
Abstract
Fabrication and characterization of polymer infiltrated ceramic networks (PICNs) were undertaken for use in dentistry. Calcia-stabilized tetragonal zirconia (7-tCSZ) nanoparticles were synthesized via a modified co-precipitation. The composition and particle size of the prepared nanoparticles were studied using X-ray diffraction (XRD) and transmission electron microscopy (TEM). Densification parameters of porous ceramic samples, with varying amounts of 7-tCSZ, 0, 5, 10, and 15 wt.%, and PICN materials were examined using the Archimedes principle. The flexural strength and elastic modulus of PICNs were measured using a three-point bending strength test. The microhardness and fracture toughness were determined using Vickers microhardness and V-notched bars tests, respectively. The microstructure was investigated for selected materials before and after polymer infiltration using scanning electron microscopy (SEM). The results revealed that the samples with 10 and 15 wt.% of nano-tetragonal zirconia showed the highest elastic modulus (8.24 GPa) and fracture toughness (1.82 MPa m1/2), respectively, which is attributed to transformation toughening of zirconia. The flexural strength and microhardness of PICNs were in the range 66.14–71.72 MPa, 0.18–0.19 GPa, respectively, whilst the brittleness index was maximally 0.21 μm−½, which is significantly below the ultimate brittleness index (4.3 μm−1/2) allowing machinability. Conclusively, the inclusion of nanometric zirconia posed a dramatic enhancement of the mechanical properties without affecting the machinability of these biomimetic materials, making them similar to natural dentin in terms of the elastic modulus and fracture toughness along with ease of machinability, which renders them promising as indirect dental restorative materials.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
There are many dental ceramics and a great selection of resin-based composites (RBCs) in the market. Dental ceramics are stiffer and harder than natural tooth structures [1], whereas RBCs show lower values [2]. These factors reveal the need for a material that brings together the advantages of ceramics with resin composites with special attention to avoidance of natural tooth wear. This makes it essential for ideal material with enhanced performance to have elastic modulus close to dentin and hardness between enamel and dentin.
There is a limitation on the extensive industrial application of most advanced ceramic materials because of their poor fracture toughness [3]. On the other hand, tetragonal zirconia polycrystals (TZP) exhibit superior fracture toughness to most ceramic materials, in particular, yttria-stabilized zirconia polycrystals (7.5–15.5 MPa m1/2) [4, 5].
Principally, nano-based structures are defined as materials into which their size scale is less than 100 nm. The enhanced properties of these materials in terms of the physical, chemical, mechanical, and functional parameters compared to other traditional ones with a higher size scale inspired tremendous research to focus on their synthesis and characterization, particularly nanocrystalline ceramics. It has been acknowledged that the conversion from microstructural length scale to nanometer level can bring about a profound impact on the mechanical and physical properties of materials [6]. On the microscopic level, nano-grain sizes lead to most atoms near the grain boundary regions [7]. The reduction in grain sizes is expected to cause a considerable enlarge in mechanical properties [7, 8].
In recent years, zirconia became one of the most significant ceramic materials as a result of its attractive properties, which are strongly related to its crystalline polymorphs. Zirconia stabilized in tetragonal form is appropriate for use as structural applications in dentistry owing to its high fracture toughness and strength [5]. In addition, the production of partially stabilized zirconia (PSZ) in nanosized grains leads to more translucent, higher strength dental ceramics with more resistance to low-temperature degradation [9, 10]. The synthesis of PSZ nano-crystals was accomplished via different routes [11,12,13,14]. Among them, the co-precipitation method received great concern as it can be scaled up using low-cost accessories [15, 16].
Polymer-infiltrated ceramic networks (PICNs) materials are graded into phase interpenetrating composites with two connecting phases. PICNs materials possess dual structures, both of which have properties related to ceramics, long-lasting esthetics, and polymers for simulation of dentin from the viewpoint of elastic modulus [17, 18]. The PICN materials consist of a sintered ceramic matrix structure (71 vol.%) infiltrated with a polymer matrix (29 vol.%) [19]. Such a class of materials is already known on the commercial level as Vita Enamic CAD/CAM blocks. The indications are minimally invasive restorations and posterior crowns, veneers, inlays, and onlays for posterior teeth [20, 21]. Earlier studies on the fabrication of experimental PICN materials paid attention to the effect of different sintering temperatures on the mechanical properties [17, 18, 20,21,22]. None of them inspected the consequence of the inclusion of different percentages of calcia-stabilized tetragonal zirconia as a toughening component of the ceramic network and its effect on the mechanical properties of PICNs. Moreover, there was a recommendation from a previous report [22] that this class of materials is still in need of further substantive research.
The present work aims to synthesize tetragonal zirconia (t-ZrO2) nanoparticles via a modified co-precipitation method with different amounts of calcia (0, 3, 7, and 12wt.%). Additionally, varying amounts of the tetragonal zirconia nanoparticles (0, 5, 10, and 15 wt.%) on the mechanical properties and microstructure of the PICN materials were investigated.
2 Materials and methods
2.1 Synthesis of calcia-stabilized tetragonal zirconia (tCSZ)nanoparticles
The t-ZrO2 nanoparticles were prepared by a modified co-precipitation method [12, 16, 23], which was partially stabilized by different percentages of calcium oxide. CaO amounts equivalent to ~ 0, 3, 7, and 12 wt.% were prepared by dissolving different concentrations of calcium chloride dihydrate (CaCl2.2H2O) in distilled water and used as oxide stabilizer to the partially stabilized tetragonal phase of zirconia nanoparticles. Zirconyl oxychloride octahydrate (ZrOCl2.8H2O) was used as the source of zirconia. Solutions of ZrOCl2.8H2O and CaCl2.2H2O (equivalent to CaO ~ 0, 3, 7, and 12 wt.%) were prepared by dissolving in distilled water. Zirconium and calcium-containing solutions were then mixed under continuous stirring on a hot plate stirrer at room temperature for 30 min. After that, ammonia solution (25% NH4OH) was added drop-wise into the stirring solution using a graduated burette until the pH reached and remained at 10.5. The stirring was continued for an additional 2 h at this pH. Then, the formed precipitate was left to settle down over ~ 24 h, filtered and washed with a mixture of ethanol (99%) and distilled water (30:70) while stirring for 30 min. The washing procedure was repeated twice. Then, the washed precipitate was dried at 120 °C for 24 h in a drying oven. Finally, the powder was divided into two parts; one was calcined at temperature 500 °C and the other at 700 °C using an electric furnace at a heating rate of 10 °C/min and 1 h soaking time. The obtained powders were characterized by X-ray diffraction (XRD) and transmission electron microscopy (TEM). Chemicals that were used in the preparation of t-ZrO2 are shown in Table 1.
2.2 Preparation of porous sintered ceramics
Sodium aluminum silicate/ calcia stabilized zirconia (7-tCSZ) compositions with the porous structure were prepared by mixing sodium aluminum silicate powder (82%SiO2, 9.5%Al2O3, and 8% Na2O) with different amounts of the 7-tCSZ nanoparticles (0, 5, 10 and 15 wt.%). It was added 15 wt.% at the expense of aluminum silicate content with a constant increment of 5 wt.% (0, 5, 10, and 15; hence, such samples are named Z0, Z5, Z10, and Z15). The aqueous solution of polyvinyl alcohol (PVA) (3 wt.%) was employed as a binder. 1 ml of the PVA solution was added for each powder mixture (3 g). After ensuring good mixing, the mixed powders were uniaxially pressed at 157.79 MPa for 1.5 min followed by drying at 120 °C for 24 h then sintered at 700 °C in an electric furnace with a heating rate of 5 °C/min until 525 °C then soaked for 30 min and lastly, the temperature was raised to 700 °C with the same heating rate followed by soaking for 1 h.
2.3 Fabrication of PICN materials
Tri(ethyleneglycol) dimethacrylate (TEGDMA) (Sigma-Aldrich Chemical Co., USA, STBH2136) and Bisphenol A glycerolate dimethacrylate (Bis-GMA) (Sigma Aldrich Chemical Co., USA, MKCD8912) were consistently mixed at a ratio of 1:1 as the polymer matrix. Benzoyl peroxide (BPO) (Sigma Aldrich Chemical Co., USA, MKCD4518) as initiator was mixed first with TEGDMA at a mass ratio of 1 wt.%, then Bis-GMA was added and mixed. Sodium aluminum silicate /tetragonal zirconia (7-tCSZ) ceramic networks were immersed in the monomers mixture for infiltration. The liquid was infiltrated into the porous ceramic networks by capillary action at room temperature for 24 h in dark containers. Then, PICN composites were obtained after the polymerization is triggered by heat treatment at 70 °C for 2 h and 110 °C for 2 h under the atmospheric pressure. The materials of PICN materials, incorporating different loadings of 7-tCSZ nanoparticles (0, 5, 10, and 15wt.%), are termed PZ0, PZ5, PZ10, and PZ15, respectively.
2.4 X-ray diffraction (XRD) analysis
The phase composition and average crystallite size of the synthesized nanoparticles and sintered ceramic networks were investigated using X-ray diffractometer (Model Bruker D8 Advance (Bruker AXS, Madison, WI, USA), Cu Kα1 (λ = 1.54058 Å) radiation with secondary monochromator at a scanning speed of 1°/min) and (Phillips X’Pert PW 3710, The Netherlands)operating at room temperature with Cu (Kα) radiations of wavelength (λ = 1.5406 Å), generated at 30 kV & 30 mA respectively. The obtained XRD patterns were compared with model patterns on the Joint Committee on Powder Diffraction Standard (JCPDS) database. Crystallite size of powder samples was calculated from broadening of XRD peaks based on the Scherrer equation: D = kλ /(β Cosθ) [24], where D is crystallite size, k is the dimensionless shape factor which has a typical value of about (0.94), λ is the wavelength of the X-ray radiation (λ = 1.5406 Å), β is FWHM (the observed full width at half maximum); θ is the Bragg angle.
2.5 Transmission electron microscopy (TEM)
Transmission electron microscopy (TEM) (TEM; JEOL JEM-2100, Tokyo, Japan) was used to determine the particle size and examine the morphology of prepared nanoparticles.
2.6 Densification parameters
Bulk density (BD) and apparent porosity (AP) of the sintered ceramic samples (Z0-Z15) and PICN materials (PZ0-PZ15) were determined by Archimedes method (where n = 5) according to ASTM C373-88:2006 using Eqs. 1, 2.
where ρ is bulk density, P is apparent porosity, D is dry mass, M is saturated mass, S is the mass after immersion in water.
2.7 Scanning electron microscopy (SEM)
The microstructure of sintered ceramic composites (Z10 and PZ10) was examined before and after infiltrated by the polymer using scanning electron microscopy (Quanta, FEG 250, Japan).
2.8 The polymer contents in different PICNs after monomer infiltration and polymerization
The polymer content in PICNs can be referred to as the amount of polymer added to the ceramic material via infiltration of monomer first into the pores of ceramic networks by capillary action at room temperature, followed by in situ polymerization at elevated temperature to yield PICN materials. Thus, it can be calculated simply by applying the relation: polymer percentage (wt.%) = [(WT − Wo) / WT] × 100, where; WT: weight of the PICN materials and Wo is original weight of sintered porous ceramic networks.
2.9 Mechanical properties
Testing bars were cut out of the cured discs (PZ0, PZ5, PZ10, and PZ15). Bars with dimensions of 2.2 × 2.2 × 15mm3 and 4.2 × 3.2 × 18 mm3 were cut from the above mentioned cured discs using an appropriate machine (IsoMet 4000 Buehler Germany) for flexural strength and fracture toughness tests, respectively. Concerning Vickers microhardness, the fractured parts of the flexural strength test were tested.
2.9.1 Evaluation of flexural strength
Five bars with dimensions 2.2 × 2.2 × 15 mm3 (n = 5) were prepared from each PICN material; PZ0, PZ5, PZ10, and PZ15. Thus, the samples were finished with abrasive paper to the final dimensions; 2 × 2 × 15mm3 then subjected to polishing using coarse and fine (TexMetTMP 6 μm and MicroClothTM 1μm, Buehler Ltd. Illinois, USA) wet polishing clothes sequentially, followed by fine polishing paste; (MetaDi Ultra1um Buehler Ltd. Illinois, USA) until no visible scratches could be detected on the surface. The flexural strength (σf) was calculated from Eq. 3 by employing a three-point bending test [25] using the Instron universal testing machine (Model 3345- England):
where F is the maximum load at the point of fracture; l is the distance of the roller span; b is the width, and h is the height of the specimen. The supporting span was 10 mm, and the crosshead speed was 0.5 mm/min.
2.9.2 Determination of the elastic modulus
The elastic modulus, Ef, was calculated from the results of three-point bending tests [20, 26] using Eq. 4:
where l is the distance of the roller span; b is the width, and h is the height of the specimen; (F/m) is the slope of the force–displacement curve.
2.9.3 Measurement of fracture toughness
Fracture toughness (KIC) was investigated using the V-notched beam method. Five bars with dimensions of 4.2 × 3.2 × 18 mm3 (n = 5) were sanded with abrasive paper to the final dimensions; of 4 × 3 × 18 mm3 for fracture toughness testing. Then, they were polished as undertaken in case of flexural strength testing. After that, a notch with a length of 1.3 mm through the width 4 mm was sawed in the middle of each bar by the aid of stainless steel mold containing V-notch depth 1.3 mm using a safe-sided diamond disc with a low speed cutting machine for standardization. For the sharpness of the V-notch tip, a razor blade sprinkled with diamond paste (3 μm) was used in back and forth motion with some impact. The samples were cleaned in a sonic (BandelinSonorex, Bandelin, Berlin, Germany) containing ethanol for 5 min. The bar was centralized on the supporting roller of three-point flexure in a universal test machine so that the surface having notch was subjected to tension mode. The distance (So) between the center of the roller was 16 mm at a crosshead speed of 0.5 mm/min till failure. The V-notch depths were measured from two faces of the sample using a stereomicroscope (Nikon MA 100, Japan) with 50 × magnification then averaged. Equations 5 and 6 were used to calculate the KIC (MPa m1/2) [19, 27], Fig. 1.

Digital image showing v-notched sample loaded on a universal testing machine for evaluation of the fracture toughness
where g = g(α/w),
Pmax is the load to failure (N), and B is the sample thickness (m), W is the sample width, α is the average value of V-notch depths, and relative α/W was approximately 0.3.
2.9.4 Vickers microhardness
The surface microhardness (MPa) of the samples, PZ0, PZ5, PZ10, and PZ15 (where n = 5) was determined using digital display Vickers microhardness tester (TUCON1102 Wilson hardness tester, Buehler, Germany). The load (200 gf) is applied smoothly, without impact, forcing the indenter into the test specimen. The indenter is held in place for dwell time 20 s. Three indentations were equally placed on the surface of each specimen, not closer than 0.5 mm to each other. The Vickers microhardness (HV) was calculated using Eq. 7 [27, 28]:
P is the force acting on the indenter in kilograms-force, and d is the diagonal of the impression in millimeters.
2.9.5 Brittleness index
The ease of machinability can be estimated quantitatively from the brittleness index. It was used for each specimen using the ratio between values of Vickers hardness to fracture toughness, as demonstrated in Eq. 8 [29, 30].
BI is the brittleness index in μm−1/2, Hv is the hardness in GPa and K1c is the fracture toughness in MPa m1/2.
Their concept considers the basic idea that brittleness measures the relative susceptibility to two competing mechanical responses, deformation and fracture. While the former is being measured by the hardness and the latter by the fracture toughness.
2.10 Statistical analysis
One-way ANOVA followed by Tukey post hoc test was used to compare the data between different samples; PZ0, PZ5, PZ10, and PZ15 in the same test; flexural strength, elastic modulus, microhardness, fracture toughness, and the brittleness index. Pearson test was used to examine the correlation between different tests. The significance level was set at P ≤ 0.05. Statistical analysis was performed with IBM® SPSS® Statistics Version 20 for Windows.
3 Results and discussion
3.1 Synthesized tetragonal zirconia nanoparticles
3.1.1 X-ray diffraction (XRD)
Figure 2a and b show the XRD patterns of the synthesized zirconia nanoparticles calcined at 500 °C and 700 °C for 1 h. At 500 °C, all the firm peaks appeared at 2θ = 30.2°, 35.1°, 50.2°, 60.2o can be indexed to the crystal planes of t-ZrO2 structure, namely, (101), (110), (112), (211), respectively, according to the JCPDS card No. 079-1771 as shown in (Fig. 2a). Besides, no other phases were detected in patterns, which indicate the high purity of the synthesized tetragonal zirconia nanoparticles. Zirconia exhibits three polymorphs phases depending on temperature: monoclinic (m) phase up to 1197 °C, tetragonal (t) phase to 2300 °C, and beyond this cubic (c) phase. The monoclinic phase of zirconia is a stable phase at room temperature. However, in the current study, the tetragonal zirconia was the only detected phase for all obtained XRD patterns at 500 °C, due to the tiny crystallite size of the synthesized zirconia nanoparticles. The average crystallite size for four nanoparticles (0, 3, 7, and 12wt.% CaO) was 8.3 nm, 10.65 nm, 8.2 nm, 4.4 nm, respectively. In the case of the sample prepared without any CaO (0wt.%CaO), calcined at 700 °C in (Fig. 2b), new diffraction peaks of monoclinic zirconia were detected at 2θ = 24.05°, 24.45°, 28.17°, 31.46°, 34.14°, 41.1°, in addition to weak diffraction peaks of tetragonal zirconia. The new crystalline monoclinic phase of zirconia might be justified by the crystal growth occurring with increasing the calcination temperature. It is known that the stability of t-ZrO2 is a function of the particle size of zirconia [31]. For the samples with 3, 7 and 12 wt.% of CaO, only the tetragonal phase was detected at 2θ = 30.2°, 35.3°, 50.5°, and 60.3o which are indexed to the crystal plane of tetragonal ZrO2;(101), (110), (112) and (211) respectively. This is due to the effect of CaO addition, which stabilized the tetragonal phase of zirconia at 700 °C. No secondary phase of monoclinic zirconia was detected in the patterns, which indicates the purity of synthesized tetragonal zirconia. Accordingly, a small addition of a stabilizer to pure zirconia will partially stabilized its structure into a tetragonal phase. Usually, such partially stabilized zirconia (tetragonal phase) consists of more than 2.77 wt.% MgO, 3.81 wt.% CaO, or 5.4–7.1 wt.% Y2O3. On the other side, zirconia needs the addition of more than 7.9 wt.% CaO, 5.86 wt.% MgO, or 13.75 wt.%Y2O3 to be stabilized in cubic phase [32]. Based on that, it can be deduced that 7 wt.% of CaO (7-tCSZ) is sufficient to stabilize t-ZrO2 nanoparticles at 700 °C. Consequently, the partially stabilized zirconia becomes toughened material via a transformation [32]. The peak intensities of XRD patterns increased progressively as the temperature of calcination increased from 500 °C to 700 °C, owing to the grain growth. The average crystallite size of four nanoparticle samples (0, 3, 7, and 12 wt.%) prepared by calcination at 700 °C was found to be in the range of 18.3 nm, 16.3 nm, 10.12 nm, and 8.6 nm, respectively.

a XRD patterns of the synthesized nanoparticles with different percentages of CaO (0, 3, 7, 12 wt.%) after calcination at 500; b XRD patterns of the synthesized nanoparticles with different percentages of CaO (0, 3, 7, 12 wt.%) after calcination at 700 °C
3.1.2 Investigation with transmission electron microscopy (TEM)
The surface morphology and particle sizes of the prepared 7-tCSZ nanoparticles calcined at 700 °C were examined by TEM, as displayed in (Fig. 3a–d). Figure 3a–c reveals that 7-tCSZ nanoparticles are spherical in shape, uniform in size, and highly crystalline. Moreover, the particle size can be estimated in the range 5.47–14.22 nm, which is in line with the size determined through XRD analysis; 10.12 nm. The high-resolution TEM image (HR-TEM) image of 7-tCSZ nanoparticles presented in selected area electron diffraction (SAED), as shown in (Fig. 3d) clarifies the presence of well-organized lattices fringe arrangement, which is an indication of the high degree of crystallinity of t-ZrO2 nanoparticles.

a–c HR-TEM images of 7-tCSZ nanoparticles at different magnifications; d SAED pattern
3.2 Tetragonal zirconia-based porous ceramics
3.2.1 X-ray diffraction
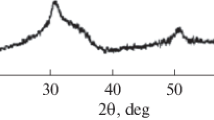
XRD patterns of Z0 and Z10 (10wt.% of 7-tCSZ nanoparticles) are exhibited in (Fig. 4a, b). For Z0, the XRD pattern signified the amorphous structure of the silicate structure, represented by a broad peak between 2θ = 20o and 30° [33], Fig. 4a, without the appearance of any sign of crystallinity. Such a broad peak of amorphous silicate is related to the high amount of silica (82% SiO2) in the sodium aluminum silicate powder and low sintering temperature (700 °C). On the other hand, the XRD pattern of Z10 showed a broad peak between 2θ = 20° and 30°, indicating the amorphous structure of silicate, in addition to sharp diffraction peaks at 2θ = 29.9°, 34.7°, 50.2°, 59.8oassociated with the crystal plane of tetragonal ZrO2;(101), (110), (112) and (211). As shown in (Fig. 4b), no monoclinic phase was observed in the XRD pattern.

XRD patterns of Z0 (a) and Z10 (b) sintered at 700 °C
3.2.2 Densification parameters
The results of bulk density (g/cm3) and apparent porosity (%) of the sintered samples are shown in (Fig. 5). It is clear that the values of bulk density (g/cm3) increased significantly with increasing the amount of 7-tCSZ nanoparticles through the samples Z0–Z15; 1.10 ± 0.01 g/cm3, 1.13 ± 0.01 g/cm3,1.18 ± 0.01 g/cm3, and 1.22 ± 0.01 g/cm3, for Z0, Z5, Z10, and Z15, respectively. At the same time, the apparent porosity decreased slightly from Z0 (52.78%) to Z10 (51.77%) and then increased in the case of Z15 (52.21%) with increasing the amount of 7-tCSZ nanoparticles. This may be attributed to the better compaction between the particles after the sintering, especially in the presence of denser crystalline zirconia nanoparticles within the amorphous silicate matrix. Before sintering during applying pressure, particles are constrained to keep in contact with each other. The presence of the nanoparticles increases the compactness between the grains by filling up the intergranular voids. During sintering, the number of grain boundaries per unit volume increases and the porosity decreases because the grains tend to combine with each other through mass transfer and diffusion [34]. Moreover, it is interesting to notice that t-ZrO2 possesses a higher true density (6.10 g/cm3) [35]. This, in turn, increases the overall density of the samples from Z5 to Z15 with increasing the content of nanosized 7-tCSZ. However, the apparent porosity in case of Z15 (52.21 ± 0.47%) was slightly higher with respect to Z10. It seems that the increasing content of t-zirconia was associated with more formation of microcracks, leading the apparent porosity to increase as a result of the significant mismatching in the thermal expansion coefficients between silicate matrix and t-ZrO2 [36].

Bulk density and apparent porosity of the different ceramic networks sintered at 700 °C
3.3 Polymer-infiltrated ceramic network (PICN) materials
3.3.1 Densification parameters
Figure 6 shows the bulk density (g/cm3) and apparent porosity (%) of PICN materials. It can be suggested that after polymer infiltration, the open pores became almost filled, which was associated with a marked decrease in apparent porosity from 51.77 to 52.78% in case of the porous ceramics (Fig. 5) to 3.79–4.57% in case of PICN materials (Fig. 6). Therefore, the PICNs became denser compared to the porous ceramic and the bulk density was increased from 1.10 g/cm3 to 1.22 g/cm3 for the porous ceramics to 1.68 g/cm3–1.76 g/cm3 for the corresponding PICNs.

Bulk density and apparent porosity of the different PICNs
3.3.2 Estimation of the polymer content in PICNs
Table 2 signifies the infiltrated polymer content (wt.%) in the overall of PICN materials. It is interesting to conclude that the pores of the ceramic networks became filled after polymer infiltration into the different samples. However, infiltrated polymer content decreased from 45.36wt.% for PZ0 to 37.73wt.% for PZ10 and re-increased again to 42.98% in case of PZ15. This can be correlated with the decrease in porosity of porous ceramics from Z0 to Z10 and re-increase in case of Z15 (Fig. 5). The higher the porosity of the specimen, the greater the amount of infiltrated monomer, which is converted later to a polymer at elevated temperature [34, 37]. The decreasing porosity and the increasing polymer content are dominating the mechanical behavior.
3.3.3 The mechanical properties of polymer infiltrated ceramic network (PICN) materials
The mechanical properties of PICN materials, flexural strength, elastic modulus, microhardness, fracture toughness, and the brittleness index of experimental PICN materials were investigated. Comparing the results to natural teeth, a very promising trial toward imitating natural teeth has been realized using PICNs.
3.3.3.1 Elastic modulus and microhardness
Figure 7 shows the elastic modulus of the PICN materials. A statistically significant difference between groups (p < 0.05) can be recognized (Table 3). The difference between PZ0 and PZ5, PZ10, and PZ15 was p = 0.004, 0.005, and 0.015, respectively. Whereas there was no significant difference between PZ5 and PZ10 or PZ15, where p = 0.930 and p = 0.998, respectively. The elastic modulus increased from 5.55GPa in case of PZ0 to 8.24GPa in case of PZ10 and decreased again to 7.88GPa in case of PZ15. This can be attributed to the fact that the porosity has an opposite effect on the elastic modulus (Figs. 5, 6, and 7). Besides, the content of infiltrated polymer decreased from 45.36wt.% in case of PZ0 to 37.73wt.% in case of PZ10, then re-increased again to 42.98wt.% (PZ15), as shown in Table 2. This ensures that the weight fraction of polymer decreases, while accompanied by an increase in the weight fraction of ceramic, which can be correlated with the elevation in the elastic modulus from 5.55GPa for PZ0 to 8.24GPa for PZ10, since the elastic modulus of ceramics is higher than that of polymers [17, 34]. Accordingly, the re-increase in the polymer content infiltrated to PZ15 can be linked to the higher porosity of the original ceramic network, leading to a slight decrease in elastic modulus compared to PZ10. This was in accordance with a previous study, which revealed that the increase of the elastic modulus of vita Enamic was higher than the similar PICN materials without zirconia particles [19].

Elastic modulus of different PICNs
Among the elastic moduli of PZ5, PZ10, and PZ15 (7.83, 8.24, and 7.88 GPa, respectively), it is noticed that PZ10 (8.24) is the highest, thus considered comparable to that of adhesive luting cements (6.8–10.8 GPa) [38]. Also, it was found that the elastic modulus of PZ10 corresponds to that of dentin (8.7–25 GPa) [22, 39, 40]. This similarity of elastic modulus for PZ10, dentin, and luting cements can hold for the assumption of a proper uniform stress distribution to take place, which decreases the chance of debonding.
As the incorporation of nanosized particles in a material is common methodology to improve the mechanical properties [7], the impact of this was also extended to the microhardness, whose values of PZ5, PZ10, and PZ15 were above those of PZ0 (Fig. 8). The enhanced particle compaction by the increase of the t-ZrO2 nanoparticles as a result of more grain boundaries formed between the nanoparticles during the sintering of the ceramic matrix is thought to have contributed to higher microhardness. The microhardness of the prepared PICN materials (0.18–0.19 GPa) was lower concerning enamel (2.7–6.4 GPa) [41,42,43]. Meanwhile, the highest obtained microhardness for the different PICN materials (PZ5 and PZ10; 0.19 GPa) was more comparable to that of dentin (0.12–0.67 GPa) [39, 43]. This might give an advantage to the PICN materials, PZ5 and PZ10 of the current study, as there is no expected excessive wear of opposing natural teeth. This is consistent with the fact that nanocrystalline containing materials are inherently stronger and harder than their microcrystalline counterparts. Furthermore, the increase in hardness might be caused by a reduction in grain size; which is expected from the Hall–Petch relation: [6, 44]

Microhardness of different PICNs
where Hv is the Vickers hardness, and d is the grain diameter. The indenter can cause microcracking, which leads to plastic deformation. The plastic deformation can be lessened with a decrease in grain size. This ensures that the amount of deformation which results from Vickers indenter at given stress should decrease with grain refinement [6]. The increase in elastic modulus was accompanied by an increase in hardness as both are related to density. This is in compliance with another study [17], in which the elastic moduli and hardness values were found to upgrade with increasing the ceramic ratio of PICN materials, considering the corresponding different particle sizes and firing temperatures of the ceramics.
3.3.3.2 Estimation of fracture toughness and brittleness index
The fracture resistance of brittle materials and their clinical performance is often demonstrated by the fracture toughness property. The single-edge-V-notched-beam (SEVNB) test is one of the recommended tests because its results are the most reliable and reproducible. Figures 9 and 10 reveal the fracture toughness and brittleness indices of the samples, PZ0-PZ15. The increase in the content of 7-tCSZ nanoparticles up to 15 wt.% gives rise to a significant enhancement of the fracture toughness, as shown in Fig. 9 and Table 3. It might be ascribed to the shielding of the crack tip due to stress-induced transformation of tetragonal to monoclinic ZrO2, which is accompanied by a volume increase (4–5%). This unique feature is known as transformation toughening [4, 5, 31, 45]. The highest mean value was acquired by PZ15 (1.82 ± 0.54 MPa m1/2), while the smallest mean value was noticed for PZ0 composite (1.03 ± 0.44 MPa m1/2). The significant improvement in fracture toughness can be correlated with the increase in the fracture surface energy owing to a reduction of crack tip stress intensity in the plastic zone ahead of the crack by energy dissipation through relaxation occurring in the grain boundary phase. Similar enhancements were obtained in a work carried out by Montazerian et al. [46] for a glass–ceramic reinforced with different contents of yttria-PSZ.

Fracture toughness of different PICNs

Brittleness index of different PICNs
From another side, the low brittleness index of PICN materials (Fig. 10) dictates good machinability. The corresponding index declined from 0.21um−½ to 0.15 um−½ for samples PZ0 and PZ10, respectively. A strong negative relationship between fracture toughness and brittleness index (r = −0.811) can be identified, as shown in Figs. 9 and 10, which indicates that an increase in the fracture toughness is accompanied by a diminishing in the brittleness index. It seems real as the brittleness index is calculated from the ratio of Vickers hardness and fracture toughness. This is in accordance with a previous study [47], where it was found that the addition of more crystalline phase besides increasing fracture toughness, it was beneficial to enhance the ease of machinability, reflected quantitatively from the lower value of brittleness index.
3.3.3.3 Flexural strength
Figure 11 displays the flexural strength of the PICN materials, in which it decreases with increasing the content of 7-tCSZ from PZ0 to PZ10. The particles compaction during sintering is associated with the densification of the overall network and porosity development. Such porosity decreases as more grain boundaries form [48]. The addition of nano t-zirconia to the ceramic matrix causes a decrease of the apparent porosity due to the sintering temperature for nano-ceramics (t-ZrO2), which is generally lower than their conventional counterparts [6]. This, in turn, reduces the monomer infiltration into the pores by capillary action. PICNs reached greater strains than brittle ceramics when loaded to fracture due to the polymer ratio [17]. A lower ceramic fraction of PICNs implies a lower elastic modulus accompanied by increased flexural strength because the flexural strength of polymers is higher than that of porous ceramic [17]. This explains the slight decrease in the flexural strength with decreasing the amount of infiltrated polymer up to PZ10. This is more evidenced by the re-increase in flexural strength for PZ15, which can be simply linked to a higher amount of polymer infiltrated due to the increased porosity (Figs. 5 and 6). This trend was similar to that obtained elsewhere [17], where the flexural strength values of PICN materials were directly proportional to ceramic porosity, which was filled as a result of polymer infiltration.

Flexural strength of different PICNs
3.3.4 Microstructure
The microstructures of Z10 and PZ10 are exposed before and after infiltration, respectively (Figs. 12 and 13) at different magnifications. Figure 12a reveals the uniform distribution of the minor nanoparticles of t-ZrO2 within the alumina silicate matrix, while the higher magnification indicated the developed porosity became apparent after sintering at 700 °C (Fig. 12b, c). Micropores were formed parallel with the established grain growth of t-ZrO2 ~ 644 nm (Fig. 12d). Such a change might cause decreasing in porosity and contribute to material strengthening. There were no cracks found at a sintering temperature of 700 °C.

a–d SEM microphotographs of Z10 before polymer infiltration at different magnifications

a–c SEM microphotographs of PZ10 after polymer infiltration at different magnifications
On the other hand, SEM microphotographs of the fractured sample of PZ10 (Fig. 13a–c) show the darker grey areas represent the polymer phase while the lighter zones reveal silicate ceramic network containing zirconia nanoparticles. It is highly recognized the widely distributed white particles, demonstrating zirconia nanoparticles. Their size range was 763–782 nm. As expected, a considerable combination of ceramic and polymer matrixes is identified, except the areas where the polymer underwent shrinkage (~ 5%) [49], causing defects, in addition to the existence of fewer and smaller voids (Fig. 13c). These defects can weaken the mechanical properties. There were no propagating cracks present in the PZ10 sample, indicating little damage caused by machining and milling. The SEM micrographs show a great extent of polymer infiltration into the porous structure of the partially sintered ceramic network. Such a combination could contribute to advance the mechanical properties. The surface topography of PZ10 disclosed rough marks, which requires high energy for milling, which is again a sign of high toughness (Fig. 13a).
4 Conclusion
Tetragonal zirconia nanoparticles in the range of 10 nm were successfully synthesized via a modified co-precipitation method and stabilized using 7wt.% of CaO at 700 °C. Both elastic modulus and fracture toughness as high as 8.24 GPa and 1.82 MPa.m1/2 were reached for PICN materials, incorporated with 10 and 15 wt.% of tetragonal zirconia nanoparticles, respectively, could be mainly attributed to the tetragonal to monoclinic phase transformation of zirconia particles in a zone at the tip of crack propagation, which leads to the transformation toughening. The PICN materials are acquiring similar elastic modulus and fracture toughness compared to dentin, which opens the door for broadened dental applications. Interestingly, the remarkable mechanical properties improvement associated with the inclusion of t-zirconia was not accompanied by any worsening in the ease of machinability, which makes these types of materials promising materials for indirect restorative material applications.
References
M. Montazerian, E.D. Zanotto, History and trends of bioactive glass-ceramics. J. Biomed. Mater. Res. Part A 104(5), 1231–1249 (2016)
N.B. Cramer, J.W. Stansbury, C.N. Bowman, Recent advances and developments in composite dental restorative materials. J. Dent. Res. 90(4), 402–416 (2011)
A.S. Verma, D. Kumar, A.K. Dubey, A review of an innovative concept to increase the toughness of the ceramics by piezoelectric secondary phases. Ceram. Int. 44(14), 16119–16127 (2018)
J.U. Jansen, N. Lümkemann, B. Sener, B. Stawarczyk, Comparison of fracture toughness measurements for zirconia materials using two test methods. Dent. Mater. J. 38(5), 806–812 (2019)
M. Maleki, S.M. Sheikh-Al-Eslamian, E. Hasani, A. Ghasemi, Comparative study on the microstructure and mechanical behavior of monolithic ceramic and laminated composite of high strength 3Y-TZP and high fracture toughness 12Ce-TZP. J. Alloys. Compd. 776(5), 166–171 (2019)
A. Mukhopadhyay, B. Basu, Consolidation–microstructure–property relationships in bulk nanoceramics and ceramic nanocomposites: a review. Int. Mater. Rev. 52(5), 257–288 (2007)
S. Maitra, J. Roy, Nano ceramic matrix composites: types, processing and applications, in Advances in Ceramic Matrix Composites, 2nd edn, ed. by I.M. Low. (Woodhead Publishing is an imprint of Elsevier, United Kingdom, 2018), pp. 27–48. ISBN: 9780081021668
A. Mukhopadhyay, B. Basu, Bulk nanoceramic composites for structural applications: a review. Proc. Indian. Natn. Sci. Acad. 72(2), 97–111 (2006)
S. Zang, N. He, X. Sun, M. Sun, W. Wu, H. Yang, "Influence of additives on the purity of tetragonal phase and grain size of ceria-stabilized tetragonal zirconia polycrystals (Ce-TZP). Ceram. Int. 45(1), 394–400 (2019)
R. Shahmiri, O.C. Standard, J.N. Hart, C.C. Sorrell, Optical properties of zirconia ceramics for esthetic dental restorations: a systematic review. J. Prosth. Dent. 119(1), 36–46 (2018)
Z.J. Ming, J.K. Lee, H.K. Hong, J.J. Ho, Synthesis and thermal stability of nanocrystalline tetragonal zirconia by hydrolysis with ethylene diamine. J. Nanosci. Nanotechnol. 18(2), 1241–1245 (2018)
H. Shokry, M. Elkady, H. Hamad, Synthesis and characterization of stabilized tetragonal nano zirconia by precipitation method. J. Nano. Res. 56, 142–151 (2019)
B. Fan, F. Zhang, J. Li, H. Chen, R. Zhang, Synthesis and crystallization behavior of 3 mol% yttria partically stabilized zirconia (3Y-PSZ) nanopowders by microwave pyrolysis process. J. Mater. Sci. Eng. 6(2), 1–4 (2017)
S. Ghorbani, R.S. Razavi, M.R. Loghman-Estarki, A. Alhaji, Development of MgO–Y2O3 composite nanopowder by Pechini sol-gel method: effect of synthesis parameters on morphology, particle size, and phase distribution. J. Cluster. Sci. 28(3), 1523–1539 (2017)
Z. Huang, W. Han, Z. Feng, J. Qi, D. Wu, N. Wei, Z. Tang, Y. Zhang, J. Duan, T. Lu, The effects of precipitants on co-precipitation synthesis of yttria-stabilized zirconia nanocrystalline powders. J. Sol-Gel. Sci. Technol. 90(2), 359–368 (2019)
Y.T. Foo, A.Z. Abdullah, B.A. Horri, B. Salamatinia, Optimised co-precipitation synthesis condition for oxalate-derived zirconia nanoparticles. Ceram. Int. 45(17), 22930–22939 (2019)
A. Coldea, M.V. Swain, N. Thiel, Mechanical properties of polymer-infiltrated-ceramic-network materials. Dent. Mater. 29(4), 419–426 (2013)
J.C. Facenda, M. Borba, P.H. Corazza, A literature review on the new polymer-infiltrated ceramic-network material (PICN). J. Esthet. Restor. Dent. 30(4), 281–286 (2018)
A. Della Bona, P.H. Corazza, Y. Zhang, Characterization of a polymer-infiltrated ceramic-network material. Dent. Mater. 30(5), 564–569 (2014)
H. Wang, B. Cui, J. Li, S. Li, Y. Lin, D. Liu, M. Li, Mechanical properties and biocompatibility of polymer infiltrated sodium aluminum silicate restorative composites. J. Adv. Ceram. 6(1), 73–79 (2017)
M. Petrini, M. Ferrante, B. Su, Fabrication and characterization of biomimetic ceramic/polymer composite materials for dental restoration. Dent. Mater. 29(4), 375–380 (2013)
M.V. Swain, A. Coldea, A. Bilkhair, P.C. Guess, Interpentrating network ceramic-resin composite dental restorative materials. Dent. Mater. 32(1), 34–42 (2016)
A. Zarkov, A. Stanulis, J. Sakaliuniene, S. Butkute, B. Abakeviciene, T. Salkus, S. Tautkus, A.F. Orliukas, S. Tamulevicius, A. Kareiva, On the synthesis of yttria-stabilized zirconia: a comparative study. J. Solgel. Sci. Technol. 76(2), 309–319 (2015)
M. Hajizadeh-Oghaz, R.S. Razavi, M. Barekat, M. Naderi, S. Malekzadeh, M. Rezazadeh, Synthesis and characterization of Y2O3 nanoparticles by sol–gel process for transparent ceramics applications. J. Solgel. Sci. Technol. 78(3), 682–691 (2016)
B. Cui, R. Zhang, F. Sun, Q. Ding, Y. Lin, L. Zhang, C. Nan, Mechanical and biocompatible properties of polymer-infiltrated-ceramic-network materials for dental restoration. J. Adv. Ceram. 9(1), 123–128 (2020)
B. Cui, J. Li, H. Wang, Y. Lin, Y. Shen, M. Li, X. Deng, C. Nan, Mechanical properties of polymer-infiltrated-ceramic (sodium aluminum silicate) composites for dental restoration. J. Dent. 62, 91–97 (2017)
S.E. Elsaka, A.M. Elnaghy, Mechanical properties of zirconia reinforced lithium silicate glass-ceramic. Dent. Mater. 32(7), 908–914 (2016)
G. Zamfirova, A. Dimitrova, Some methodological contributions to the Vickers microhardness technique. Polym. Test. 19(5), 533–542 (2000)
D. Geetha, P.J. Sophia, D. Arivuoli, Evaluation of microindentation properties of epitaxial 3C–SiC/Si thin films. Phys. B 490, 86–89 (2016)
A.R. Boccaccini, Machinability and brittleness of glass-ceramics. J. Mater. Process. Technol. 65(1–3), 302–304 (1997)
S. Shukla, S. Seal, Mechanisms of room temperature metastable tetragonal phase stabilisation in zirconia. Int. Mater. Rev. 50(1), 45–64 (2005)
F. Cardarelli, Materials Handbook: A Concise Desktop Reference, 2nd edn. (Springer-Verlag, London Limited, New York, London, 2008), pp. 593–689. ISBN: 978-1-84628-668-1
N.D. Lestari, R. Nurlaila, N.F. Muwwaqor, S. Pratapa, Synthesis of high-purity zircon, zirconia, and silica nanopowders from local zircon sand. Ceram. Inter. 45(6), 6639–6647 (2019)
J. Li, X.H. Zhang, B.C. Cui, Y.H. Lin, X.L. Deng, M. Li, C.W. Nan, Mechanical performance of polymer-infiltrated zirconia ceramics. J. Dent. 58, 60–66 (2017)
B. Basu, Toughening of yttria-stabilised tetragonal zirconia ceramics. Int. Mater. Rev. 50(4), 239–256 (2005)
S. Ghasemi-Kahrizsangi, H.G. Dehsheikh, M. Boroujerdnia, Effect of micro and nano-Al2O3 addition on the microstructure and properties of MgO-C refractory ceramic composite. Mater. Chem. Phys. 189(1), 230–236 (2017)
J.F. Nguyen, D. Ruse, A.C. Phan, M.J. Sadoun, High-temperature-pressure polymerized resin-infiltrated ceramic networks. J. Dent. Res. 93(1), 62–67 (2014)
E. Magni, M. Ferrari, R. Hickel, N. Ilie, Evaluation of the mechanical properties of dental adhesives and glass-ionomer cements. Clin. Oral. Invest. 14, 79–87 (2010)
J.H. Kinney, M. Balooch, S.J. Marshall, G.W. Jr Marshall, T.P. Weihs, Hardness and Young’s modulus of human peritubular and intertubular dentine. Arch. Oral. Biol. 41(1), 9–13 (1996)
D. Ziskind, M. Hasday, S.R. Cohen, H.D. Wagner, Young’s modulus of peritubular and intertubular human dentin by nano-indentation tests. J. Struct. Biol. 174(1), 23–30 (2011)
J.L. Cuy, A.B. Mann, K.J. Livi, M.F. Teaford, T.P. Weihs, Nanoindentation mapping of the mechanical properties of human molar tooth enamel. Arch. Oral. Biol. 47(4), 281–291 (2002)
S. Habelitz, S.J. Marshall, G.W. Marshall, M. Balooch, Mechanical properties of human dental enamel on the nanometre scale. Arch. Oral. Biol. 46(2), 173–183 (2001)
H.H. Xu, D.T. Smith, S. Jahanmir, S.E. Romberg, J.R. Kelly, V.P. Thompson, E.D. Rekow, Indentation damage and mechanical properties of human enamel and dentin. J. Dent. Res. 77(3), 472–480 (1998)
A.G. Sheinerman, R.H. Castro, M.Y. Gutkin, A model for direct and inverse Hall-Petch relation for nanocrystalline ceramics. Mater. Lett. 260, 126886 (2020)
M. Turon-Vinas, M. Anglada, Strength and fracture toughness of zirconia dental ceramics. Dent. Mater. 34(3), 365–375 (2018)
M. Montazerian, P. Alizadeh, B.E. Yekta, Processing and properties of a mica–apatite glass–ceramic reinforced with Y-PSZ particles. J. Eur. Ceram. Soc. 28(14), 2693–2699 (2008)
H.N. Yoshimura, C.C. Gonzaga, P.F. Cesar, W.G. Miranda, Relationship between elastic and mechanical properties of dental ceramics and their index of brittleness. Ceram. Int. 38(6), 4715–4722 (2012)
M.N. Rahaman, Sintering of Ceramics, 1st edn, (CRC Press, United States of America, 2007), pp. 1–43. ISBN: 9780849372865
Q. Chen, Y. Zhao, W. Wu, T. Xu, H. Fong, Fabrication and evaluation of Bis-GMA/TEGDMA dental resins/composites containing halloysite nanotubes. Dent. Mater. 28(10), 1071–1079 (2012)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mohammed, N.Y., Wahsh, M.M.S., Motawea, I.T. et al. Fabrication and characterization of polymer-infiltrated ceramic network materials based on nano-tetragonal zirconia. J. Korean Ceram. Soc. 58, 359–372 (2021). https://doi.org/10.1007/s43207-020-00102-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43207-020-00102-4