
Abstract
Materials science is crucially involved in researching advanced materials for energy conversion and storage. The discovery process has been accelerated by computational simulations, which have become a powerful tool for predicting the properties of new materials in recent years. This study examines the structural, electronic, and optical properties of double perovskites Cs2NaInX6 (X = Br, Cl, and I), emphasizing their potential use in energy conversion. Relevant results for optoelectronic applications have been obtained through a comparative study of the studied halide double perovskites. The compounds exhibit a high absorption coefficient of over 106 cm−1 in the ultraviolet, as demonstrated by the obtained results. In addition, these compounds are able to absorb light with a maximum absorption of 105 cm−1 in the visible range. Cs2NaInX6 double perovskite compounds exhibit direct semiconductor behavior with optical bandgap energy values of around 2.46, 3.44, and 1.64 eV for Cs2NaInBr6, Cs2NaInCl6, and Cs2NaInI6, respectively. The reached results led to the conclusion that Cs2NaInX6 compounds can be used as a promising candidate for optoelectronic and solar cell applications. Our understanding is that this is the first theoretical prediction of the electro-optical properties of these compounds that has not yet been confirmed experimentally.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Energy shortages have become a major challenge worldwide, over the past few years. The consumption process of conventional fossil fuel storage results in severe environmental pollution due to its finite, non-renewable resource. Solar energy is known to be a clean, inexhaustible, and perpetual energy source. Perovskite solar cells have drawn a great deal of attention compared to conventional crystalline silicon solar cells. This is due to their photoelectric properties, simple manufacturing process, and low cost [1, 2]. Solar cell efficiency has improved significantly for perovskite solar cells (PSCs) over the last decade. An increase in power cell efficiency (PCE) can be achieved by adding MAPbI3 to power cells with an increase from 3.8 to 25.2% in FAPbI3 PSCs [1]. In addition, these materials can easily spontaneously decompose and become unstable when exposed to prolonged exposure to air and moisture. Also, MAPbI3 and FAPbI3-based PSCs contain Pb2+ cations, which pose a serious threat to both human and aquatic life. This has hindered the creation and commercialization of organic halide perovskite solar cells. Identifying perovskite materials that are stable, lead-free, and have a high light absorption in the visible range is crucial [1, 3, 4]. The unique crystal structure and potential applications in energy conversion and storage have made double perovskites a subject of significant attention in recent years. These materials have the general formula A2BB’X6, where A, B, B’ are different cations and X is a different anion. It is typical for the lead-based perovskite structure to consist of divalent ions. The divalent ion is replaced by a trivalent and a monovalent ion (B3 + and B' +). Halogen (Br, Cl, and I) is represented by X, while alkaline-metal or rare-earth is represented by A. In our scenario, A, B, and B' correspond to Cs, K, and In, respectively. The variety of structures and properties available in these materials is extensive. Outstanding electronic dimensionality, excellent reliability, eco-friendliness, promising solar cell applications, and tunable optoelectronic properties are the features of the lead-free double perovskite [5,6,7,8,9]. Sol–gel synthesis and colloidal synthesis are both methods to synthesize these materials. The design of double lead-free perovskite was made because of the ineffective ability to maintain chemical compatibility and stability of single lead-free perovskite. In addition, this is facilitating the improvement of chemical compatibility and electrical conductivity, which can be employed in various applications, such as photocatalytic and photovoltaic applications [7]. The first principal calculations of the Ag-based double perovskite display significant optoelectronic properties. Their direct bandgap energies are characterized by a semiconductor behavior, with X ranging from Cl to I, exhibiting direct bandgap energies ranging from 2.956 to 0.029 eV. The three compounds exhibit high optical absorption coefficients of around 105 cm−1 [10, 11]. Silver-based double perovskite is a highly synthesized and characterized product that can be utilized in photoluminescence and photovoltaic applications [10]. The effect of replacing the Ag element with the Na element will be investigated using density functional theory calculations in this investigation. Our goal with this manuscript is to examine the structural, electronic, and optical characteristics of Na-based double perovskites for optoelectronic and photovoltaic applications. The outcomes of this study are expected to be useful in the development of lead-free double perovskites. As far as we know, this is the first theoretical prediction about the electro-optical properties of these compounds, which are still unconfirmed by experimenters.
2 Computational methods
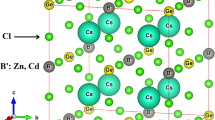
The physical properties of new lead-free double perovskites are investigated based on the density functional theory (DFT) [12]. The first principal calculations are implemented in the WIEN2K package, using the full potential linearized augmented plane wave method (FP-LAPW) [2, 13]. The exchange correlation energy is calculated using the Generalized Gradient Approximation (GGA) [14]. The energy convergence criterion is set at 104 Ry, for RMT \(\times \) Kmax = 7, the convergence force at 1 mRy/a.u., and the 10 \(\times \) 10 \(\times \) 10 k-point in the irreducible wedge of the Brillion zone for optimization. The optimization of cell parameters is achieved to identify the ideal lattice parameters for each compound, that crystallized in the cubic structure with space group of Fm-3 m (N°225); as a result, the cell parameter a of Cs2NaInX6 double perovskite compounds take values of 11.55, 10.91, and 12.50 Å for Cs2NaInBr6, Cs2NaInCl6, and Cs2NaInI6, respectively (see Fig. 1).

Crystalline structure of Cs2NaInX6 (X ≡ Br, Cl, and I)
3 Results and discussion
The cell parameters of the new lead-free double perovskites Cs2NaInBr6, Cs2NaInCl6, and Cs2NaInI6 are optimized using the GGA-PBE approximation [14]. The obtained results are illustrated in Fig. 2 and summarized in Table 1.

Total energy of Cs2NaInX6 (X ≡ Br, Cl, and I) compounds as a function of the cell volume
Figure 2 shows the variation of total energy of the studied double perovskite compounds as a function of the cell volume leading to conclude that Cs2NaInI6 is more stable compared to the others double perovskites. And this is due to the lower energy of Cs2NaInI6 [15]. Likewise, we investigate the structural stability of Cs2NaInX6 compounds based on the calculation of formation energy ΔHf [16, 17].
The formation energy is calculated using the following equation:
It is considered one of the most critical parameters to identify the structural stability of a compound. This is calculated for each compound and presented in Table 1. The negative sign of the studied compounds indicates the structural stability of Cs2NaInX6 compounds, and they can be synthesized experimentally. According to the calculated values of the formation energies, we can assume that Cs2NaInI6 is most stable compared to Cs2NaInBr6 and Cs2NaInCl6, as mentioned before in Fig. 2 (ΔHf (X ≡ Br) = − 5.305 eV, ΔHf (X ≡ Cl) = − 5.351 eV, ΔHf (X ≡ I) = − 5.427 eV).
This section aims to examine the electronic properties of compounds Cs2NaInX6 with (X = Br, Cl, and I) by analyzing their band structure and density of state. Figure 3 displays the calculated band structures of the Cs2NaInX6 compounds and allows the conclusion of the semiconductor behavior with a direct bandgap. This is supported by the fact that the valence band maximum (VB) and conduction band minimum (CB) are at the same point (Γ) in all the double perovskite samples investigated. According to the obtained electronic bandgap energies, it has been determined that Cs2NaInBr6, Cs2NaInCl6, and Cs2NaInI6, respectively, have energies of 1.96, 3.07, and 0.91 eV. The results obtained here are in good agreement with those reported for silver-based perovskites, Cs2AgInX6, and Cs2AgSbX6 [10, 18].

Band structures calculations of the Cs2NaInX6 compounds calculated via GGA-PBE approximation: a Cs2NaInBr6, b Cs2NaInCl6, c Cs2NaInI6
To analyze and explain the origin of the calculated band structure illustration, the density of the state is analyzed. The total and partial densities of state calculations for the studied double perovskites Cs2NaInX6 show that the P-type semiconductor behaves as described in the band structure plots. The total and partial densities of states are depicted in Fig. 4 (a, b, and c). Moreover, these findings were asserted via the band structure calculations. It can be seen from Fig. 4 that the density of state contribution is made via the s-p hybridization between s-In, s-Na, and p-Cs orbits in the CB for the three Na-based double perovskites. In addition, the density of state contribution in the VB is made via the s-p hybridization between s-In, p-Br, and p-Cs orbitals for Cs2NaInBr6, between s-In, p-Cl, and p-Cs for Cs2NaInCl6 and, between s-In, p-I, and p-Cs orbitals for Cs2NaInI6.

Total and partial densities of state of the Cs2NaInX6 compounds calculated via GGA-PBE approximation: a Cs2NaInBr6, b Cs2NaInCl6, c Cs2NaInI6
The optical properties of the Cs2NaInX6 with (X = Br, Cl, I) compounds are also investigated in this manuscript. Based on the complex dielectric function, which is composed of both real and imaginary parts, we examine the absorption coefficient α(\(\omega \)), the reflectivity R(\(\omega \)), and refractive index n(\(\omega \)). The optical properties α(\(\omega \)), R(\(\omega \)), and n(\(\omega \)) are calculated as follows:
where \(k(\omega )\), \({\varepsilon }_{1}\left(\omega \right)\), and, \({\varepsilon }_{2}\left(\omega \right)\) are the extinction index, the real parts, and imaginary parts of the dielectric function, respectively.
\(k(\omega )\), \({\varepsilon }_{1}\left(\omega \right)\), and \({\varepsilon }_{2}\left(\omega \right)\) are described using the Ehrenreich and Cohen formalism as follows.
where M signifies the matrix elements for the transitions between the valence and the conduction bands, e and m are the charge and the mass of the electrons, respectively. ω denotes the frequency of the incident photon, fi is the Fermi distribution function for the state, Ei is the energy of the electron in the ith state, while i and j represent the initial and final states, respectively.
The optical properties α(\(\omega \)), R(\(\omega \)), and n(\(\omega \)) of Cs2NaInX6 compounds are presented as a function of the wavelength λ in the range of [100–1000 nm] in Figs. 5, 7, and 8, respectively.

Absorption spectrums of the Cs2NaInX6 (X ≡ Br, Cl, and I) compounds as a function of wavelength λ
The spectrums of optical absorption are depicted in Fig. 5. In the ultraviolet region, the absorption coefficient of Cs2NaInX6 compounds is high, exceeding 106 cm−1, as shown in Fig. 5. Also, they exhibit a high absorption coefficient of more than 105 cm−1 in the visible spectrum.
The calculation of the optical bandgap energies (Eg) was achieved by the Tauc equation [19, 20] (Fig. 6):
where A is a constant of proportionality depending on the nature of the material, and n is equal to 1/2 for a direct allowed transition or 2 for an indirect one. The studied materials present a direct bandgap with an energy value of 2.46, 3.44, and 1.64 eV for Cs2NaInBr6, Cs2NaInCl6, and Cs2NaInI6, respectively. These values are in good agreement with previous results [10, 18].

Tauc plot of the Cs2NaInX6 (X ≡ Br, Cl, and I) compounds
Figure 7 presents reflectivity plots of Cs2NaInX6 compounds. Based on Fig. 7, one can conclude that Cs2NaInI6 reveals a high reflection ratio compared to Cs2NaInBr6 and Cs2NaInCl6, which vary from 14 to 34% in the ultraviolet range and possesses a low reflection between 7 and 14% in the visible spectrum.

The reflectivity of the Cs2NaInX6 (X ≡ Br, Cl, and I) compounds as a function of wavelength λ
The calculated refractive index n of the Cs2NaInX6 compounds is shown in Fig. 8. The n values are increased with a slight fluctuation between 100 and 400 nm to reach maximum values of 2.50, 2.25, and 2.75 for Cs2NaInBr6, Cs2NaInCl6, and Cs2NaInI6, respectively. Then decreased until getting stable values of 1.80, 1.75, and 2.20 for Cs2NaInBr6, Cs2NaInCl6, and Cs2NaInI6, respectively.

The refractive index of Cs2NaInX6 (X ≡ Br, Cl, and I) compounds as a function of wavelength λ
4 Conclusion
We used Wien2k code to conduct first-principle calculations based on density functional theory (DFT) using the Generalized Gradient Approximation (GGA-PBE). This investigation is being carried out to study the optoelectronic properties of a novel lead-free double perovskite Cs2NaInX6 with (X = Br, Cl, I). The structural properties show that the compounds studied can be synthesized. Their behavior resembles P-type semiconductor with values ranging from 1.64 eV to 3.44 eV. Furthermore, they have a high absorption coefficient in both regions, the ultraviolet and visible spectrum, that exceeds 105 cm−1. According to the attaining results in this work and our latest work [20], we can conclude that the double perovskites Cs2AInX6 with (A ≡ K, Na and X ≡ Br, Cl, I) can be used in the photovoltaic and optoelectronic applications, due to their promising electro-optic properties.
Data availability
Not applicable.
References
D.-Y. Hu, X.-H. Zhao, T.-Y. Tang, L.-M. Lu, L. Li et al., Sol. Energy 231, 236–242 (2022)
I. Hamideddine, N. Tahiri, O. El Bounagui, H. Ez-Zahraouy, Mater. Sci. Semicond. Process. 126, 105657 (2021)
D. Liu, H. Peng, H. Zeng, R. Sa et al., J. Solid State Chem. 303, 122473 (2021)
V. Hoseinpour, Z. Shariatinia, L. Echegoyen, Mater. Res. Bull. 159, 112088 (2023)
S. Bimli, V. Manjunath, S.R. Mulani, A. Miglani, O.S. Game, R.S. Devan, Sol. Energy 256, 76–87 (2023)
K. Radja, B.L. Farah, A. Ibrahim, D. Lamia, I. Fatima, B. Nabil et al., J. Phys. Chem. Solids 167, 110795 (2022)
S.S. Nair, L. Krishnia, A. Trukhanov, P. Thakur, A. Thakur, Ceram. Int. 48, 34128–34147 (2022)
A.H. Slavney, T. Hu, A.M. Lindenberg, H.I. Karunadasa et al., J. Am. Chem. Soc. 138, 2138–2141 (2016)
F.A. Faber, A. Lindmaa, O.A. von Lilienfeld, R. Armiento, Machine learning energies of 2 million elpasolite (ABC2D6) crystals. Phys. Rev. Lett. 117, 135502 (2016)
R. Anbarasan, M. Srinivasan, R. Suriakarthick, H. Albalawi, J.K. Sundar, P. Ramasamy et al., J. Solid State Chem. 310, 123025 (2022)
M. Li, H. Chen, S. Ming, B. Wang, J. Su, Chem. Phys. 559, 111520 (2022)
I. Hamideddine, H. Zitouni, N. Tahiri, O. El Bounagui, H. Ez-Zahraouy, Appl. Phys. A 127, 443 (2021)
I. Hamideddine, N. Tahiri, O.E. Bounagui, H. Ez-Zahraouy, J. Korean Ceram. Soc. 59, 350–358 (2022)
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77, 3865–3868 (1996)
H. Jebari, N. Tahiri, M. Boujnah, O. El Bounagui, M. Taibi, H. Ez-Zahraouy, Phase Transit. 94, 1–12 (2021)
I. Hamideddine, H. Jebari, N. Tahiri, O. El Bounagui, H. Ez-Zahraouy, Int. J. Energy Res. 46, 20755–20765 (2022)
Y. He, G. Galli, Chem. Mater. 26, 5394–5400 (2014)
D.-Y. Hu, X.-H. Zhao, T.-Y. Tang, L.-M. Lu, L. Li et al., Mater. Sci. Semicond. Process. 152, 107077 (2022)
H. Jebari, N. Tahiri, M. Boujnah, O. El Bounagui, L. Boudad, M. Taibi et al., Appl. Phys. A 128, 842 (2022)
I. Hamideddine, H. Jebari, H. Ez-Zahraouy, Physica B 677, 415699 (2024)
Funding
No funding was received for this work.
Author information
Authors and Affiliations
Contributions
H. Jebari: investigation, writing the draft, writing—review and editing, literature analysis; I. Hamideddine: investigation, writing—review and editing, H. Ez-zahraouy: review and editing; project administration, supervision, and validation.
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jebari, H., Hamideddine, I. & Ez-Zahraouy, H. Exploring the optoelectronic properties of novel lead-free double halide perovskites Cs2NaInX6 (X = Br, Cl, I) for photovoltaic applications: DFT study. J. Korean Ceram. Soc. 61, 599–604 (2024). https://doi.org/10.1007/s43207-024-00395-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43207-024-00395-9