
Abstract
The requirement for biomaterials with superior properties, used in bone treatment applications, is inevitable due to escalated bone tissue defects. Baghdadite (BAG) is a calcium silicate that benefits from the presence of zirconium (Zr) in its structure and has attracted huge attention in recent years. In this study, a modified sol–gel route was proposed to synthesize BAG by dissolving Zr precursor separately and using optimum amounts of solvent and chelating agent. Due to thermal gravimetric analysis and differential thermal analysis (TGA–DTA), X-ray diffraction (XRD), and Fourier Transform Infrared Spectroscopy (FTIR) results, the BAG nanoparticles were successfully synthesized using this modified approach for the first time, and they were comprehensively characterized in terms of physicochemical, mechanical, and biological properties. During synthesis, a transparent sol without any insoluble Ca or Zr precursors and/or no premature gelation was observed, unlike samples that we produced using the conventional sol–gel method in the literature. The crystalline BAG nanoparticles with semi-spherical shapes demonstrated ~ 20% weight loss after 28 days during the biodegradability test, extensive bioactivity, and enhanced mechanical strength (~4 MPa). Moreover, BAG powder was biocompatible with no cytotoxic effect and osteoinductive in the absence of an osteogenic medium. We believe that the synthesized BAG nanoparticles through this modified sol–gel route could serve as a promising biomaterial for cancellous bone defect treatment applications.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The chemical composition of biomaterials plays a significant role in the ultimate success of scaffolds, used in bone tissue engineering (BTE), and bone grafts to treat bone defects. It is possible to modify the properties of either scaffolds or grafts using different materials and satisfy the requirements needed for the proper performance of these bone healing approaches [1]. Calcium silicates (CaSi) are biocompatible materials that have been widely used as alternatives to the other well-known bioceramics in biomedical applications, such as calcium phosphates (CaP) and bioactive glasses (BG) [2]. CaSi materials possess bioactive and osteoconductive nature, and they contribute to osteogenic activities of cells and bone formation [3, 4]. In addition, a higher degradation rate makes them a preferable choice over some CaP ceramics, such as hydroxyapatite (HA), in some applications [5]. However, low mechanical properties, brittleness, and long-lasting stability of CaSi on account of the high dissolution rate are considered serious issues, especially in load-bearing applications [6].
One approach to attenuate the drawbacks of CaSi is an incorporation of the third element into their structure [7]. Elements like Mg, Zn, and Ti are examples that have been used to promote the mechanical properties, chemical stability, or biological features of CaSi [8,9,10]. The incorporation of zirconium (Zr) into the structure of CaSi led to the formation of a chemical compound, named baghdadite (BAG) by the formula of Ca3ZrSi2O9 or Ca3Zr(Si2O7)O2, whose natural mineral had been discovered in Iraq for the first time [11].
BAG usage appeared in biomedical fields after the findings of Ramaswamy et al. [12], who demonstrated better cell spread, proliferation, and even differentiation of human osteoblast cells in BAG-containing ceramics. Later, other studies also confirmed non-cytotoxicity and the promoted influence of BAG on cell proliferation and osteogenic differentiation [13]. BAG has been mainly applied to fabricate scaffolds for tissue engineering, notably BTE. Furthermore, its applications have been extended to coating metallic implants and developing bone cement and filler materials [14]. Not only in vitro studies [15] exhibited promoted osteogenic differentiation through the expression of elevated bone morphogenetic protein-2 (BMP2), osteocalcin, or runt-related transcription factor 2 (RUNX2), but also in vivo evaluation demonstrated successful bone formation in large animal models [16] or greater bone ingrowth in scaffolds with BAG compared to tricalcium phosphate (TCP)/HA [17].
Sol–gel method is the major route that has been employed in studies for the synthesis of BAG [18]. Nevertheless, some studies used a direct solid-state route, which is time-consuming and needs higher sintering temperatures [19]. Furthermore, a solid-state method could inhere drawbacks, such as chemical inhomogeneity, the presence of impurities during ball milling, and coarser particle sizes [20]. Concerning the sol–gel technique, the complete dissolution of Si, Ca, and Zr precursors inside a solvent (ethanol) and obtaining a clear solution (sol) is a necessary step prior to the gelation of the mixture. However, upon trying to reproduce the BAG by following the proposed sol–gel procedure in the literature [21, 22], we faced premature gel formation before the complete dissolving of Zr precursor, and the larnite with PDF # 33-0302 was the primary phase after the sintering process. Therefore, in this study, the BAG powder was synthesized through the slightly modified sol–gel method, where all precursors were dissolved completely to have a clear solution (sol). Later, synthesized powders were characterized to determine their physicochemical, mechanical, and biological properties for BTE and bone restoration applications. To shed light on the various aspects of this research, thermal, structural, microstructural, particle size, mechanical, and biological properties of these powders were analyzed with the help of Differential Thermal Analysis–Thermogravimetric Analysis (DTA–TGA), Fourier Transform Infrared Spectroscopy (FTIR), X-Ray Diffraction (XRD), Scanning Electron Microscopy (SEM), Particle Size Analyzer (PSA), universal tensile testing system, in vitro biodegradability and bioactivity, and in vitro cell culture test machines/equipments as an in-depth work.
2 Materials and methods
2.1 Synthesis of BAG powder
The modified sol–gel route was employed for the synthesis of BAG powder, in which two different features were changed compared to conventional sol–gel methods: (a) dissolving Zr precursor separately in ethanol, and (b) using different TEOS:ethanol: HNO3 molar ratio. Zirconium [IV] oxynitrate hydrate (ZrO(NO3)2·xH2O, Aldrich, USA), tetraethyl orthosilicate (TEOS, Si(C2H5O)4, Merck, Germany), and calcium nitrate tetrahydrate (Ca(NO3)2·4H2O, Merck, Germany) were used as Zr, Si, and Ca precursors, respectively. The amount of each precursor was calculated according to 1:2:3 molar ratios of Zr:Si:Ca in the BAG chemical formula. Briefly, 2.335 g of zirconium [IV] oxynitrate hydrate were dissolved in 50 ml pure ethanol (EMSURE®, Merck, Germany) using the stirrer. Meanwhile, 1 ml nitric acid (HNO3, 2 M, Isolab, Turkey), as a chelating agent, was added into the 50 ml ethanol (EMSURE®, Merck, Germany) that had 4.45 ml TEOS, and obtained mixture of TEOS:ethanol:HNO3 with molar ratios of 1:42:1 mixed in a separate beaker on the stirrer for hydrolysis. Nitric acid, in addition to the modifier of the molecular structure of sol, acts as a chelating agent and stabilizes the sol by reducing the alkoxide reactivity [23]. After complete dissolving of Zr precursor in ethanol, this solution and 7.084 g of calcium nitrate tetrahydrate were added into the mixture of TEOS:ethanol:HNO3, and a clear solution (sol) was obtained after 5 h (h) of stirring before gelation. The prepared sol was heated at 60 °C for 24 h and at 100 °C for another 24 h inside the oven to have dried gel as a powder. Finally, the dried powder was heat-treated and sintered at 1150 °C for 3 h in the air. Moreover, different ratios of TEOS:ethanol:HNO3 (1:8:0.16, 1:21:1, 1:30:1, and 1:42:1) were examined to figure out the optimum value for TEOS:ethanol:HNO3 mixture. Clarity of the sol (ethanol after dissolving all precursors) and phase structure of the powder after heat treatment and sintering at 1150 °C for 3 h were two criteria that were used to control the quality of the final product. After the naked eye visual inspection of the solution’s turbidity, the pH values were measured by a standard pH meter.
2.2 Heat treatment
Heat treatment gives the opportunity to manipulate the properties of the materials by controlling the rate of diffusion and the cooling rate within the microstructure. To synthesize BAG powder, first gel powder was dried at 60 °C for 24 h in the air and subsequently dried at 100 °C for 24 h in the air again on the hot plate. Then, the obtained samples were placed in the furnace for drying at 300 °C for 30 min with a heating rate of 5 °C/min, oxidizing at 600 °C for 30 min, and sintering at 1150 °C for 3 h in the air. After the sintering process, the sample was left to cool in the furnace.
2.3 Physicochemical characterization
The thermal properties of dried gel were analyzed with thermogravimetry analysis–differential thermal analysis (TGA–DTA, Setaram Labsys), to determine reaction types of powders and provide a convenient process regime at a scan rate of 5°/min from 25 to 1200 °C. Fourier Transform Infrared Spectroscopy (FTIR, Bruker IFS66/S) was used to identify functional groups in the synthesized BAG. FTIR spectrum was recorded within the frequency range of 4000–400 cm−1.
X-ray diffraction (XRD, Rigaku Ultima IV) was used to determine the BAG phase purity, lattice parameters, and cell unit volume of the material. The XRD test was operated at 40 kV and 30 mA utilizing CuKα radiation on a step size of 0.1° and a step time of 1 s, and a diffractogram was obtained in the range of 10°–70° of 2θ angles. Using XRD data, the modified Scherrer Eq. (1) was applied to estimate the size of crystallite of BAG [24], where D is the size of crystallite, k is constant that depends on crystallite morphology and its value is ~ 0.9; λ is the wavelength of X-ray (1.5405 Ȧ); β is the value of full width at half-maximum (FWHW) of the peaks of the (\(202\)), (113), (023), (032), (\(213\)), (\(242\)) planes; and θ is the diffraction angle corresponding to peaks in radian.
Lattice parameters and cell volume unit of material were derived by refinement of XRD data using Cell software of Holland and Redfern [25]. Moreover, the crystallinity percentage of BAG was calculated using XRD data by a ratio of the area of crystalline peaks to the total area of all amorphous and crystalline peaks together using OriginLab software. Inductively coupled plasma mass spectrometry (ICP-MS, Perkin Elmer DRC II model) was used to analyze the elemental and chemical composition of synthesized BAG powder. Moreover, the microstructure and morphology of the powder were examined by scanning electron microscopy (QUANTA 400F Field Emission SEM, U.S.). The gold (Au) sputtering was applied to coat the surface of the sample before taking SEM images. The particle size distribution of BAG was measured by laser diffraction particle size analyzer (Mastersizer 2000, Malvern, UK). The particles were passed through the sieve with an orifice size of 212 µm before measuring their size by laser diffraction particle size analyzer.
For measuring the density, BAG was sieved through a sieve with the orifice of 212 μm, the powder was pressed in the shape of a disk (~13 mm diameter), using an automatic hydraulic cold press device (Carver, USA). The pressing force of ~22.24 kN was imposed for 3 min, and the disks were sintered at 1150 °C for 3 h before measurement. The density (ρ) and porosity of samples were measured by applying Archimedes principles and Eqs. (2) and (3) [26, 27], where wtair is the weight of dry disk in the air, wtwet is the weight of water-saturated wet disk in the air, and wtsub is the weight of submerged disk in the water. The measured density was divided by the theoretical density of BAG (ρth = 3.84 g/cm3) [28] to calculate the relative density of samples.
2.4 Mechanical tests
Diametral tensile strength of BAG samples (n = 6) was measured by subjecting disks to compression load using the universal testing system (Instron 5944, USA). Note that in the course of compression, surface diameters of the disk that were perpendicular to the compressive force direction are where the maximum tensile force would be created across. The samples were prepared and tested according to ASTM Standard—D3967-08 and Eq. (4) were used to calculate the tensile strength [29], where S and F are tensile strength (MPa) and failure force (N), respectively, while D is the sample's diameter (mm), and t was disk thickness (mm):
Similar to density, sample disks were prepared beforehand for measuring mechanical properties. The microhardness test device (HMV-2, Shimadzu, Japan) was employed to measure the Vicker’s hardness (HV) of samples. Three samples were prepared and 10 measurements were performed on each disk to have a total number of 30 measurements. Diamond indenter applied a force of 490.3 mN for 15 s and the optical microscope measured the indent diameter. The hardness value HV (GPa) was calculated using Eq. (5), in which P was applied load (N), and d was the length of indent (mm) [30, 31].
2.5 Biological studies
2.5.1 In vitro biodegradability and bioactivity
Weight loss of samples in acetic acid and sodium acetate buffer solution (0.1 M, pH = 5.5) was measured to examine in vitro biodegradability of BAG. Briefly, the dry BAG disks (n = 3) were weighed (wbi), then, submerged in the prepared buffer solution inside polystyrene falcon tubes, and incubated at 37 °C shaking water bath for 28 days. The buffer solutions were changed each day to minimize the pH change in solutions. At each time point of 1, 7, 14, 21, and 28 days, after removing the buffer solution, the samples were rinsed 3 times with distilled water (dH2O), and their weights were measured after air-drying disks (wad). The formula (6) was used to calculate weight losses:
Kokubo and Takadama’s protocol [32] was applied to study in vitro bioactivity of BAG. The disk samples (n = 3) were kept in simulated body fluid (SBF) with a pH of 7.4 for specified incubation periods at 37 °C. The volume of used SBF was ~ 31 ml, which was 1/10 of the surface area of the specimen (~310 mm2). To evaluate bioactivity, the samples were gently rinsed with dH2O and air-dried using the fume hood at the end of each incubation period (1, 4, 7, and 14 days). The formation, morphology, and Ca/P ratios of apatite deposition on the surface of BAG disks were examined by Scanning Electron Microscopy and Energy Dispersive X-ray analysis (SEM/EDX, QUANTA 400F Field Emission SEM, U.S.). Furthermore, changes in the concentration of Ca, P, and Si of the filtrate were determined using ICP-MS (Perkin Elmer DRC II model), and the pH of the filtrate was measured using a pH meter (Thermo Orion 3 star).
2.5.2 In vitro cell culture studies
The Saos-2 was used throughout in vitro cell culture studies. Saos-2 is a human osteosarcoma cell line that exhibits several characteristics of osteoblastic cells, like having high alkaline phosphatase activity and producing a mineralized matrix, which makes them an attractive source of bone-related molecules for research [33]. The cells were cultured in a growth medium (GM) consisting of Dulbecco’s Modified Eagle’s medium (DMEM), 10% fetal bovine serum (FBS), and 100 U penicillin/streptomycin and maintained in a humidified atmosphere of 95% air and 5% CO2 (5215 Shel Lab., Cornelius, OR, USA) at 37 °C. Saos-2 cells reached almost 80% confluency before subculturing them with 0.1% of the trypsin/EDTA solution to evaluate the cytotoxicity of BAG powders. BAG particles were sterilized at 200 °C for 2 h before adding to the cell growth medium. The cells were seeded at the initial density of 1.0 × 104 cells per well in 96-well plates. After 24 h, they were exposed to the cell growth medium containing different concentrations of BAG particles (25, 12.5, 6.25, and 3. 125 µg/ml) and incubated for 24 and 48 h. Cell viability was measured using MTT assay at each time point [34]. Briefly, the growth media were discarded, wells were rinsed with phosphate-buffered saline (PBS, 0.1 M, pH = 7.4), and 200 μl MTT solution (0.5 mg/ml in DMEM without phenol red) was added to each well. Then, the plate was maintained in the incubator at 37 °C for 4 h. Afterward, MTT solutions were vacated and 200 μl dimethyl sulfoxide (DMSO) was added to each well for dissolving precipitated formazan crystals formed inside the cells. Finally, the optical density (OD) of the solutions in wells was read at 570 nm (n = 3) using Microplate Reader (μQuantTM, BioTek, Winooski, VT, USA). Cells were also cultured in the plastic base of the plate (TCPS) and exposed to growth medium without the BAG particles and served as the positive control group. Cell viability (%) was calculated using the following formula (7), where the viability of the positive control was taken as 100% [35]:
Alkaline phosphatase (ALP) activity and intracellular calcium quantification assays were used to examine the influence of BAG on the osteogenic activity of Saos-2 cells. The initial density of seeded cells (passage 22) was 3 × 104 cells/well in 24-well plate (n = 3). After 24 h, the GM in the wells was replaced with an osteogenic medium (OM) that included BAG particles with different concentrations of 50, 25, 12.5, 6.25, and 3.125 µg/ml. Plates were incubated for 7 and 14 days in a humidified atmosphere of 95% air and 5% CO2 (5215 Shel Lab., Cornelius, OR, USA) at 37 °C. OM consisted of GM, 50 µg/ml ascorbic acid, 10 mM β-glycerophosphate, and 10–8 M dexamethasone (DEX), which was changed every two days. At the end of each incubation period, the medium (OM + BAG) was removed from the wells, and discs were washed with PBS. Afterward, wells were filled with 600 µl fresh cell lysis buffer (Triton X100-1%, carbonate buffer, 1:9 v/v) and the cells on the discs were burst by the two cycles of the freeze–thaw process. To measure ALP activity, 50 µl of the cell lysate was incubated in a 100 µl para-nitrophenyl phosphate (PNPP) working solution at 37 °C for 60 min, and absorbance of the solution was measured at 405 nm by the microplate reader [36].
Intracellular calcium content of the cell lysates was measured through o-cresolphthalein dye assay [37]. Briefly, the lysates in 100 µl aliquots were mixed with 100 µl Ca2+ indicator dye (o-cresolphthalein complexone, 8-hydroxyquionone, hydrochloride acid, and ethanolamine) using shaking on an orbital shaker for 5 min, and the intensity of color was measured by reading absorbance at 570 nm using the microplate reader. The calcium amounts were calculated using the calibration curve constructed with different concentrations of calcium chloride (Merck, Germany) in distilled water. To normalize ALP activity and intracellular calcium, protein amounts of the cell lysates were determined using Bicinchoninic Acid assay (BCA) [38]. The cell lysate was mixed with BCA stain (1:7 v/v) and incubated at 37 °C for 1 h. The absorbance was then measured at 562 nm with the microplate reader. A calibration curve constructed with different concentrations of bovine serum albumin was used to quantitate the total protein amount in the lysates. Moreover, the ALP activity of cells cultured in the absence of an osteogenic medium (OM), where GM merely contained 10–8 M DEX and 12 µg/ml BAG, was measured by following the same steps (n = 5).
2.6 Statistical analyses
One-way analysis of variance (ANOVA) with Tukey’s post hoc tests was employed for statistical analysis of data. SPSS software (version: 21, IBM, USA) was used to determine the differences between the mean values of groups, and a p-value ≤ 0.05 was considered significant.
3 Results and discussion
3.1 Solution (sol) characteristics
Once BAG was synthesized by following the proposed sol–gel procedure in many studies [13, 22, 39] with TEOS:ethanol:HNO3 at a molar ratio of 1:8:0.16, the sol started gelling before the complete dissolution of Zr precursor, and a transparent solution (sol) could not be obtained. Thus, we increased the amount of ethanol, as the solvent, nitric acid (HNO3, 2 M), as the chelating agent, and dissolved zirconium [IV] oxynitrate hydrate in ethanol separately. Different ratios of TEOS:ethanol:HNO3 were tried during optimization to obtain crystalline BAG powder. No premature gelation or insoluble zirconium [IV] oxynitrate hydrate powder was observed in samples that had a higher amount of ethanol than conventional sol (TEOS:ethanol:HNO3 = 1:8:0.16). However, as pictured in Fig. 1, after dissolving Ca, Si, and Zr precursors, the clarity of the solution (sol) became clearer (lower turbidity) as the amount of ethanol increased. The lowest turbidity was observed in the sol of the sample with TEOS:ethanol: HNO3 at molar ratio of 1:42:1. It was a quality that we are desiring during sol–gel methods and it means higher dissolution of salts like calcium nitrate tetrahydrate and zirconium [IV] oxynitrate hydrate.

The solutions (sol) after dissolving Ca, Zr, and Si precursors for 5 h on the stirrer. a TEOS:ethanol:HNO3 = 1:21:1, b TEOS:ethanol:HNO3 equal to 1:30:1, and c TEOS:ethanol:HNO3 = 1:42:1
Generally speaking, the key factor for achieving a good transparent solution is the solubility of powder-based precursors. The clarity indicates that the precursors are completely dissolved in the solvent, which is reducing the possibility of possible different structures in the synthesis of the full stoichiometric BAG structure as Ca3ZrSi2O9 or Ca3Zr(Si2O7)O2. Bear in mind that if the turbidity was high, it would indicate undissolved powder precursors in solution, therefore the fully stoichiometric BAG structure would not be produced.
The pH value of the solution in which the precursors were dissolved was measured to be approximately 2 on account of the NO3− and C2H5O−1 anions coming from Zr-, Ca-, and Si-based precursors in the solution. In addition to NO3− and C2H5O− anions, ethanol, and HNO3 added to the solution is another factor affecting the pH value. These results emphasize the importance of the choice of processing procedures. Typically, the gel structure, which depends on the pH of the solution, regulates further tailorable characteristics, such as phase structure, microstructure, mechanical, and biological properties. This facilitates the formation of pure phases in further processes.
3.2 Thermal behavior
Thermal behavior of BAG-dried gel was evaluated using TGA–DTA, which is depicted in Fig. 2 The first endothermic peak occurred at around 60 °C, which is related to the removal of ethanol [40], whilst the second one, which was observed around 100 °C, is assigned to water evaporation [41]. The nitrate removal from zirconium nitrate happens at about 500 °C [42], and the removal of nitrate from the calcium nitrate occurs between 550 and 600 °C [43]. Nitrate removal from our sample is observed at 560 °C in Fig. 2. The exothermic peak at 950 °C corresponds to the formation of baghdadite. However, the exothermic peak at 875 °C is due to the crystallization of calcium silicate (Ca2SiO4) [44], or wollastonite (CaSiO3) phases [45]. Unlike the study that reported lower sintering temperature (800 °C) for the synthesis of crystalline BAG nanoparticles using P123 surfactant [46], we had crystalline BAG particles without using any surfactant at ≥ 900 °C, compared to conventional sol–gel (˃1100 °C) [47], or solid-state (~1350 °C) [48] techniques.

TGA and DTA curves of BAG gel dried at 100 °C for 24 h
These results contributed to determining suitable drying, oxidation/annealing, and sintering temperatures/regimes. Depending on thermal analyses, it was found appropriate to apply the heating process at 60 °C for 24 h as well as extra 24 h at 100 °C for drying, 30 min at 300 °C for heat treatment, 30 min at 600 °C for annealing, and ultimately at 1150 °C for 3 h under the air in the furnace. Heating the gel to higher temperatures is necessary to obtain more stable phases. Gels in their first form, before heat treatment, do not have the chemical composition that a technical ceramic should have. With the heat treatment, the matter composing the solid network undertakes a crystallographic reorganization, and the pore texture changes. This deformation cannot be the most stable thermodynamic phase, therefore metastable structures emerge. The temperature range approximately between 100 and 1000 °C is called the intermediate temperature range and mostly produces a crystalline transition phase. These metastable phases eventually transform into a stable thermodynamic phase like Ca3ZrSi2O9 or Ca3Zr(Si2O7)O2 at high temperatures (1150 °C).
3.3 FTIR analysis
In conjunction with the TGA–DTA analysis, the FTIR results clearly showed the effect of all reactions on the performance of the final material production, starting at room temperature with gel structure, and continuing to oxide formation and perovskite formation temperatures until the desired phase is obtained. This influence can be seen in FTIR spectra of BAG samples (Fig. 3), which were heat-treated at 50, 200, 400, 600 °C for 30 min, and 1150 °C for 3 h in the air. The result was obtained in the range of wavenumbers of 4000–400 cm−1. FTIR spectrum reveals that there are functional groups related to BAG. In Fig. 3, those curves that represent the FTIR spectra of the gel, which was heat-treated at 50, 200, and 400 °C, contain strong symmetric and antisymmetric stretching in-plane bending of NO3− at approximately 840–880 cm−1 [49]. Whilst those bands disappeared when the dried gel was heat-treated at 600 °C and above, which is in agreement with TGA–DTA results. In the same curves, the bands at 1360–1480 cm−1 represent the vibration mode of the carboxylic group (COO), and =CH3− structure [50].

FTIR spectra of BAG powders heat-treated at 50, 200, 300, 400, and 600 °C for 30 min, as well as sintered at 1150 °C for 3 h in the air
In addition, there is a peak around 1626 cm−1 which belongs to the hydroxyl (OH) group [49]. Symmetrical and asymmetrical expansions, namely OH expansions, are ascribed by the peaks in the range of 2800–3800 cm−1 [51]. It is highly expected that these bonds are being seen as a result of FTIR analysis in the structure of gel and dried/heat-treated samples that have not yet been annealed, and phases have not formed until 500 °C. In the FTIR spectrum (top curve) of BAG samples sintered at1150 °C for 3 h in the air, it is seen that the functional groups containing C, H, and N were removed from the dried gel owing to the high temperature. In fact, after the burning of organic structures, NO group was decomposed, and oxide structures were formed. For instance, the analysis results displayed that bands in the frequency range of 800–950 cm−1 are assigned to isolated SiO4 [52, 53]. The absorbance bands in the range 450–600 cm−1 wavenumbers correspond to Ca–O [54], while the absorbance band at ~1009 cm−1 is associated with Si–O–Zr, and those in the range of 600–750 cm−1 belong to Si–O–Si vibration [55, 56].
3.4 Phase analysis and microstructure
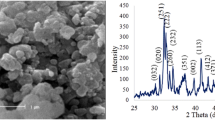
As far as the crystal structures and phase of the BAG powders are concerned, XRD was thoroughly studied for the powder samples sintered at 1150 °C for 3 h in the air (Fig. 4). It is obvious that despite the presence of BAG phase, the major phase was larnite with PDF # of 33-0302 in samples with TEOS:ethanol:HNO3 equal to 1:8:0.16 and 1:21:1. In the sample with 1:30:1 ratio of TEOS:ethanol:HNO3, the intensity of BAG peaks increased but the crystal plane with the greatest intensity belonged to larnite. However, in our proposed ratio (TEOS:ethanol:HNO3 = 1:42:1), the BAG was a major phase with high purity and consistency with the standard in terms of atomic planes in the crystal lattice. Nevertheless, the CaZrO3 phase existed in all samples but with a lower amount in our material. Therefore, we continued our characterization tests with the purest synthesized powder with optimum TEOS:ethanol:HNO3, which is equal to 1:42:1.

XRD patterns of prepared BAG with different TEOS:ethanol:HNO3 ratios. Note that B stands for baghdadite and L stands for larnite (PDF No. 33-0302) with the formula Ca2SiO4
The main reason for the presence of a major larnite phase in samples with TEOS:ethanol:HNO3 rather than 1:42:1 ratio could lie beneath TEOS hydrolysis and the fact that the Zr precursor did not dissolve completely in ethanol because the amount of solvent (ethanol) and HNO3 were not sufficient in the synthesis conditions other than the optimum proposed value of 1:42:1 for TEOS:ethanol:HNO3. Then, the gelation happened when the composition of the sol was not equal to 1:2:3 in terms of molar ratio of Zr:Si:Ca, which led to the presence of another Ca–Si phase like larnite.
Using a higher volume of ethanol and nitric acid lets to have sufficient amounts of solvent and chelating agent, which are needed to complete the hydrolysis of TEOS based on two formulas below [47]:
Moreover, Zr source (zirconium [IV] oxynitrate hydrate) was dissolved in ethanol separately and later was added with Ca source to TEOS solution before forming a polymeric network of huge silica molecules and condensation. In this way, we succeeded in obtaining a clear solution (sol) with completely dissolved zirconium [IV] oxynitrate hydrate and calcium nitrate tetrahydrate and before gelation as the next step toward the formation of BAG.
As just mentioned, Fig. 4 also represents the detailed XRD pattern of optimum BAG after sintering at 1150 °C for 3 h. Herein, the logic of sintering BAG at 1150 °C was based on the fact that we wanted to restrict our variable features to the modified procedure of synthesis merely. Then, in terms of sintering temperature, we kept consistency with other literature studies that used the sol–gel route. The observed peaks of the powder complied with the peaks of the standard synthetic baghdadite with JCPDS No. 47-1854, which has unit cell parameters of a = 10.45 Å, b = 10.18 Å, c = 7.36 Å, β = 90.87°, volume = 782.8 Å3 with space group P21/c that describes the symmetry of the crystal. The crystallinity degree of our synthesized BAG was ~87%, Its crystallite size was 16 nm based on the modified Scherrer equation, in which error sources have been minimized using the least-squares technique. The size of crystallite (grain) was smaller than previously reported crystallite sizes (38–52 nm) [47], which were prepared using the conventional sol–gel method. Moreover, its monoclinic crystal possessed a unit cell parameters of a = 10.44 Å, b = 10.19 Å, c = 7.34 Å, β = 90.87°, with volume = 780.7 Å3. The sintered samples were not 100% pure and there were a small fraction of other phases, like calcium zirconium oxide (JCPDS No. 35-0790), calcium silicate called larnite (JCPDS No. 33-0302), and wollastonite (JCPDS No. 27-0088). The minor phases, such as CaZrSi2O7, have also been observed in other studies [47, 57]. The presence of these impurities might be due to using hydrated Zr and Ca precursors in the synthesis of BAG as reported by Lee [58].
It is worth pointing out that the surface morphology of sol–gel-derived BAG powders essentially depends on solution properties, sol–gel parameters, and heating regimes [59]. The surface morphology and particle size of these sol–gel-derived BAG powder sintered at 1150 °C for 3 h in the air were depicted in Fig. 5. It is clear that the semi-spherical crystallites of BAG powder agglomerated after sintering at 1150 °C. Each agglomerate included many crystals that formed an irregular shape, which was also reported in other studies [18, 22, 60]. However, Mehrafzoon et al. [46] observed needle-like particles when surfactant was added during the sol–gel synthesis process.

a SEM micrograph, b particle size distribution of BAG powder sintered at 1150 °C for 3 h in the air, c, d SEM micrographs of BAG disk sample sintered at 1150 °C for 3 h in the air and used in mechanical tests
The particle size of sintered samples changed between 1 and 100 µm as depicted in Fig. 5. Approximately, 61% of particles had a size between 10 and 50 µm, while 24% of them were ≤ 10 µm, and only about 15% of particles were larger than 50 µm. Knowing the particle size distribution of synthesized BAG could help researchers promote the quality of their products. In a study by Zadorozhnaya et al. [61], different wear resistance as a function of particle size distributions was reported in Zr-containing materials. Recently, Averardi et al. [62] demonstrated the importance of particle size distribution on the strength and quality of products fabricated using additive manufacturing, as one of the advanced developments in bone treatment applications.
Elemental analysis of BAG powder confirmed the presence of Ca, Zr, and Si in the samples sintered at 1150 °C. Table 1 represents the theoretical and measured molar ratio of elements in BAG. Regarding the chemical composition formula of BAG (Ca3ZrSi2O9), The theoretical molar ratio of Ca:Zr:Si is equal to 3:1:2. The ICP-MS results indicated that the Ca value matches the theoretical molar ratio, whilst Zr and Si measured values are slightly smaller than the theoretical ones. However, Zr/Si ratio (0.47) is almost the same as the theoretical value (0.5) in the synthetic sample. Furthermore, the Zr/Si ratio (~0.5) seems an influential element in particle size distribution. Now that our results comply with a study, conducted by Fu et al. [55], where they reported similar particle distribution with the same Zr/Si ratio. The existence of other calcium silicate phases, like larnite, as a minor phase, demonstrates that part of Si source reacts quickly with Ca source, which is highly soluble even before mixing with Zr source and forming other calcium silicate phase. In the presence of zirconia, the calcium oxide can form CaZrO3 [63] and therefore, a tiny quantity of Zr ion failed to be located in the BAG lattice and reacted with Ca to form a small fraction of CaZrO3 phase. Other studies brought the presence of likewise minor phases as a reason behind lower molar ratios in the experiment, especially when Ca/Si ratio of the theoretical stoichiometric formula is higher than one [64]. As mentioned previously, the hydrating nature of precursors of Zr and Ca facilitates the formation of these impurities and leads to the measurement of lower experimental Ca:Zr:Si ratios in BAG powders.
3.5 Mechanical properties
Mechanical properties are the key factors for the clinical application of BAG in the human body [65]. Using the universal testing machine, the diametral tensile strength of BAG samples (n = 6) was measured by subjecting disks to compression load. A diametral compression test involves applying a stress load or force to the point where a material object is split in half. This test indirectly measures the tensile property of the BAG sample, as the particles of the material are pushed apart in opposite directions, similar to what happens to particles in a direct tensile strength test [66]. In this case, the average strength was measured 3.86 ± 0.19 MPa, which is higher than other Ca–Si, such as wollastonite [67], and even compressive strength of BAG (0.28 ± 0.02 MPa or 1.6 MPa, respectively) available in the literature [57, 68]. Mechanical properties of BAG were studied by Schumacher et al. [48] for the first time, where they measured the bending strength and hardness of the samples. Later, Najafinezhad et al. [52] demonstrated that the compressive strength of BAG was higher than diopside and akermanite, two other members of Ca–Si materials. BAG coating exhibited superior microhardness and modulus than even HA due to a study by Pham et al. [69]. BAG also enhanced the compressive strength of samples when it is used as a secondary component [70, 71].
The BAG samples had an average microhardness value of 0.47 ± 0.05 GPa. In other studies, the microhardness value was reported among 6–8 GPa [19, 48, 69]. In fact, porosity (density) has been addressed as the main factor that affects the strength of materials [28, 72], while crystallinity, grain size, and morphology are other factors that play minor roles [73,74,75]. The relation between densification and mechanical properties could be explained more by changes that occur in the structure of silicates by adding elements. It seems that the incorporation of some elements, like Mg, inhibits the movement of calcium atoms in the structure and makes it more stabilized [52]. Considering that the BAG ceramic is sorosilicate with two silicate tetrahedrons linked by one oxygen ion [76], it is hypothesized that the ZrO6 octahedral and Si2O7 group can form a more stable network that ionically binds the Ca ions [77]. This may improve the chemical stability of calcium silicate-based ceramics. Therefore, the different porosity (density) can be addressed as the main reason for observing the difference between our results and other studies. To illustrate this, the less porous samples (15%) [48] had higher hardness values than our study, where the porosity of samples was about 34% (Table 2). Similarly, the samples with higher porosity (78%)[68] had lower compressive strength than our samples. Furthermore, the smaller grain size of our sample might be a minor reason for its enhanced mechanical strength [78]. The microstructure of the disk samples that have been sintered at 1150 °C for 3 h is represented in Fig. 5c, d. Generally, the particles are composed of several grains of varying smaller sizes. The grain size, derived from SEM images, sometimes matches the crystallite size, which is figured out from XRD data, but grains are often constructed due to the combination of several crystallites and are larger than them. Figure 5c shows that our BAG disk samples have been made of 1 µm-sized particles that are packed together almost evenly with small holes (white triangular marks) in between. Like microstructure of the powder (Fig. 5a), those particles are made up of many smaller grains (hundreds in nm), which in turn are made of 16 nm long crystalline material. As mentioned above smaller grain sizes resulted in more grain boundaries that prevent the development of defects and led to enhanced mechanical strength in our samples. The ultimate strength and microhardness of cancellous human bone have been reported in the range of 1.5–9.3 MPa [79] and 0.46–0.49 GPa [80]. It implies that BAG (Table 2) could withstand loads imposed on them in bone treatments like bone grafts or BTE.
3.6 Biological properties
3.6.1 In vitro bioactivity and biodegradability
In vitro bioactivity is referred to the formation of apatite on the surface of materials, which are submerged in a medium. In fact, in vitro bioactivity test serves as a tool to predict the bonding of materials to the surrounding host tissue through CaP apatite formation during in vivo and clinical tests [81]. Different media types, sample geometry, and immersion conditions are factors that affect in vitro testing [72]. Herein, we used SBF with ionic concentration similar to the inorganic part of human blood plasma, proposed by Kokubo, as a well-known protocol used in many studies [82]. Figure 6 demonstrates the BAG sample before soaking in SBF and after soaking. There is no apatite-like material on the sample before soaking, which was confirmed by EDX where no P was detected in the chemical composition of the sample. However, apatite formation was observed on the surface of the BAG sample after 24 h of soaking. Tiny spherical agglomerated apatite particles started to cover the surface of the BAG sample after 1 day of soaking in SBF. The chemical composition of these particles contains CaP, which is the main component of apaptites. The texture of the formed apatite layer, which included loosely packed agglomerated particles on the first day, started to change after soaking in SBF for a longer time. On day 7 apatite ball-like particles were more like flake crystals than smooth particles. After two weeks of soaking in SBF, flake-like apatites covered the surface of the BAG sample extensively. The change in morphology is assigned to the crystallinity degree of formed apatites [83]. The amorphous apatites converted to more crystalline particles during immersion time that gave rise to those shapes.

SEM micrographs and EDX of BAG disk’s surface after immersion in SBF for different time periods
Changes in the concentrations of Ca, P, and Si elements in SBF, and pH of SBF filtrates were measured throughout the bioactivity test (Fig. 7). As soon as the BAG was immersed, the Ca and Si concentrations in the SBF solution raised sharply due to the hydrolyze of BAG in the medium (Fig. 7a, b). The rate of calcium ion diffusion was higher than Si ions in SBF. Immediately positively charged Ca2+ ions reacted with PO43− negative ions (available in SBF), which were attached to the sample’s surface, and formed amorphous calcium phosphates during the first day of soaking, which could be detected through a steep reduction of P concentration from SBF [84] (Fig. 7c). These calcium phosphates are more like carbonated HA, rather than stoichiometric ones [85]. Later, Si ions, released from BAG, made silanol groups on the surface of the sample that changed the surface’s charge to negative and caused further deposition of Ca ions on the surface [47]. The concentration of the Zr was less than 0.02 mg/l in SBF. However, the presence of Zr element in the EDX (Fig. 6) confirms that Zr–OH groups participated in apatite formation similar to Si–OH groups [52]. The EDX also indicated Si-rich apatites on disks. This cycle continued until day 14, where amorphous apatites eventually were converted to more stable crystalline apatites and the balance between precipitated and diffused ions among the disk’s surface and SBF solution was sustained. The pH changes of SBF filtrate as a function of incubation time (Fig. 7 d) indicated a steep increase in value during the first 4 days, where the pH of SBF increased from 7.4 to 8.6. The ionic exchange between H+ ions of SBF and Ca2+ and Zr4+ ions from the disk surface lies beneath this event. However, this parameter tended to stabilize after 14 days because the precipitate reduces ion exchange between SBF and sample [60]. Eventually, the apatite formation mechanism on the surface of BAG follows the nucleation and growth mechanism of an apatite-like layer on ceramic materials [86].

Element concentration profiles of a Ca, b Si, c P, and d pH changes of SBF filtrate during 14 days of incubation (n = 3) in the water bath at 37 °C; as well as e weight loss percentage (in vitro biodegradability) of BAG samples (n = 3) that incubated in acetic acid and sodium acetate buffer for 28 days at 37 °C
In vitro biodegradability of the BAG was investigated by measuring weight loss (n = 3) in acetic acid and sodium acetate buffer solution (AcOH–AcONa, pH = 5.5), which simulates osteoclastic resorption of bone [87]. The degradation rate of our samples increased from almost 1% at 1 day to a maximum value of 21% after about 1 month of soaking in the buffer at 37 °C (Fig. 7e). The slower degradation rate of samples during the initial days (<2% after 7 days) was followed by the faster rate throughout the 2nd, 3rd, and 4th weeks of immersion. A quadratic equation (Fig. 7e) could predict the degradation rate with an admissible accuracy.
Degradability, also referred to as dissolution rate, plays a major role in the further success of biomaterials during in vivo and clinical applications. Initially, an appropriate dissolution rate contributes to having adequate ions like Ca and Si, which leads to higher bioactivity of materials and enhanced cellular proliferation or differentiation [72]. Herein, the initial dissolution rate of our prepared BAG seems enough to trigger apatite formation on the surface of samples (Fig. 6). More importantly, maintaining the balance between the degradation rate of implanted samples and the healing process of bone is necessary to support the gradual transfer of loads from the implant to the newly formed bone and avoid any implant failure. Slower degradation of our BAG samples in the initial days coincides with the primary stage of implantation, which is accompanied by inflammation phenomenon in the patient’s body and the beginning of bone formation. Therefore, a synthetic implant tolerates loads and prevents excessive force on newly formed bone. Later, by the higher degradation rate of our samples during the 3rd and 4th weeks, a fraction of loads transfer on newly formed bone and that is essential for the natural growth of bone. Degradation of our BAG samples (~20 wt%) after 28 days was less than those analyzed by Sadeghzadeh et al. [68] with degradation of 51 wt% and was greater than samples of Roohani et al. [17] (~10 wt%) at the end of 28 days. Impressively, our results showed excellent compatibility with the appropriate strengthening of newly formed bone tissue and degradation percentages of implants in terms of time [88].
3.7 In vitro cell viability and osteogenic activity
Figure 8 represents the results of the MTT assay that was performed with the direct contact of various concentrations of material with Saos-2 cells to evaluate the cytocompatibility of BAG. A significant time-dependent increase in absorbance was observed for both the positive control group (P.C.) and BAG samples, which indicates that cells proliferated (Fig. 8a). Additionally, relative cell viabilities were calculated for BAG-containing groups, where the viability of cells in the control group was taken 100%. All BAG-containing groups exhibited relative cell viability greater than 80% with respect to the positive control group (TCPS without any BAG particles) after 24 and 48 h (Fig. 8b). Although there was no significant difference when the relative viability of samples was compared between 24 and 48, the confluency over 80% could be the reason for the slight decrease of cell viability in two groups with higher BAG particle concentrations (12 and 25 µg/ml) after 48-h incubation at 37 °C, compared to 24 h (supplementary optical microscope images are available). Our results confirmed that BAG particles had no cytotoxic effect on in vitro cultured Saos-2 cells. Ca, Si, and Zr ions, released from BAG, did play significant roles in cell spreading and proliferation [12, 89, 90]. For example, silica provides negatively charged silanol groups that could interact with functional groups to facilitate cell spreading and growth [91]. Similar results were reported in recent studies, where non-cytotoxic effect and improved cell attachment and viability have been observed in Saos-2 osteosarcoma cells [21], and MG6 osteoblast cells [22, 53].

a MTT assay results of Saos-2 cells after direct contact with various concentrations of BAG powder (µg/ml) for 24 and 48 h of incubation (n = 3); b relative cell viability of Saos-2 cells after direct contact with various concentrations of BAG powder (µg/ml) with respect to the control group without any BAG particles after 24 and 48 h of incubation (n = 3); c ALP activity (n = 3) and, d intracellular calcium ion deposition of cells incubated in osteogenic medium containing different concentrations of BAG particles (n = 3); e ALP activity of Saos-2 during 14 days of incubation in the osteogenic medium (without ascorbic acid and β-glycerophosphate) containing BAG particles (12 µg/ml) (n = 5). Note that Asterisk (*) indicates significant differences between the groups (p < 0.05), and there is no significant difference (p < 0.05) between groups at both 7 and 14 days for ALP and intracellular calcium deposition of cells in the osteogenic medium with the ascorbic acid and β-glycerophosphate
The alkaline phosphatase activity of Saos-2 cells in direct contact with different concentrations of BAG particles is depicted in Fig. 8c. Statistically, there was no difference (p < 0.05) among groups at both 7 and 14 days. The same pattern was observed for intracellular Ca ion deposition after 7 and 14 days (Fig. 8d). It seems that the presence of ascorbic acid and β-glycerophosphate in the osteogenic medium (OM) triggered osteogenic activity and mineralization of Saos-2 cells cultured in TCPS as a blank group [92]. A similar result was reported by Tsigkou et al. [93] when they tested the ALP activity of 45S5 Bioglass® (with the linear formula of CaO:SiO2:P2O5) in the presence of OM. We believe that the presence of ascorbic acid and β-glycerophosphate masks or affects the influence of BAG dissolution products. Furthermore, Zhang et al. [94] observed no difference in ALP activity between BAG and blank groups when the concentration was ≤ 6 µg/ml. Nevertheless, our result of ALP activity in the absence of ascorbic acid and β-glycerophosphate revealed that cells in wells with 12 µg/ml of BAG particles had higher osteogenic activity (ALP) than cells without any BAG material in their medium (Fig. 8e). Ionic products of BAG (Ca, Si, and Zr ions) and their influences on the activation of osteogenic-related gene expression have been reported to promote osteogenic activity in cells [15, 94]. This finding indicated that the osteogenic feature of BAG could contribute to its performance as a promising candidate for bone defect treatments.
4 Conclusion
In the present study, a modified sol–gel route was proposed to synthesize baghdadite (BAG) bioceramic, and various properties of produced BAG were comprehensively characterized to examine its suitability for bone defect treatments. Our proposed sol–gel approach led to the preparation of a transparent solution (sol), which indicates the complete dissolution of precursors and is essential for the quality of the final product, using optimum amounts of solvent (ethanol) and chelating agent (nitric acid), as well as dissolving Zr source (zirconium [IV] oxynitrate hydrate) separately. Physicochemical characterization results demonstrated that our prepared BAG had a highly crystalline structure and acceptable purity with minor impurities. The enhanced mechanical strength (~4 MPa), the biodegradation rate of ~20% after a month, as well as promoted bioactivity and osteoconductivity—due to the extensive formation of apatite on samples in SBF—were observed. Furthermore, in vitro cell culture tests revealed the non-cytotoxic feature and the promoted osteoinductive effect of our material in the absence of an osteogenic medium. Thus, our synthesized BAG satisfies the essential requirements, such as bioactivity, biodegradation rate, mechanical strength, biocompatibility, and having osteogenic effect to serve as a promising candidate for bone defect treatment applications.
References
G. Turnbull, J. Clarke, F. Picard, P. Riches, L. Jia, F. Han, B. Li, W. Shu, 3D bioactive composite scaffolds for bone tissue engineering. Bioact. Mater. 3, 278–314 (2018). https://doi.org/10.1016/j.bioactmat.2017.10.001
H. Jodati, B. Yılmaz, Z. Evis, A review of bioceramic porous scaffolds for hard tissue applications: Effects of structural features. Ceram. Int. 46, 15725–15739 (2020). https://doi.org/10.1016/j.ceramint.2020.03.192
P. Feng, P. Wei, P. Li, C. Gao, C. Shuai, S. Peng, Calcium silicate ceramic scaffolds toughened with hydroxyapatite whiskers for bone tissue engineering. Mater. Charact. 97, 47–56 (2014). https://doi.org/10.1016/J.MATCHAR.2014.08.017
Y. Zhu, M. Zhu, X. He, J. Zhang, C. Tao, Substitutions of strontium in mesoporous calcium silicate and their physicochemical and biological properties. Acta Biomater. 9, 6723–6731 (2013). https://doi.org/10.1016/J.ACTBIO.2013.01.021
A. Zheng, L. Cao, Y. Liu, J. Wu, D. Zeng, L. Hu, X. Zhang, X. Jiang, Biocompatible silk/calcium silicate/sodium alginate composite scaffolds for bone tissue engineering. Carbohydr. Polym. 199, 244–255 (2018). https://doi.org/10.1016/J.CARBPOL.2018.06.093
H. Mohammadi, M. Hafezi, N. Nezafati, S. Heasarki, A. Nadernezhad, S.M.H. Ghazanfari, M. Sepantafar, Bioinorganics in bioactive calcium silicate ceramics for bone tissue repair: Bioactivity and biological properties. J. Ceram. Sci. Technol. 5, 1–12 (2014). https://doi.org/10.4416/JCST2013-00027
G.C. Wang, Z.F. Lu, H. Zreiqat, Bioceramics for skeletal bone regeneration, in Bone substitute biomaterials. ed. by K. Mallick (Woodhead Publishing, Cambridge, 2014), pp.180–216
T. Luo, C. Wu, Y. Zhang, The in vivo osteogenesis of Mg or Zr-modified silicate-based bioceramic spheres. J. Biomed. Mater. Res. Part A 100, 2269–2277 (2012). https://doi.org/10.1002/jbm.a.34161
C. Wu, Y. Ramaswamy, A. Soeparto, H. Zreiqat, Incorporation of titanium into calcium silicate improved their chemical stability and biological properties. J. Biomed. Mater. Res. - Part A 86, 402–410 (2008). https://doi.org/10.1002/jbm.a.31623
C. Wu, Y. Ramaswamy, J. Chang, J. Woods, Y. Chen, H. Zreiqat, The effect of Zn contents on phase composition, chemical stability and cellular bioactivity in Zn-Ca-Si system ceramics. J Biomed. Mater. Res. Part B Appl. Biomater. 87, 346–353 (2008). https://doi.org/10.1002/jbm.b.31109
H.M. Al-Hermezi, D. McKie, A.J. Hall, Baghdadite, a new calcium zirconium silicate mineral from Iraq. Mineral. Mag. 50, 119–123 (1986). https://doi.org/10.1180/minmag.1986.050.355.15
Y. Ramaswamy, C. Wu, A. Van Hummel, V. Combes, G. Grau, H. Zreiqat, The responses of osteoblasts, osteoclasts and endothelial cells to zirconium modified calcium-silicate-based ceramic. Biomaterials 29, 4392–4402 (2008). https://doi.org/10.1016/J.BIOMATERIALS.2008.08.006
A. Arefpour, M. Kasiri-Asgarani, A. Monshi, A. Doostmohammadi, S. Karbasi, Fabrication, characterization and examination of in vitro of baghdadite nanoparticles for biomedical applications. Mater. Res. Express. 6, 095411 (2019). https://doi.org/10.1088/2053-1591/ab318c
H. Jodati, B. Yilmaz, Z. Evis, Calcium zirconium silicate (baghdadite) ceramic as a biomaterial. Ceram. Int. 46, 21902–21909 (2020). https://doi.org/10.1016/j.ceramint.2020.06.105
Z. Lu, G. Wang, I. Roohani-esfahani, Baghdadite ceramics modulate the cross talk between human adipose stem cells and osteoblasts. Tissue Eng. Part A 20, 992–1003 (2014). https://doi.org/10.1089/ten.tea.2013.0470
J.J. Li, A. Akey, C.R. Dunstan, M. Vielreicher, O. Friedrich, D.C. Bell, H. Zreiqat, Effects of material–tissue interactions on bone regeneration outcomes using baghdadite implants in a large animal model. Adv. Healthc. Mater. 7, 1–9 (2018). https://doi.org/10.1002/adhm.201800218
S.I. Roohani-Esfahani, C.R. Dunstan, B. Davies, S. Pearce, R. Williams, H. Zreiqat, Repairing a critical-sized bone defect with highly porous modified and unmodified baghdadite scaffolds. Acta Biomater. 8, 4162–4172 (2012). https://doi.org/10.1016/J.ACTBIO.2012.07.036
S. Sadeghzade, R. Emadi, F. Tavangarian, A. Doostmohammadi, In vitro evaluation of diopside/baghdadite bioceramic scaffolds modified by polycaprolactone fumarate polymer coating. Mater. Sci. Eng. C 106, 110176 (2020). https://doi.org/10.1016/j.msec.2019.110176
T.C. Schumacher, A. Aminian, E. Volkmann, H. Lührs, D. Zimnik, D. Pede, W. Wosniok, L. Treccani, K. Rezwan, Synthesis and mechanical evaluation of Sr-doped calcium-zirconium-silicate (baghdadite) and its impact on osteoblast cell proliferation and ALP activity. Biomed. Mater. 10, 055013 (2015). https://doi.org/10.1088/1748-6041/10/5/055013
Y.P. Fu, Y.H. Su, C.H. Lin, Comparison of microwave-induced combustion and solid-state reaction for synthesis of LiMn2−xCrxO4 powders and their electrochemical properties. Solid State Ionics 166, 137–146 (2004). https://doi.org/10.1016/J.SSI.2003.09.018
A. Arefpour, M. Kasiri-Asgarani, A. Monshi, S. Karbasi, A. Doostmohammadi, Baghdadite/Polycaprolactone nanocomposite scaffolds: preparation, characterisation, and in vitro biological responses of human osteoblast-like cells (Saos-2 cell line). Mater. Technol. 00, 1–12 (2019). https://doi.org/10.1080/10667857.2019.1692161
V. Abbasian, R. Emadi, M. Kharaziha, Biomimetic nylon 6-baghdadite nanocomposite scaffold for bone tissue engineering. Mater. Sci. Eng. C 109, 110549 (2020). https://doi.org/10.1016/j.msec.2019.110549
M.K. Ahmad, M. Rusop, Influence of glacial acetic acid and nitric acid as a chelating agent in sol-gel process to the nanostructured titanium dioxide thin films. AIP Conf. Proc. 1136, 339 (2009). https://doi.org/10.1063/1.3160160
A. Monshi, M.R. Foroughi, M.R. Monshi, Modified Scherrer equation to estimate more accurately nano-crystallite size using XRD. World J. Nano Sci. Eng. 02, 154–160 (2012). https://doi.org/10.4236/wjnse.2012.23020
Y. Kaya, H. Jodati, Z. Evis, Effects of biomimetic synthesis route and sintering temperature on physicochemical, microstructural, and mechanical properties of hydroxyapatite. J. Aust. Ceram. Soc. 57, 1117–1129 (2021). https://doi.org/10.1007/s41779-021-00609-x
Z. Evis, B. Yilmaz, M. Usta, S. LeventAktug, X-ray investigation of sintered cadmium doped hydroxyapatites. Ceram. Int. 39, 2359–2363 (2013). https://doi.org/10.1016/J.CERAMINT.2012.08.087
R. Moonesi Rad, D. Atila, E.E. Akgün, Z. Evis, D. Keskin, A. Tezcaner, Evaluation of human dental pulp stem cells behavior on a novel nanobiocomposite scaffold prepared for regenerative endodontics. Mater. Sci. Eng. C 100, 928–948 (2019). https://doi.org/10.1016/J.MSEC.2019.03.022
H. Kariem, M.-I. Pastrama, S.I. Roohani-Esfahani, P. Pivonka, H. Zreiqat, C. Hellmich, Micro-poro-elasticity of baghdadite-based bone tissue engineering scaffolds: A unifying approach based on ultrasonics, nanoindentation, and homogenization theory. Mater. Sci. Eng. C. 46, 553–564 (2015). https://doi.org/10.1016/J.MSEC.2014.10.072
ASTM Standard, D3967–08 Standard test method for splitting tensile strength of intact rock core specimens (West Conshohocken ASTM International, 2008).
ASTM International, E384–17 Standard test method for microindentation hardness of materials (ASTM International, West Conshohocken, 2017)
H. Jodati, B. Güner, Z. Evis, D. Keskin, A. Tezcaner, Synthesis and characterization of magnesium-lanthanum dual doped bioactive glasses. Ceram. Int. (2020). https://doi.org/10.1016/j.ceramint.2020.01.050
T. Kokubo, H. Takadama, Simulated body fluid (SBF) as a standard tool to test the bioactivity of implants. Handb. Biominer. Biol. Asp. Struct. Form. 3, 97–109 (2008). https://doi.org/10.1002/9783527619443.ch51
E. Murray, D. Provvedini, D. Curran, B. Catherwood, H. Sussman, S. Manolagas, Characterization of a human osteoblastic osteosarcoma cell line (SAOS-2) with high bone alkaline phosphatase activity. J. Bone Miner. Res. 2, 231–238 (1987). https://doi.org/10.1002/jbmr.5650020310
A.E. Pazarçeviren, A. Tezcaner, D. Keskin, S.T. Kolukısa, S. Sürdem, Z. Evis, Boron-doped biphasic hydroxyapatite/β-tricalcium phosphate for bone tissue engineering. Biol. Trace Elem. Res. 199, 968–980 (2020). https://doi.org/10.1007/s12011-020-02230-8
Y. Hu, J. Chen, T. Fan, Y. Zhang, Y. Zhao, X. Shi, Q. Zhang, Biomimetic mineralized hierarchical hybrid scaffolds based on in situ synthesis of nano-hydroxyapatite/chitosan/chondroitin sulfate/hyaluronic acid for bone tissue engineering. Colloids Surf. B Biointerfaces 157, 93–100 (2017). https://doi.org/10.1016/j.colsurfb.2017.05.059
A.Z. Alshemary, A. EnginPazarceviren, A. Tezcaner, Z. Evis, Fe3+/SeO42− dual doped nano hydroxyapatite: A novel material for biomedical applications. J. Biomed. Mater. Res. Part B Appl. Biomater. 106, 340–352 (2018). https://doi.org/10.1002/jbm.b.33838
A.E. Pazarçeviren, Z. Evis, D. Keskin, A. Tezcaner, Resorbable PCEC/gelatin-bismuth doped bioglass-graphene oxide bilayer membranes for guided bone regeneration. Biomed. Mater. 14, 035018 (2019). https://doi.org/10.1088/1748-605X/AB007B
A.E. Pazarçeviren, A. Tahmasebifar, A. Tezcaner, D. Keskin, Z. Evis, Investigation of bismuth doped bioglass/graphene oxide nanocomposites for bone tissue engineering. Ceram. Int. 44, 3791–3799 (2018). https://doi.org/10.1016/j.ceramint.2017.11.164
A. Doostmohammadi, Z. KarimzadehEsfahani, A. Ardeshirylajimi, Z. Rahmati Dehkordi, Zirconium modified calcium-silicate-based nanoceramics: An in vivo evaluation in a rabbit tibial defect model. Int. J. Appl. Ceram. Technol. 16, 431–437 (2019). https://doi.org/10.1111/ijac.13076
L.C.R. Quiroga, J.A.P. Balestieri, I. Ávila, Thermal behavior and kinetics assessment of ethanol/gasoline blends during combustion by thermogravimetric analysis. Appl. Therm. Eng. 115, 99–110 (2017). https://doi.org/10.1016/j.applthermaleng.2016.12.051
K. Lawson-wood, I. Robertson, Study of the decomposition of calcium oxalate monohydrate using a hyphenated thermogravimetric analyser - FT-IR system (TG-IR), PerkinElmer Inc. 1–3 (2016). https://resources.perkinelmer.com/lab-solutions/resources/docs/app-DecompositionCalcium-oxalate-monohydrate-013078-01.pdf
J.S. Alzahrani, I.H. Midala, H.M. Kamari, N.M. Al-Hada, C.K. Tim, N.N.S. Nidzam, Z.A. Alrowaili, M.S. Al-Buriahi, Effect of calcination temperature on the structural and optical properties of (ZnO)0.8 (ZrO2)0.2 nanoparticles. J. Inorg. Organomet. Polym. Mater. 32, 1755–1765 (2022). https://doi.org/10.1007/S10904-022-02238-8/FIGURES/9
A. Zhao, B. Xiong, Y. Han, H. Tong, Thermal decomposition paths of calcium nitrate tetrahydrate and calcium nitrite. Thermochim. Acta. 714, 179264 (2022). https://doi.org/10.1016/J.TCA.2022.179264
J.M. Coldrey, M.J. Purton, Application of thermal analysis in investigations on calcium silicate bricks. J. Appl. Chem. 18, 353–360 (2007). https://doi.org/10.1002/jctb.5010181203
R. Lakshmi, V. Velmurugan, S. Sasikumar, Preparation and phase evolution of Wollastonite by sol-gel combustion method using Sucrose as the fuel. Combust. Sci. Technol. 185, 1777–1785 (2013). https://doi.org/10.1080/00102202.2013.835308
S. Mehrafzoon, S.A. Hassanzadeh-Tabrizi, A. Bigham, Synthesis of nanoporous Baghdadite by a modified sol-gel method and its structural and controlled release properties. Ceram. Int. 44, 13951–13958 (2018). https://doi.org/10.1016/J.CERAMINT.2018.04.244
S. Sadeghzade, F. Shamoradi, R. Emadi, F. Tavangarian, Fabrication and characterization of baghdadite nanostructured scaffolds by space holder method. J. Mech. Behav. Biomed. Mater. 68, 1–7 (2017). https://doi.org/10.1016/J.JMBBM.2017.01.034
T.C. Schumacher, E. Volkmann, R. Yilmaz, A. Wolf, L. Treccani, K. Rezwan, Mechanical evaluation of calcium-zirconium-silicate (baghdadite) obtained by a direct solid-state synthesis route. J. Mech. Behav. Biomed. Mater. 34, 294–301 (2014). https://doi.org/10.1016/j.jmbbm.2014.02.021
M. Yurddaskal, E. Celik, Effect of halogen-free nanoparticles on the mechanical, structural, thermal and flame retardant properties of polymer matrix composite. Compos. Struct. 183, 381–388 (2018). https://doi.org/10.1016/J.COMPSTRUCT.2017.03.093
X. Chen, J. Yu, S. Guo, S. Lu, Z. Luo, M. He, Surface modification of magnesium hydroxide and its application in flame retardant polypropylene composites. J. Mater. Sci. 44, 1324–1332 (2009). https://doi.org/10.1007/S10853-009-3273-6/FIGURES/11
E. Spahiu, ATR-FTIR evaluation of structural and functional changes on murine macrophage cells upon activation and suppression by immuno-therapeutic oligodeoxynucleotides, Middle East Technical University (2015). https://etd.lib.metu.edu.tr/upload/12618753/index.pdf.
A. Najafinezhad, M. Abdellahi, H. Ghayour, A. Soheily, A. Chami, A. Khandan, A comparative study on the synthesis mechanism, bioactivity and mechanical properties of three silicate bioceramics. Mater. Sci. Eng. C 72, 259–267 (2017). https://doi.org/10.1016/J.MSEC.2016.11.084
H.R. Bakhsheshi-Rad, E. Hamzah, A.F. Ismail, M. Aziz, Z. Hadisi, M. Kashefian, A. Najafinezhad, Novel nanostructured baghdadite-vancomycin scaffolds: In-vitro drug release, antibacterial activity and biocompatibility. Mater. Lett. 209, 369–372 (2017). https://doi.org/10.1016/J.MATLET.2017.08.027
F. Pahlevanzadeh, H.R. Bakhsheshi-Rad, E. Hamzah, In-vitro biocompatibility, bioactivity, and mechanical strength of PMMA-PCL polymer containing fluorapatite and graphene oxide bone cements. J. Mech. Behav. Biomed. Mater. 82, 257–267 (2018). https://doi.org/10.1016/j.jmbbm.2018.03.016
C.J. Fu, Z.W. Zhan, M. Yu, S.M. Li, J.H. Liu, L. Dong, Influence of Zr/Si molar ratio on structure, morphology and corrosion resistance of organosilane coatings doped with zirconium(IV) n-propoxide. Int. J. Electrochem. Sci. 9, 2603–2619 (2014)
D.M. Pickup, G. Mountjoy, G.W. Wallidge, R.J. Newport, M.E. Smith, Structure of (ZrO2)(x)(SiO2)(1–x) xerogels (x =0.1, 0.2, 0.3 and 0.4) from FTIR, 29Si and 17O MAS NMR and EXAFS. Phys. Chem. Chem. Phys. 1, 2527–2533 (1999). https://doi.org/10.1039/a901401b
A. Khandan, E. Karamian, M. Mehdikhani-Nahrkhalaji, H. Mirmohammadi, A. Farzadi, N. Ozada, B. Heidarshenas, K. Zamani, Influence of spark plasma sintering and baghdadite powder on mechanical properties of hydroxyapatite. Procedia Mater. Sci. 11, 183–189 (2015). https://doi.org/10.1016/J.MSPRO.2015.11.087
Y.L. Lee, W.H. Wang, F.H. Lin, C.P. Lin, Hydration behaviors of calcium silicate-based biomaterials. J. Formos. Med. Assoc. 116, 424–431 (2017). https://doi.org/10.1016/J.JFMA.2016.07.009
S. Arya, P. Mahajan, S. Mahajan, A. Khosla, R. Datt, V. Gupta, S.-J. Young, S.K. Oruganti, Review—Influence of processing parameters to control morphology and optical properties of Sol-Gel synthesized ZnO nanoparticles. ECS J. Solid State Sci. Technol. 10, 023002 (2021). https://doi.org/10.1149/2162-8777/ABE095
S. Sadeghpour, A. Amirjani, M. Hafezi, A. Zamanian, Fabrication of a novel nanostructured calcium zirconium silicate scaffolds prepared by a freeze-casting method for bone tissue engineering. Ceram. Int. 40, 16107–16114 (2014). https://doi.org/10.1016/J.CERAMINT.2014.07.039
O.Y. Zadorozhnaya, T.A. Khabas, O.V. Tiunova, S.E. Malykhin, Effect of grain size and amount of zirconia on the physical and mechanical properties and the wear resistance of zirconia-toughened alumina. Ceram. Int. 46, 9263–9270 (2019). https://doi.org/10.1016/j.ceramint.2019.12.180
A. Averardi, C. Cola, S.E. Zeltmann, N. Gupta, Effect of particle size distribution on the packing of powder beds: A critical discussion relevant to additive manufacturing. Mater. Today Commun. 24, 100964 (2020). https://doi.org/10.1016/J.MTCOMM.2020.100964
Z. Evis, Reactions in hydroxylapatite–zirconia composites. Ceram. Int. 33, 987–991 (2007). https://doi.org/10.1016/J.CERAMINT.2006.02.012
M. Martín-Garrido, M. Teresa Molina-Delgado, S. Martínez-Ramírez, A comparison between experimental and theoretical Ca/Si ratios in C-S–H and C–S(A)–H gels. J. Sol-Gel Sci. Technol. 94, 11–21 (2020). https://doi.org/10.1007/S10971-019-05097-X/FIGURES/5
A.J. Wagoner Johnson, B.A. Herschler, A review of the mechanical behavior of CaP and CaP/polymer composites for applications in bone replacement and repair. Acta Biomater. 7, 16–30 (2011). https://doi.org/10.1016/J.ACTBIO.2010.07.012
B. Ranjkesh, F. Isidor, M. Dalstra, H. Løvschall, Diametral tensile strength of novel fast-setting calcium silicate cement. Dent. Mater. J. 35, 559–563 (2016). https://doi.org/10.4012/DMJ.2015-390
S. Kunjalukkal Padmanabhan, F. Gervaso, M. Carrozzo, F. Scalera, A. Sannino, A. Licciulli, Wollastonite/hydroxyapatite scaffolds with improved mechanical, bioactive and biodegradable properties for bone tissue engineering. Ceram. Int. 39, 619–627 (2013). https://doi.org/10.1016/j.ceramint.2012.06.073
S. Sadeghzade, R. Emadi, T. Ahmadi, F. Tavangarian, Synthesis, characterization and strengthening mechanism of modified and unmodified porous diopside/baghdadite scaffolds. Mater. Chem. Phys. 228, 89–97 (2019). https://doi.org/10.1016/j.matchemphys.2019.02.041
D.Q. Pham, C.C. Berndt, U. Gbureck, H. Zreiqat, V.K. Truong, A.S.M. Ang, Mechanical and chemical properties of Baghdadite coatings manufactured by atmospheric plasma spraying. Surf. Coat. Technol. 378, 124945 (2019). https://doi.org/10.1016/j.surfcoat.2019.124945
D.A. Samani, A. Doostmohammadi, M.R. Nilforoushan, H. Nazari, Electrospun polycaprolactone/graphene/baghdadite composite nanofibres with improved mechanical and biological properties. Fibers Polym. 20, 982–990 (2019). https://doi.org/10.1007/s12221-019-1161-5
Y.J. No, I. Holzmeister, Z. Lu, S. Prajapati, J. Shi, U. Gbureck, H. Zreiqat, Effect of baghdadite substitution on the physicochemical properties of brushite cements. Materials (Basel) 12, 1–15 (2019). https://doi.org/10.3390/MA12101719
S. Punj, J. Singh, K. Singh, Ceramic biomaterials: Properties, state of the art and future prospectives. Ceram. Int. 47, 28059–28074 (2021). https://doi.org/10.1016/J.CERAMINT.2021.06.238
F.P. Knudsen, Dependence of mechanical strength of Brittle polycrystalline specimens on porosity and grain size. J. Am. Ceram. Soc. 42, 376–387 (1959). https://doi.org/10.1111/J.1151-2916.1959.TB13596.X
G. Zhao, C. Huang, H. Liu, B. Zou, H. Zhu, J. Wang, Preparation of in-situ growth TaC whiskers toughening Al2O3 ceramic matrix composite. Int. J. Refract. Met. Hard Mater. 36, 122–125 (2013). https://doi.org/10.1016/J.IJRMHM.2012.08.003
D. Basu, A.N. Banerjee, Dynamic mechanical thermal analysis - crystallinity and tensile-strength of PBT/ABS blends: their interdependency and variations with ABS content, Indian. J. Chem. Technol. 1, 31–34 (1994)
Y.C. Tsui, C. Doyle, T.W. Clyne, Plasma sprayed hydroxyapatite coatings on titanium substrates. Part 2: Optimisation of coating properties. Biomaterials 19, 2031–2043 (1998). https://doi.org/10.1016/S0142-9612(98)00104-5
Y. Liang, Y. Xie, H. Ji, L. Huang, X. Zheng, Excellent stability of plasma-sprayed bioactive Ca3ZrSi2O9 ceramic coating on Ti–6Al–4V. Appl. Surf. Sci. 256, 4677–4681 (2010). https://doi.org/10.1016/J.APSUSC.2010.02.071
M. Naghizadeh, H. Mirzadeh, Effects of grain size on mechanical properties and work-hardening behavior of AISI 304 austenitic stainless steel. Steel Res. Int. 90, 1–9 (2019). https://doi.org/10.1002/srin.201900153
Y.H. An, R.A. Draughn (eds.), Mechanical testing of bone and the bone–implant interface, 1st edn. (CRC Press, Boca Raton, 2000)
G. Boivin, Y. Bala, A. Doublier, D. Farlay, L.G. Ste-Marie, P.J. Meunier, P.D. Delmas, The role of mineralization and organic matrix in the microhardness of bone tissue from controls and osteoporotic patients. Bone 43, 532–538 (2008). https://doi.org/10.1016/j.bone.2008.05.024
T. Kokubo, H. Takadama, How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 27, 2907–2915 (2006). https://doi.org/10.1016/j.biomaterials.2006.01.017
B. Yilmaz, A.E. Pazarceviren, A. Tezcaner, Z. Evis, Historical development of simulated body fluids used in biomedical applications: A review. Microchem. J. 155, 104713 (2020). https://doi.org/10.1016/j.microc.2020.104713
P. Siriphannon, Y. Kameshima, A. Yasumori, K. Okada, S. Hayashi, Formation of hydroxyapatite on CaSiO3 powders in simulated body fluid. J. Eur. Ceram. Soc. 22, 511–520 (2002). https://doi.org/10.1016/S0955-2219(01)00301-6
X. Liu, S. Tao, C. Ding, Bioactivity of plasma sprayed dicalcium silicate coatings. Biomaterials 23, 963–968 (2002). https://doi.org/10.1016/S0142-9612(01)00210-1
W.C. Lepry, S. Naseri, S.N. Nazhat, Effect of processing parameters on textural and bioactive properties of sol–gel-derived borate glasses. J. Mater. Sci. 52, 8973–8985 (2017). https://doi.org/10.1007/s10853-017-0968-y
X. Liu, C. Ding, Morphology of apatite formed on surface of wollastonite coating soaked in simulate body fluid. Mater. Lett. 57, 652–655 (2002). https://doi.org/10.1016/S0167-577X(02)00848-0
A. Ito, K. Senda, Y. Sogo, A. Oyane, A. Yamazaki, R.Z. LeGeros, Dissolution rate of zinc-containing β-tricalcium phosphate ceramics. Biomed. Mater. 1, 134–139 (2006). https://doi.org/10.1088/1748-6041/1/3/007
G. Chandra, A. Pandey, Biodegradable bone implants in orthopedic applications: a review. Biocybern. Biomed. Eng. (2020). https://doi.org/10.1016/j.bbe.2020.02.003
P.J. Marie, The calcium-sensing receptor in bone cells: A potential therapeutic target in osteoporosis. Bone 46, 571–576 (2010). https://doi.org/10.1016/j.bone.2009.07.082
J.-Y. Sun, T.-S. Yang, J. Zhong, D.C. Greenspan, The effect of the ionic products of Bioglass® dissolution on human osteoblasts growth cycle in vitro Jun-Ying. J. Tissue Eng. Regen. Med. 1, 281–286 (2007). https://doi.org/10.1002/term
M. Diba, M. Kharaziha, M.H. Fathi, M. Gholipourmalekabadi, A. Samadikuchaksaraei, Preparation and characterization of polycaprolactone/forsterite nanocomposite porous scaffolds designed for bone tissue regeneration. Compos. Sci. Technol. 72, 716–723 (2012). https://doi.org/10.1016/J.COMPSCITECH.2012.01.023
F. Langencach, J. Handschel, Effects of dexamethasone, ascorbic acid and β-glycerophosphate on the osteogenic differentiation of stem cells in vitro. Stem Cell Res. Ther. 4, 117–123 (2013). https://doi.org/10.1089/ten.teb.2011.0199
O. Tsigkou, J.R. Jones, J.M. Polak, M.M. Stevens, Differentiation of fetal osteoblasts and formation of mineralized bone nodules by 45S5 Bioglass® conditioned medium in the absence of osteogenic supplements. Biomaterials 30, 3542–3550 (2009). https://doi.org/10.1016/j.biomaterials.2009.03.019
X. Zhang, P. Han, A. Jaiprakash, C. Wu, Y. Xiao, A stimulatory effect of Ca3ZrSi2O9 bioceramics on cementogenic/osteogenic differentiation of periodontal ligament cells. J. Mater. Chem. B 2, 1415–1423 (2014). https://doi.org/10.1039/c3tb21663b
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they did not receive any funds, grants, or other support from any organization and have no competing interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jodati, H., Tezcaner, A., Evis, Z. et al. Synthesis of baghdadite using modified sol–gel route and investigation of its properties for bone treatment applications. J. Korean Ceram. Soc. 60, 381–398 (2023). https://doi.org/10.1007/s43207-022-00275-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43207-022-00275-0