
Abstract
Chinese hamster ovary (CHO) cells are widely used in biopharmaceuticals because of their high-density suspension culture, high safety, and high similarity between expressed exogenous proteins and natural proteins. However, the level of exogenous protein expression decreases with increasing culture time; this phenomenon occurs due to the recombination of foreign genes into chromosomes through random integration. The present study integrated the foreign genes into a specific chromosomal site for stable expression based on CRISPR–Cas9 technology. The results showed that the exogenous proteins enhanced green fluorescent protein (EGFP) and human serum albumin (HSA) were successfully integrated into the vicinity of base 1969647 on chromosome NW_003613638.1 of CHO-K1 cells. The obtained positive monoclonal cell lines expressed all the corresponding exogenous proteins after 60 consecutive passages, and no significant differences in expression levels were observed. This study might provide a feasible method to construct a CHO cell line with long-term stable expression of exogenous proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Chinese hamster ovary (CHO) cell line is the most widely used cell line in biopharmaceuticals and has numerous advantages over other cell lines [1]. For example, CHO cells can be cultured on a large scale in a serum-free medium with limited chemical composition. In addition to that, these cells are highly safe and do not act as a host for the replication of human pathogenic viruses. Their proteins expressed after post-translational modification are similar to human proteins [2]. However, a serious concern regarding the use of these cells is unstable protein expression during long-term production [3]. According to Food and Drug Administration (FDA), stable cell lines should maintain protein expression levels of ≥ 70% after 70 generations of cell division. The expressed protein should not have any clinically significant differences from reference products in terms of structure, function, purity, physical and chemical properties, and biological activity [4, 5]. In actual industrial production, selected monoclonal cell lines need 60 generations of amplified culture from the main cell bank (MCB) to the working cell bank (WCB) [6]. Therefore, the stability of 60 generations or more of cell lines is crucial to the commercialization of products. However, recombinant cell lines constructed by traditional methods are difficult to meet this requirement, and according to Mark Leonard et al., 8 ~ 63% of recombinant CHO cell lines show unstable expression of target proteins within 90 days of continuous culture [7].
There are three main reasons for the expression of unstable target proteins in CHO cells. First, the genome of a CHO cell is unstable and prone to rearrangement [8]. Second, the traditional method of constructing engineered CHO cells involves the random integration of foreign genes into the cell genome, which results in the gene location effect [9]. Third, the type of host cell system, the number of transgene copy numbers, gene silencing, and transcriptional inactivation may also affect the expression stability of CHO cells [4]. Site-specific integration (SSP) is an effective method to achieve the stable expression of exogenous genes by inserting the target gene into a specific position on the CHO cell genome [10]. CRISPR–Cas9 technology has developed rapidly due to its convenience, precision, and efficiency [11], and is widely used to construct recombinant CHO cell lines [12].
During the construction of a recombinant CHO cell line by site-specific integration, the initial site selection is an important aspect of the entire process. The sequencing results of the first, second, and third generations of CHO cells have been published [12,13,14]. Some potential stable sites were detected, such as the C12orf35 site in the telomere region of chromosome 8 of CHO cells [15], the HPRT site in CHO cells [16], the Hipp11 gene in CHO-S cells [17], and the Kcmf1 gene in CHO-K1 cells [18]. Some stationary sites were also found in other cell lines, including the HPRT and GRIK1 sites of the human fibrosarcoma cell line HT1080 [19, 20]. However, stable and highly expressing integration sites have not yet been successfully used to produce engineered CHO cell lines. Therefore, it is imperative to detect and verify stable expression sites in the CHO cell genome and to establish a method of engineering cell construction based on site-specific integration. In the early stage, our research group used CHO-K1 as the starting cell line, combined with the lentivirus tracer-reporter gene (ZsGreen) technology and chromosome stepping technology, and we found six loci with the sustainable expression of reporter genes [20]. In the present study, the stability of one of the stable expression sites was confirmed; this site was located in an intron region of the Cdk6 gene on the CHO-K cell chromosome NW_003613638.1. Using CRISPR–Cas9-mediated homology-directed repair, an integration strategy was designed to integrate the enhanced green fluorescent protein (EGFP) reporter gene into this site. Subsequently, the human serum albumin (HSA) gene was cloned by the same method. The study findings revealed that exogenous genes can be stably integrated and expressed at this loci.
Materials and methods
Cell culture
The adherent cells were grown in Ham’s F-12 K (Kaighn’s) medium containing 10% FBS (Thermo Fisher Scientific, Waltham, MA) in a 37 °C, 5% CO2 incubator. Suspension cells were grown in a shaker in a CD CHO medium (Irvine, USA) supplemented with 8 mM l-glutamax at 110 rpm, 37 °C, 5% CO2.
Construction of targeted plasmids and validation of site editability
The specific integration site of the Zsgreen1 gene in 3 g10 [20]cells was located in an intron region of Cdk6 gene at base 1,969,647 of chromosome NW_003613638.1. CCTOP CRISPR–Cas9 online prediction system was used to analyze the upstream and downstream gene sequences of the Zsgreen1 gene integration site in 3 g10 cells. A sequence 5' -AACATTCTGAGGACTGCTCAAGG-3 ‘with a predicted editing efficiency of 0.69 (Score < 0.56 is low efficiency, 0.56 ≤ Score ≤ 0.74 is medium efficiency, and > 0.74 is high efficiency) was selected as the target sequence [21]. The primers sgRNA-F and sgRNA-R were annealed in the following systems: sgRNA-F 4 µL, sgRNA-R 4 µL, NEBuffer2 2 µL, ddH2O 10 µL, in a water bath at 95 ℃ for 5 min. pSK-u6-gRNA plasmid (presented by Lu Daru's research group, Fudan University) was digested by BbsI-HF (NEB Company, USA) at 37 ℃ water bath for 4 h, and the fragment was recovered using SanPrep column PCR product purification kit. Finally, the annealed fragment of sgRNA was ligated to the pSK-u6-gRNA vector by T4 ligase (Promega, USA) at 4 ℃ overnight.
The constructed sgRNA plasmid and CD513B-Cas9 plasmid (donated by the research group of Lu Daru, Fudan University) were transfected into CHO-K1 cells with Lipofectamine™ 3000 reagent (Invitrogen Company, USA) at a ratio of 1.8:1. The cells were collected 72 h later. The genomes were extracted and amplified by PCR with 3 g10-F and 3 g10-R. The PCR products were digested with T7 endonuclease I (NEB Company, USA). The PCR products were sent to Suzhou GENEWIZ Biotechnology Co., Ltd. for further sequencing verification. Primer sequences are shown in Table 1.
Construction of donor plasmid and recombinant monoclonal screening
After confirming that the sgRNA plasmid could effectively edit the target site, the upstream and downstream 600-bp DNA sequences of the PAM site were selected as the 5' homologous arm and the 3' homologous arm, respectively. The 5' and 3' homologous arms were successively constructed into EGFP donor plasmid and HSA donor plasmid by enzyme digestion and ligating method. Primer sequences are shown in Table 2. EGFP donor plasmid, green fluorescent gene expression box (hPGK-EGFP-SV40 polyA), and puromycin gene expression box (EF-1α -PuroR-SV40 polyA) were designed between 5' homologous arm and 3' homologous arm. A red fluorescent gene expression box (CMV-mCherry-SV40 polyA) was designed upstream of the 5 'homologous arm. The HSA donor plasmid is based on EGFP donor plasmid; human serum albumin expression box (CMV-HSA-SV40 polyA) is added between 5' homologous arm and green fluorescent gene expression box. The plasmid map is shown in Figure S1.
Genomic DNA was extracted using a mini-extraction kit of genomic DNA (Beyotime, Shanghai, China) from a monoclonal cell line for junction PCR amplification verification. All PCR amplifications were performed using Phantom Max Super-Fidelity DNA Polymerase (Vazyme, Nanjing, China). The specific amplification procedure for 5’/3’ junction PCR was as follows: 95 ℃ for 3 min; 95 ℃ for 15 s, 68 ℃ for 15 s, 72 ℃ for 75 s, 30 × ; 72 ℃ for 5 min. The PCR products were verified by sequencing. Out–out junction PCR was used to confirm further whether the cell line was homozygous or heterozygous according to the following condition: 95 ℃ for 3 min; 95 ℃ for 15 s, 67 ℃ for 15 s, 72 ℃ for 7 min, 30 × ; 72 ℃ for 5 min. All primers involved are shown in Supplementary Table 3.
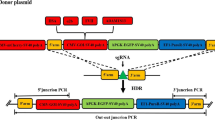
The constructed sgRNA plasmid, CD513B-Cas9 plasmid, and donor plasmid (EGFP plasmid/HSA donor plasmid) were transfected into CHO-K1 cells by LipofectamineTM 3000 at the molar ratio of 1.8:1:1.8. After 48 h culture, cells were screened with 10 µg/mL puromycin pressure (Sangon Biotech (Shanghai) Co., Ltd.). After all the cells in the control group died, the cells were collected for flow separation. Flow cytometry was used to separate monoclonal cells expressing only green fluorescence into 96-well plates with 1 cell in each well. After 6–7 days of culture, cells in the well with only one cell cluster were selected for expansion culture. The genome was extracted and confirmed by PCR. 5ʹ junction PCR and 3ʹ junction PCR were used to identify the site-specific integration of the target gene. Out–out PCR was used to identify whether the monoclonal cells were homozygous or heterozygous. Primer sequences are shown in Table 3. The fixed-point integration is shown in Fig. 1.

Site-specific integration of foreign genes into CHO-K1 genome
Detection of target protein
The expression of EGFP gene in cell lines with site-specific integration can be detected by flow cytometry. And, with CHO-K1 blank cells as negative control, the average green fluorescence intensity of 20,000 cells was detected by BD FACS AriaIII flow sorter, and the results were analyzed by FlowJo V10 software. Dot blot and Western blot analysis can detect the expression of the HSA gene in cell lines with site-specific integration. Dot blot was performed as follows: 5 µL of cell culture supernatant was spotted on NC membrane (Pall, Shanghai, China) and air dried and blocked with 5% skim milk for 2 h at room temperature. The membranes were washed three times with TBST and incubated with HSA antibody (Abcam, Shanghai, China) overnight at 4 ℃. After this, the membranes were again washed three times with TBST and incubated with goat anti-mouse antibody (Abcam, Shanghai, China) for 1.5 h at room temperature and exposed. Western blot was performed as follows: 20 µL of denatured cell culture supernatant was separated by 10% SDS-PAGE. The protein on the gel was transferred to an NC membrane at 110 V for 70 min. The subsequent operations were carried out similarly to the dot blot operations.
Determination of cell line stability verification and HSA protein production capacity
The stability of recombinant cell lines includes subculture stability and cryopreservation stability, which refers to the stability of product expression in continuous cell subculture, and cryopreservation stability refers to the stability of product expression after the resuscitation of cells subjected to liquid nitrogen cryopreservation periodically [22]. The EGFP gene was used as the reporter gene, and the CHO-K1 cell line with the EGFP gene was used for site stability verification. Three CHO-K1-EGFP cell lines were randomly selected for continuous suspension culture for 60 generations and frozen every 10 generations. After passage, cells of all generations were resuscitated, and changes in the average fluorescence intensity of suspended cell lines were detected by flow cytometry to evaluate the stability of suspended cell lines.
Based on the Western blot results, three cell lines expressing intact HSA protein were selected for suspension acclimatization. The cell lines, successfully acclimated in suspension, were continuously passed for 60 generations and frozen every 20 generations. At the end of the passage, cells of all generations were resuscitated, and batch experiments were conducted. Each cell line was inoculated in 50 mL CD CHO medium containing L-glutamine 8 mmol/L, and the initial inoculation density was 1 × 106 cells/mL. Three groups of parallel experiments were performed. Cell density, viability, glucose content, and lactate content were determined by daily sampling. The experiment was terminated when the cell viability was less than 80%. The urinary microalbumin kit (SMMIND, Shanghai, China) was used to detect HSA protein in the fermentation broth.
Results
Determination of the 3 g10 cell multiplication time
To detect whether the insertion of an exogenous gene at the intron region of the Cdk6 gene in CHO-K cell chromosome NW_003613638.1 would affect cell growth, the total number of cells was detected by the MTT method. The cell growth curve was plotted based on the absorbance reading at 560 nm (Fig. 2). CHO-K1 cells normally divide every 20 to 24 h. The results of the MTT assay showed that the proliferation time of cell line 3 g10 was approximately the same as that of CHO-K1 cells. Therefore, 24 h was used as cell doubling time in this study.

Growth rate of 3 g10 cells
Stability of the green fluorescence reporter protein ZsGreen1 expressed in 3 g10 cells
By detecting the average green fluorescence intensity of the CHO-K1 (3g10) cell line with the randomly integrated tracer gene ZsGreen1, it was preliminarily determined that the 1969647 bases of chromosome NW_003613638.1 could stably express foreign proteins. Figure 3A shows the fluorescence microscopy images of 3g10 cells in the continuous passage process. All the cells exhibited green fluorescence. Figure 3B shows the average fluorescence brightness of cells in the fluorescence field analyzed by ImageJ software. Although the average fluorescence intensity of 3g10 cells in different generations was slightly different, the expression levels of ZsGreen1 were the same. As shown in Fig. 3C, the flow cytometry detection results revealed that the fluorescence intensity of 3g10 cells in different generations remained at approximately 55,000. The stability of passage and cryopreservation of 3g10 adherent cells were confirmed, and the NW_003613638.1 site preliminarily proved to be stable.

Stability of ZsGreen1 reporter gene expression in 3 g10 cells. The expression of the ZsGreen1 protein in adherent 3 g10 cells is shown in Fig. 3A–D. A Inverted fluorescence microscopy results of adherent 3 g10 cells at 10d, 20d, 30d, 40d, and 50d. B ImageJ software analysis results of the average fluorescence intensity of cells in fluorescence field photographs in (A). C Quantitative analysis of fluorescent protein expression in cells by flow cytometry. D Mean fluorescence intensity of each generation of cells in (C). Flow cytometry analysis was performed using CHO-K1 as a negative control
Confirmation of sgRNA editing efficiency
To detect the actual editing of the designed sgRNA in CHO-K1 cells, the targeted plasmid and Cas9 plasmid were transfected into CHO-K1 cells. If the gene editing event occurred in the cells, DNA homologous directed repair would occur at the opening site mismatch. The upstream and downstream sequences of the Cas9 cleavage site in the transfected cell pool were amplified by PCR and digested by T7 endonuclease I. Because T7 endonuclease I can recognize and cut mismatched DNA double strands, if a gene editing event occurs, two bands of 750 bp and 260 bp will appear in agarose gel electrophoresis after enzyme digestion, as shown in Figure S2A. The PCR products were sequenced. The results (Fig. S2B) showed a significant peak nesting after the fourth base upstream of the PAM sequence, and the position recognized by the Cas9 complex was between three and four bases upstream of the PAM sequence. These results indicated that the designed sgRNA could guide the Cas9 protein to the fixed location in the CHO-K1 genome where gene editing events occurred.
Screening and identification of monoclonal cell lines with EGFP gene integration
According to the designed site-specific integration strategy (Fig. 1), cell lines with successful site-specific integration events expressed the EGFP gene and showed only green fluorescence. The cell lines with random integration events expressed both the EGFP gene and the mCherry gene. Cell lines without integration events showed no fluorescence. The transfected cells were collected and sorted by flow cytometry. The results are shown in Fig. 4A. Taking K1 cells as the negative control, the proportion of cells with only green fluorescence was 68.8%; this finding indicated that 68.8% of the cells had site-directed integration events. Green-only monoclonal cells were then sorted into 96-well plates at a density of 1 cell per well.

CHO-K1 cell sorting and identification results of site-specific integration of the EGFP gene. A Flow cytometric sorting of CHO-K1 cells with the EGFP gene introduced by site-directed integration. B Results of 5ʹ junction PCR, M: DL 2000 DNA Marker, 1: K1-EGFP-29, 2: K1-EGFP-39, 3: K1-EGFP-40, 4: K1-EGFP-41, 5: K1-EGFP-44. C Results of 3ʹ junction PCR, M: DL 2000 DNA marker, 1: K1-EGFP-29, 2: K1-EGFP-39, 3: K1-EGFP-40, 4: K1-EGFP-41, 5: K1-EGFP-44. D Results of out–out PCR, M: DL 5000 DNA marker, 1: K1-EGFP-29, 2: K1-EGFP-39, 3: K1-EGFP-40, 4: K1-EGFP-41, 5: K1-EGFP-44. E Fluorescence intensity was detected by flow cytometry
The genome of the monoclonal cell line was extracted for PCR analysis, as shown in Fig. 4B and C. The PCR products of 5ʹ-homologous arm and 3ʹ-homologous arm were around 1800 bp in size, which was consistent with our expectations. Five positive monoclonal strains (1: K1-EGfP-29, 2: K1-EGFP-39, 3: K1-EGFP-40, 4: K1-EGFP-41, 5: K1-EGfP-44) were screened by 5ʹ junction PCR and 3ʹ junction PCR. The out–out PCR results (Fig. 4D) indicated that the obtained positive monoclonal cells were heterozygous. Sequencing results showed that the 4800-bp band contained the site-integrated complete EGFP gene, and the 1500-bp band contained the unintegrated chromosomal DNA; this finding was consistent with our expectations. All positive monoclonal cell lines showed green fluorescence in the flow cytometry analysis. These results indicate that CRISPR–Cas9 technology can be used to integrate foreign genes into this site and achieve stable expression.
EGFP expression of site-specific integrant cell lines in 60 consecutive passages
The cell lines K1-EGFP-29, K1-EGFP-39, and K1-EGFP-40 that had been successfully acclimatized in suspension were continuously passaged for 60 generations, and the expression of the EGFP gene was detected by flow cytometry every 10 generations. As shown in Fig. 5, due to the differences in EGFP expression levels in the initial cell line, the fluorescence intensity of each cell line was different in the continuous passage process; however, all monoclonal cell lines expressed EGFP in the continuous passage of 60 generations. These results indicated that EGFP could be stably expressed at this site.

Flow cytometry analysis of average cell fluorescence intensity. A–C Flow cytometry analysis results of CHO-K1-EGFP-29 suspension cells, CHO-K1-EGFP-39 suspension cells, and CHO-K1-EGFP-40 suspension cells. D Results of the average fluorescence intensity of three CHO-K1-EGFP suspension cell lines
Site-specific integrated expression of HSA
To confirm whether the integration screening strategy can be used for site-specific integration of secreted proteins at this site, the secreted HSA gene was integrated into this site using the above method. Eighteen cloned cell lines expressing HSA were obtained by flow cytometry and Dot blot analysis (Fig. 6A, B). PCR was used to confirm the integration of exogenous genes, and the Western blot assay was used to detect the secretion and expression of HSA in the culture supernatant. The electrophoretic diagram of PCR products (Fig. 6C, D) showed that the PCR products of 5ʹ-homologous arm and 3ʹ-homologous arm were around 4100 bp and 5500 bp, respectively; this result was consistent with our expectations. The out–out PCR product (Fig. 6E) had bands around 7000 bp and 2200 bp; the 7000 bp band contained the site-directed integration of the HSA gene, while the 1500-bp band was K1 chromosomal DNA. The results of the Western blot assay (Fig. 6F) showed that the target band appeared at around 70 kDa, which was consistent with the HSA protein of 68 kDa. These results indicate that the methods mentioned above can achieve site-specific integration and expression of HSA at this site.

CHO-K1 cell sorting and identification results of site-specific integration of the HSA gene. A Flow cytometric sorting of CHO-K1 cells with the HSA gene introduced by site-directed integration; B Dot blot HSA gene expression profile in adherent cells. C Results of 5ʹ junction PCR, M: DL 2000 DNA marker, 1: HSA-B21, K1-HSA-A12, K1-HSA-14. D Results of 3ʹ junction PCR, M: DL 2000 DNA marker, 1: HSA-B21, K1-HSA-A12, K1-HSA-14. E Results of out–out PCR, M: DL 50,000 DNA marker, 1: HSA-B21, K1-HSA-A12, K1-HSA-14. F Western blot assay results of HSA gene expression in adherent cells, M: molecular mass marker, HSA-B21, K1-HSA-A12, K1-HSA-14, Exposure time: 30 s
HSA expression of site-specific integrant cell lines in 60 consecutive passages
The suspension-acclimated K1-HSA-A12 and K1-HSA-14 cell lines were continuously passaged for 60 generations, and batch experiments were performed every 20 generations to detect the expression of HSA. As shown in Fig. 7, the 2 monoclonal cell lines expressed HSA in 60 consecutive passages, with an expression level of approximately 6 mg/L. The expression level of HSA was the same between the two monoclonal cell lines, but a minor difference was observed between the individual cells. These results suggest that this site can be used for site-specific integration and stable expression of HSA.

Identification of HSA gene expression and batch culture of CHO-K1-HSA suspension cell lines. K1-HSA-14 cell density changes (A) and cell viability changes during batch culture (B). K1-HSA-A12 cell density changes (C) and cell viability changes (D) during batch culture. (E) Quantitative analysis of HSA protein expression during cell culture
Discussion
In the present study, a method based on CRISPR–Cas9 technology was established. The EGFP gene and the HSA gene were successfully integrated into the CHO-K1 cell chromosome NW_0036136338.1 near the 1969647th base (Cdk6 gene intron region). The monoclonal cell lines (K1-EGFP-29, K1-EGFP-39, K1-EGFP-40, K1-HSA-A12, and K1-HSA-14) screened by flow cytometry were successfully acclimated in suspension. The cells were subcultured for 60 passages, and the expression of exogenous proteins (EGFP and HSA) was detected every 20 passages. The results showed that all monoclonal cell lines expressed the corresponding foreign proteins. Therefore, the region near the 1969647th base of chromosome NW_0036136338.1 in CHO-K1 cells is an integration site that can be used to express foreign proteins stably.
Currently, in the field of biopharmaceuticals, expression cell lines are being constructed through the random integration of the target gene [3]. Compared to traditional methods, the protocol established in the present study can effectively solve the problem of long-term unstable expression of recombinant cell lines and greatly reduce the construction time. According to incomplete statistics, it takes 6–12 months on an average to construct a recombinant cell line by traditional methods [3]; however, the present study took only 2 months to obtain a recombinant CHO cell line that can stably express foreign proteins for a long time. More importantly, according to the requirements of the FDA and other drug review centers, non-productive cell clones should not exceed 30% during the scale-up and production process from the MCB to the WCB [4]. Therefore, cell strains constructed by site-specific integration are easier to meet this requirement and more consistent with GMP requirements in pharmaceutical production.
Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
Fischer S, Handrick R, Otte K. The art of CHO cell engineering: a comprehensive retrospect and future perspectives. Biotechnol Adv. 2015;33(8):1878–96.
Kim JY, Kim YG, Lee GM. CHO cells in biotechnology for production of recombinant proteins: current state and further potential. Appl Microbiol Biotechnol. 2012;93(3):917–30.
Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat Biotechnol. 2004;22(11):1393–8.
Dahodwala H, Lee KH. The fickle CHO: a review of the causes, implications, and potential alleviation of the CHO cell line instability problem. Curr Opin Biotechnol. 2019;60:128–37.
Knoben J. An overview of the FDA publication approved drug products with therapeutic equivalence evaluations. Am J Hosp Pharm. 1990;47:2696–700.
Schiff LJ. Review: Production, characterization, and testing of banked mammalian cell substrates used to produce biological products. In Vitro Cell Dev Biol Anim. 2005;41(3):65–70.
Mark Leonard MH, Cecilia C, Kevin M Kerstin C, Robin Heller-Harrison A. Managing cell line instability and its impact during cell line development. Biopharm Int. 2009. 2009:(4).
Derouazi M, Martinet D, Besuchet Schmutz N, Flaction R, Wicht M, Bertschinger M, Hacker DL, Beckmann JS, Wurm FM. Genetic characterization of CHO production host DG44 and derivative recombinant cell lines. Biochem Biophys Res Commun. 2006;340(4):1069–77.
Yang Y, Mariati M, Chusainow J, Yap MG. DNA methylation contributes to loss in productivity of monoclonal antibody-producing CHO cell lines. J Biotechnol. 2010;147(3–4):180–5.
Krämer O, Klausing S, Noll T. Methods in mammalian cell line engineering: from random mutagenesis to sequence-specific approaches. Appl Microbiol Biotechnol. 2010;88(2):425–36.
Kawahara AH, Yu H, Ota S, Taimatsu K. Site-specific integration of exogenous genes using genome editing technologies in zebrafish. Int J Mol Sci. 2016;17(5):727.
Wurm FM, Hacker D. First CHO genome. Nat Biotechnol. 2011;29(8):718–20.
Xu X, Nagarajan H, Lewis NE, Pan S, Cai Z, Liu X, Chen W, Xie M, Wang W, Hammond S, et al. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat Biotechnol. 2011;29(8):735–41.
Lewis NE, Liu X, Li Y, Nagarajan H, Yerganian G, O’Brien E, Bordbar A, Roth AM, Rosenbloom J, Bian C, et al. Genomic landscapes of Chinese hamster ovary cell lines as revealed by the Cricetulus griseus draft genome. Nat Biotechnol. 2013;31(8):759–65.
Zhao M, Jiaxian W, Manyu L, Han L, Meiqi Z, Lei H, Mengxiao Z, Hui Y, Yueqing X, Hua J, et al. Rapid development of stable transgene CHO cell lines by CRISPR/Cas9-mediated site-specific integration into C12orf35. Appl Microbiol Biotechnol. 2018;102(14):6105–17.
Kawabe Y, Komatsu S, Komatsu S, Murakami M, Ito A, Sakuma T, Nakamura T, Yamamoto T, Kamihira M. Targeted knock-in of an scFv-Fc antibody gene into the hprt locus of Chinese hamster ovary cells using CRISPR/Cas9 and CRIS-PITCh systems. J Biosci Bioeng. 2018;125(5):599–605.
Chi X, Zheng Q, Jiang R, Chen-Tsai RY, Kong LJ. A system for site-specific integration of transgenes in mammalian cells. PLoS ONE. 2019;14(7): e0219842.
Zhou S, Chen Y, Gong X, Jin J, Li H. Site-specific integration of light chain and heavy chain genes of antibody into CHO-K1 stable hot spot and detection of antibody and fusion protein expression level. Prep Biochem Biotechnol. 2019;49(4):384–90.
Cheng JK, Lewis AM, Kim DS, Dyess T, Alper HS. Identifying and retargeting transcriptional hot spots in the human genome. Biotechnol J. 2016;11(8):1100–9.
Zhou S, Ding X, Yang L, Chen Y, Gong X, Jin J, Li H. Discovery of a stable expression hot spot in the genome of Chinese hamster ovary cells using lentivirus-based random integration. Biotechnol Biotechnol Equip. 2019;33(1):605–12.
Manuel S, Thomas T, del Maria SK, Joachim W, Mateo JL. CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS ONE. 2015;10:e0124633.
LauraBailey A, Diane H, Ray F, Dickson AJ. Determination of Chinese hamster ovary cell line stability and recombinant antibody expression during long-term culture. Biotechnol Bioeng. 2012;109(8):2093–103.
Acknowledgements
Thanks to Dr. Lu Daru of Fudan University and Dr. Helene F. Kildegaard of Novo Nordisk for proposing experimental plasmids. Thanks to Zhang Linpei, experimental platform of School of Bioengineering, Jiangnan University, for his help in flow analysis.
Funding
No funding.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors report no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ding, X., Chen, Y., Wu, H. et al. Construction of a CHO cell line with site-specific integration to stably express exogenous proteins using the CRISPR–Cas9 technique. Syst Microbiol and Biomanuf 3, 659–668 (2023). https://doi.org/10.1007/s43393-022-00147-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43393-022-00147-y