200 (10/300 GL) gel column for chromatography. The homogeneity of purified proteins was judged by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels.
Enzyme activity and kinetic determination
The enzyme activities of wild-type (WT) l-PpTA and its mutants were measured by monitoring the reduction rate of absorbance at 340 nm using an NADH coupled alcohol dehydrogenase (ADH) method [7]. The reaction system (250 μL) HEPES–NaOH (100 mM, pH 8.0) contained 50 mM l-threonine/l-allo-threonine, 0.5 mM NADH, 50 μM PLP, 30 U ADH, and appropriate amount of purified enzyme. Under measurement conditions, one unit of enzyme activity is defined as the amount of enzyme catalyzing the oxidation of 1 μmol NADH per minute.
The kinetic parameters were determined at varied concentrations of l-threonine/l-allo-threonine (0.1–100 mM) in HEPES–NaOH (100 mM, pH 8.0) buffer. The KM and kcat values were calculated by fitting the data to the Michaelis–Menten equation using nonlinear regression with GraphPad Prism. All experiments were performed in triplicate and the data are from the mean of three replicates with standard deviations.
The thermostability analysis by circular dichroism spectrum
The thermostability of l-PpTA and its mutants were analysed by circular dichroism (CD) spectrum using Jasco J720 spectropolarimeter (Jasco, Inc., Easton, USA). The purified protein concentration was diluted to 0.1 mg mL−1 with 10 mM phosphate buffer (pH 6.0). Scans were done every 1 °C between 20 and 80 °C at 209 nm to monitor the change of protein structure with temperature and determine the melting temperature (Tm).
Molecular docking and molecular dynamic simulations
P. putida l-TA (PDB ID: 5VYE) shared the similarity of 98.26% with WT l-PpTA in amino acid sequence. Homology structures of WT l-PpTA and its variants were constructed with Discovery Studio using the crystal structure of P. putida l-TA (PDB ID: 5VYE) as a template. The substrate molecules l-threonine/l-allo-threonine were prepared and docked into WT l-PpTA and its variants using flexible docking. The docking structure was analyzed by Pymol software (https://www.pymol.org). In addition, molecular dynamic (MD) simulations of protein–ligand complexes were carried out as follows: firstly, the CHARMm force field was applied to the protein, and the system was constructed in a cube box composed of TIP3P water molecules, which extended from the surface of the protein for 10 Å to form a buffer zone between them; Secondly, sodium ions were added to the system as counterions to build a neutral system; Thirdly, the step of energy minimization was carried out using the conjugate gradient algorithms; Finally, the protein–ligand complex simulations with 50 ns were carried out, gradually heat the system from 0 to 300 K.
Results
Possible mutation determination by sequence and structural analysis of l-PpTA
The homologous structure of l-PpTA was build using crystal structure of PLP-dependent P. putida l-TA (PDB ID: 5VYE) as a template. Molecular-docking models of l-PpTA with the substrates l-threonine/l-allo-threonine were obtained using the Discovery Studio. The structure model of l-PpTA is a tetrameric with the same four subunits, which is consistent with the known structural characteristics of l-TAs [13, 20]. In the modelled l-PpTA structure, a histidine H89, π-stacks the re face of the cofactor PLP was in the range of hydrogen bond with the hydroxyl group of substrates l-threonine/l-allo-threonine, which could initiate the C–C bond cleavage as the catalytic base [21]. In addition, three amino acid residues S10, R177 and R321 formed salt bridges with the carboxylate group of l-threonine/l-allo-threonine, which probably stabilized the produced transition state during the reaction [19]. Moreover, D93 is located at the substrate entry tunnel and could form hydrogen bonds with the amino group of substrates l-threonine/l-allo-threonine [21] (Fig. 1).

View of the active sites of l-PpTA. The enzyme backbone is represented as surface in gray, the active sites are showed in cyan sticks, and the cofactor PLP and l-threonine are exhibited in pink sticks
To gain detailed insight into the characteristics of l-TAs, sequence alignment, structure classification and conservation analysis were performed. The results indicated that l-TAs exhibited high sequence and structural homology (Fig. 2a), and the structure alignment superposition displayed that S10, H89, R177 and R321 had similar functions (Fig. 2b). except that D93 was a variable residue that could be substituted by tyrosine and phenylalanine in other l-TAs. Moreover, sequence conservation analysis showed that S10, H89, R177 and R321 were highly conserved and D93 was moderately conservative (Fig. 2c), which might provide high evolutionary selective pressure and have significant impact on protein function [18]. Therefore, the five amino acids were selected for possible mutations to improve catalytic activity and thermostability.

A The sequence and secondary structure alignment of l-TAs with Pseudomonas putida KT2440 (PDB ID: 5VYE), Thermotoga maritima (PDB ID: 1LW4), Pseudomonas. putida PSALD (PDB ID: 1V72), Aeromonas jandaei (PDB ID: 3WGC) and Escherichia coli (PDB ID: 4LNL). This image was obtained using the program Espript (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi/). B Superposition of active sites in the structurally characterized l-TAs from different sources. C Conservation analysis of l-PpTA by ConSurf (https://consurf.tau.ac.il/), the structure of the monomer presented as cartoon, and the amino acid residues are colored by their conservation grades
Identification of core variants from the saturated mutation library of five potential sites
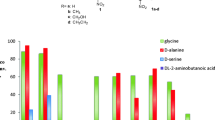
To obtain mutants with improved enzyme activity, a saturation mutation library of the five amino acid residues was constructed. The beneficial mutants were selected by measuring the enzyme activity of cleavage l-threonine to glycine and aldehyde. The relative activity of the mutant was calculated by taking the activity of the WT l-PpTA to cleave l-threonine as 100%. In the saturation mutation at S10, H89, R177, and R321 sites, most mutants completely losing activity, except that two variants of S10 and R177 retained about 5–15% activity (Fig. 3). On the contrary, most mutants of D93 showed similar enzyme activities to WT, and eight variants resulted in about 3–8-fold higher enzyme activities than that of the WT (Fig. 3c). After DNA sequencing, the mutations D93A, D93G, D93M, D93F, D93S, D93Q, D93Y and D93H were obtained. All these mutants were expressed as soluble forms in E. coli. The recombinant proteins of WT and its mutants were purified through HisTrap HP affinity, Resource Q, and Superdex 200 chromatography. The protein homogeneity was judged by SDS-PAGE, which revealed that all the enzymes were produced more than 35 mg per liter of culture.

Screening results of five active sites (S10, H89, D93, R177 and R321) saturation mutagenesis. The beneficial mutants are represented as red dots
D93H presents the highest catalytic efficiency among all the mutants
The enzyme activities of splitting l-threonine and l-allo-threonine to glycine and aldehyde were further measured. The variants showed about 2–10-fold and 1–5-fold improvement of enzyme activities toward l-threonine/l-allo-threonine, respectively (Fig. 4a). Among all mutations, D93H resulted in the highest enzyme activity with 52,214 U mg–1 and 68,898 U mg–1 toward l-threonine/l-allo-threonine, while the WT provided the specific activity with 5577 U mg–1 and 14,311 U mg–1, representing 9.4-fold and 4.8-fold higher than the WT, respectively.

A Relative enzymatic activity of WT and D93 mutants. B Ratios of kcat and kcat/KM of D93 mutants to those of WT toward l-threonine. C Ratios of kcat and kcat/KM of D93 mutants to those of WT toward l-allo-threonine. All the experiments were measured in triplicates
The kinetic parameters of WT and its mutants were also evaluated using l-threonine/l-allo-threonine as substrates at various concentrations ranging from 0.1 to 100 mM. The KM and kcat/KM values of WT for l-threonine were 24 mM and 802 s−1·mM−1, respectively. The eight variants all presented about 1–3-fold increase in the kcat values, leading to about 1–4-fold increase in the kcat/KM values toward l-threonine (Fig. 4b). In addition, The KM and kcat/KM values of WT for l-allo-threonine were 19 mM and 685 s−1·mM−1, respectively. Compared with WT, the eight mutants gave 1–6 times increase in the kcat values, resulted in 1–7-fold increase in the kcat/KM values (Fig. 4c). Among all the mutants, D93H showed the highest catalytic efficiency for l-threonine/l-allo-threonine with 2925 s−1·mM−1 and 4515 s−1·mM−1 in the kcat/KM values, which were 3.6-fold and 6.6-fold higher than WT, respectively. These results showed that the obtained variants played an important role in the substrate affinity and catalytic efficiency.
D93H obviously improves the thermostability
The apparent thermal denaturation temperatures of WT and its variants were measured by CD assay. In the thermal denaturation experiments, the recombinant protein of WT had a melting temperature (Tm) of 49.2 °C. Among all the mutations, the variant D93H had a highest Tm of 54.2 °C, which was 5 °C higher than that of the WT. However, the variant D93G had a lowest Tm of 48.6 °C, which was 0.6 °C lower than that of the WT. In addition, D93A, D93S and D93F showed slightly improvement of denaturation temperatures, affording the Tm of 50.7 °C, 50.8 °C and 51.4 °C, respectively. Furthermore, D93M, D93Q and D93Y exhibited a certain degree of increased denaturation temperatures, achieving the Tm of 52.5 °C, 52.3 °C and 53.6 °C, respectively (Fig. 5). These results indicated that the amino acid residue D93 affected the thermostability of the protein.

The thermal denaturation curve of WT and its variants. The CD spectra were recorded by measuring the ellipticity as a function of wavelength and the thermal denaturation was determined by measuring the ellipticity at 209 nm as a function of temperature at increasements of 1 °C between 20 and 80 °C
Molecular insight into the improved catalytic efficiency and thermostability by D93H mutation
To understand the molecular mechanism for improved catalytic efficiency and increased enzyme thermostability by D93H mutation, the structures of the WT l-PpTA and the D93H mutant were modeled to investigate the structural changes on the complexes of WT and D93H with l-threonine/l-allo-threonine. In these docked structures, H89 π-stacks the re face of the PLP ring, leading to form a π–π interaction between them. The distances between the imidazole group of H89 and the hydroxyl groups of l-threonine/l-allo-threonine are 3.1 Å and 3.1 Å, respectively, indicating that H89 acted as the catalytic base to extract the proton of the substrate hydroxyl group. Moreover, the amino acid residues of R177, R321 and S10 that could form salt bridges with the carboxylate group of the substrates l-threonine/l-allo-threonine to stabilize the transient state produced during the enzymatic reaction (Fig. 6). The D93H mutant shortens the distance between the imidazole group of H93 and the hydroxyl group of l-threonine from 3.2 to 2.9 Å (Fig. 6b). For l-allo-threonine, the distance between the imidazole group of H93 and the amino group of l-allo-threonine changed from 3.1 to 3.0 Å (Fig. 6c, d). Meanwhile, the imidazole group of H93 could interact with the imidazole group of H133 to form a π–π conjugation, which enhanced their attractive interactions and promoted the conformation stability (Fig. 6d). Therefore, mutant D93H exhibited the highest catalytic efficiency and obviously improved the thermostability.

The flexible docking results of l-threonine in WT (A) and D93H mutant (B). The flexible docking results of l-allo-threonine in WT (C) and D93H mutant (D). The 3D-structure of protein is represented as white cartoon; cofactor PLP, l-threonine, and l-allo-threonine are displayed in cyan sticks; active sites are indicated in grey and green sticks, and the interactions are expressed in red dashes
Discussion
β-Hydroxy-α-amino acids, a class of key precursors for the synthesis of fine chemicals and drugs, can be synthesized by TAs to catalyze the aldol condensation reaction with glycine and various aliphatic or aromatic aldehydes as substrates [22, 23]. In the synthesis of β-Hydroxy-α-amino acid, the production of highly efficient and stable l-TA is an essential work of protein engineering, which is promoted by the significance of catalytic efficiency and stability to industrial production and application [24, 25]. Therefore, it is advisable that obtain l-TA with improved characteristics by protein engineering to meet the needs of asymmetric catalytic β-hydroxy-α-amino acids in relevant industries. In this study, the key residues inside or adjacent to the substrate-binding pocket were selected for saturation mutation. The effect of variants that improved specific activity on the catalytic efficiency and thermostability was investigated by kinetic parameters assay, CD spectrum and MD simulation.
To obtain the variants with improved catalytic efficiency, based on the sequences alignment and structural analysis, five amino acid sites of S10, H89, D93, R177 and R321 were selected and mutated. As suggested previously, determination of traditional enzyme activity and functional verification were performed according to cleavage natural substrate l-threonine [26]. The saturation mutation of S10, H89, R177 and R312 lost their most of enzyme activity. Based on structural classification and conservative analysis, these amino acid residues are highly conserved and play an important role in catalytic function. Eight mutations of D93 presented higher activity, indicating that these mutants possible changed the interaction between enzyme and ligand.
Enzymatic reaction kinetics is an effective technology to reflect the enzymatic reaction rate and its influencing factors [27]. These results of kinetic parameters revealed that all the mutants displayed increased catalytic efficiency toward l-threonine and l-allo-threonine. As reported before, the side chain of Y87 in Thermotoga maritima l-TA (corresponding to D93 in P. putida l-TA) might affect the degree of stereoselectivity of l-TAs for l-threonine and l-allo-threonine [13]. And this position was a variable residue in the active sites of l-TAs and appeared that the side chain bulk at this site seems to be related to the stereospecificity [20]. These differences partly explain the reason why D93 mutants have higher selectivity toward l-threonine and l-allo-threonine than that of WT. In addition, this site also arouses the change of thermal denaturation temperature that the Tm value of D93H mutation was 5 °C higher than that of WT, suggesting that D93 site was closely related to the temperature stability of the enzyme.
The molecular mechanism of D93H mutant was further explored by studying the structural characteristics of the enzyme complexes with l-threonine and l-allo-threonine. Structural analysis in the substrate-binding pocket exhibited that these amino acid sites of S10, R177 and R321 were responsible to anchor the carboxylate group of substrate and the residue H89 was conducive to extract proton. Compared with WT, D93H mutation was more adjacent to substrate-binding pocket and could form a π–π conjugation with H133, indicating that the conformational differences have an important effect on substrate binding and protein thermostability.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
Wang LC, Xu L, Xu XQ, Su BM, Lin J. An l-threonine aldolase for asymmetric synthesis of beta-hydroxy-alpha-amino acids. Chem Eng Sci. 2020;226:115812–20.
Liu ZC, Chen X, Chen QJ, Feng JH, Wang M, Wu QQ. Engineering of l-threonine aldolase for the preparation of 4-(methylsulfonyl) phenylserine, an important intermediate for the synthesis of florfenicol and thiamphenicol. Enzyme Microb Tech. 2020;137:109551–7.
Rocha JF, Pina AF, Sousa SF. PLP-dependent enzymes as important biocatalysts for the pharmaceutical, chemical and food industries: a structural and mechanistic perspective. Catal Sci Technol. 2019;9(18):4864–76.
Liu JQ, Dairi T, Itoh N, Kataoka M, Shimizu S, Yamada H. Diversity of microbial threonine aldolases and their application. J Mol Catal B Enzym. 2000;10(1–3):107–15.
Zhao GH, Li H, Liu W, Zhang WG, Zhang F, Liu Q. Preparation of optically active beta-hydroxy-alpha-amino acid by immobilized Escherichia coli cells with serine hydroxymethyl transferase activity. Amino Acids. 2011;40(1):215–20.
Zhao WY, Yang BL, Zha RF, Zhang Z, Tang SJ, Pan YP. A recombinant l-threonine aldolase with high diastereoselectivity in the synthesis of l-threo-dihydroxyphenylserine. Biochem Eng J. 2021;166:107852–9.
Steinreiber J, Fesko K, Reisinger C, Schurmann M, van Assema F, Wolberg M. Threonine aldolases—an emerging tool for organic synthesis. Tetrahedron. 2007;63(4):918–26.
Bodkin JA, McLeod MD. The sharpless asymmetric aminohydroxylation. J Chem Soc Perkin Trans. 2002;24:2733–46.
Makino K, Goto T, Hiroki Y, Hamada Y. Stereoselective synthesis of anti-beta-hydroxy-alpha-amino acids through dynamic kinetic resolution. Angew Chem Int Ed Engl. 2004;43(7):882–4.
Zheng WL, Yu HR, Fang S, Chen KT, Wang Z, Cheng XL. Directed evolution of l-threonine aldolase for the diastereoselective synthesis of beta-hydroxy-alpha-amino acids. Acs Catal. 2021;11(6):3198–205.
Wang LC, Xu L, Su BM, Lin W, Xu XQ, Lin J. Improving the C-beta stereoselectivity of l-threonine aldolase for the synthesis of l-threo-4-methylsulfonylphenylserine by modulating the substrate-binding pocket to control the orientation of the substrate entrance. Chem Eur J. 2021;27(37):9654–60.
Jander G, Norris SR, Joshi V, Fraga M, Rugg A, Yu SX. Application of a high-throughput HPLC-MS/MS assay to arabidopsis mutant screening: evidence that threonine aldolase plays a role in seed nutritional quality. Plant J. 2004;39(3):465–75.
Salvo MLD, Remesh SG, Vivoli M, Ghatge MS, Paiardini A, D’Aguanno S. On the catalytic mechanism and stereospecificity of Escherichia coli l-threonine aldolase. FEBS J. 2014;281(1):129–45.
Fesko K. Threonine aldolases: perspectives in engineering and screening the enzymes with enhanced substrate and stereo specificities. Appl Microbiol Biotechnol. 2016;100(6):2579–90.
Chen QJ, Chen X, Cui YF, Ren J, Lu W, Feng JH. A new d-threonine aldolase as a promising biocatalyst for highly stereoselective preparation of chiral aromatic beta-hydroxy-alpha-amino acids. Catal Sci Technol. 2017;7(4):5964–73.
Park SH, Seo H, Seok J, Kim H, Kwon KK, Yeom SJ. C beta-selective aldol addition of d-threonine aldolase by spatial constraint of aldehyde binding. Acs Catal. 2021;11(12):6892–9.
Blesl J, Trobe M, Anderl F, Breinbauer R, Strohmeier GA, Fesko K. Application of threonine aldolases for the asymmetric synthesis of alpha-quaternary alpha-amino acids. ChemCatChem. 2018;10(16):3453–8.
Chen QJ, Chen X, Feng JH, Wu QQ, Zhu DM, Ma YH. Improving and Inverting C-beta-stereoselectivity of threonine aldolase via substrate-binding-guided mutagenesis and a stepwise visual screening. Acs Catal. 2019;9(5):4462–9.
Li LH, Zhang RZ, Xu Y, Zhang WC. Comprehensive screening strategy coupled with structure-guided engineering of l-threonine aldolase from Pseudomonas putida for enhanced catalytic efficiency towards l-threo-4-methylsulfonylphenylserine. Front Bioeng Biotechnol. 2023;11:1117890–900.
Kielkopf CL, Burley SK. X-ray structures of threonine aldolase complexes: structural basis of substrate recognition. Biochemistry. 2002;41(39):11711–20.
Giger L, Toscano MD, Bouzon M, Marliere P, Hilvert D. A novel genetic selection system for PLP-dependent threonine aldolases. Tetrahedron. 2012;68(37):7549–57.
Fesko K, Reisinger C, Steinreiber J, Weber H, Schurmann M, Griengl H. Four types of threonine aldolases: Similarities and differences in kinetics/thermodynamics. J Mol Catal B Enzym. 2008;52–53:19–26.
Fesko K, Strohmeier GA, Breinbauer R. Expanding the threonine aldolase toolbox for the asymmetric synthesis of tertiary alpha-amino acids. Appl Microbiol Biotechnol. 2015;99(22):9651–61.
Yi D, Bayer T, Badenhorst CPS, Wu S, Doerr M, Hohne M. Recent trends in biocatalysis. Chem Soc Rev. 2021;50(14):8003–49.
Wiltschi B, Cernava T, Dennig A, Galindo Casas M, Geier M, Gruber S. Enzymes revolutionize the bioproduction of value-added compounds: from enzyme discovery to special applications. Biotechnol Adv. 2020;40:107520–34.
Fesko K. Comparison of l-threonine aldolase variants in the aldol and retro-aldol reactions. Front Bioeng Biotechnol. 2019;7:119–31.
Callender R, Dyer RB. The dynamical nature of enzymatic catalysis. Acc Chem Res. 2015;48(2):407–13.
Funding
This project was supported by the National Key research and Development Program of China (2018YFA0900302), the National Science Foundation of China (32271487, 31970045), the National First-class Discipline Program of Light Industry Technology and Engineering (LITE2018-12), the Program of Introducing Talents of Discipline to Universities (111-2-06), and Top-notch Academic Programs Project of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
All authors have their own contributions to this study. LL conducted the research and wrote the first draft. RZ guided the project and revised the manuscript. WZ and YX provided valuable advices and helped with data analysis.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, L., Zhang, R., Zhang, W. et al. Improvement of the activity and thermostability of l-threonine aldolase from Pseudomonas putida via tailoring of the active sites lining the binding pocket. Syst Microbiol and Biomanuf 3, 440–448 (2023). https://doi.org/10.1007/s43393-023-00171-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43393-023-00171-6