
Abstract
Agrimonia pilosa (AP) and Rhus gall (RG) are traditional medicinal plants. The bioflavonoid composition standardized by HPLC analysis was named APRG64. Despite many studies reported to beneficial bioactivities of AP and RG, very limited range of toxicity tests have documented. So, we did experiment diversely on the toxicity tests of the substance APRG64. Genotoxicity (mammalian chromosomal aberration test, micronoucleus test) against APRG64, acute and sub-chronic toxicity test from rodent/non-rodent, and systemic safety pharmacology test were conducted. As a result of the test, genotoxicity against APRG64 was not observed. The NOAEL of rodents was confirmed as 2000 mg/kg/day and non-rodents was confirmed as 500 mg/kg/day. In addition, systemic safety pharmacological toxicity (effects on respiratory system, central nervous system, cardiovascular system) following administration of APRG64 was not observed. Finally, we accomplished ten potential toxicity tests and evaluated extensive safety of APRG64. Consequently, APRG64 may be a promising material for nutraceuticals and natural medicines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Traditional medicinal plants have been used for treatment of particular diseases since ancient times and widely used throughout the world. In developing countries approximately 80% of people use traditional medicines, continue to provide humans with new remedies as 50% of all drugs in clinical use in the world are derived from natural products [1]. Despite, the widespread use of plants as medicines, often the use of these resources occurs without proper scientific evidence of their pharmacological properties and toxic potential. Previous study has shown that three quarters of herbal preparations marketed do not contain safety information for adequate consumption. Even if a particular species has low toxicity, its inappropriate use associated with risk factors may lead to serious conditions that are sometimes underreported [2, 3].
Here are two traditional medicinal plants, Agrimonia pilosa (AP), also known as hairy agrimony, belongs to the Rosaceae family. It is a perennial herb and widely distributed in Korea and China. The aerial parts of AP were used in folk medicine to treat hematuria, hematemesis, leukorrhea, traumatic injury, diarrhea, stomatitis, tonsillitis, eczema, hemoptysis, hematochezia, anti-viral and gastroenteritis [4,5,6]. Apigenin (AG) is one of the main flavonoid compounds from AP, has the pharmacological functions of anti-oxidation, anti-inflammation, anti-viral and inhibition of pulmonary fibrosis. Also, it can prevent or reverse liver injury [7, 8]. The gall of Rhus javanica, Rhus gall (RG) is the excrescence produced by parasitic aphids, mainly Melaphis chinensis (Bell) Baker, on the leaf of Rhus javanica L. [9]. The herb contains Penta-galloyl glucose (PGG), one of the main polyphenolic compounds from RG, has been reported to possess antioxidant activity, anti-diabetic, anti-cancer, and effect on blocking hepatitis C virus entry [10].
Agrimonia pilosa and Rhus gall were reported to have anti-viral activity, respectively. Therefore, the antiviral effect is expected to increase more when the two extracts are mixed. The mixture of the two extracts was named APRG64.
This article purposes to evaluate the genetic toxicity, acute and sub-chronic oral toxicity and systemic safety of APRG64. Because the bioflavonoid compounds, AP and PGG of AP and RG have aroused intense interest in the development of pharmacological reagent [7, 10]. Firstly, bacterial reverse mutation test, chromosome aberration test, erythrocyte micronucleus test in mammalian cells were performed for the genetic toxicity evaluation. Secondly, Single dose oral toxicity study, 4-week repeated oral dose range finding study, Repeated dose 13-week oral toxicity and 4-week recovery study were practiced using SD rats to find the NOAEL of APRG64. Lastly, to secure more safety of APRG64, Repeated dose 4-week oral toxicity study in beagle dog was carried out, and central nervous systems, respiratory, and cardiovascular function were assessed through the functional observational battery (FOB), whole body plethysmography system in rats and implanted telemetry in dogs, respectively.
Materials and method
Preparation of extract
Sampling and identification
Dried Agrimonia pilosa (AP) and Rhus gall (RG) was provided by Kyung Dong Market (Seoul, Korea). The plant was identified and authenticated on the basis of its macroscopic and microscopic characteristics by Dr. Kang, Se Chan, Kyung Hee University (Yongin, Korea). The voucher specimen (NMR-KR-2017-01) was deposited in the Laboratory of Natural Medicine Resources in BioMedical Research Institute, Kyung Hee University.
Extraction
The samples were washed three times with distilled water to remove slats, plants, and sand, and then dry to without direct sunlight, and pulverized. Each crude extract was obtained by extracting dried AP and RG twice with 50% ethanol for 24 h, at room temperature, respectively. The extracts were concentrated for 16 h at reduced pressure and 40 °C using a rotary evaporator, and then the crude extract was lyophilized to obtain a powder. And then, each 50% ethanolic extracts mixed 6:4 (AP:RG) ratio. A standardized bioflavonoid composition was named APRG64.This mixture stored at − 20 °C until use, and when administered to experimental animals, they were orally mixed with 0.9% normal saline.
HPLC analyses
We conducted the analysis of the dosing formulations of AG and PGG from AP and RG using high performance liquid chromatography (HPLC). HPLC was performed using Shimadzu LC-20AD (Shimadzu, Kyoto, Japan) with and Waters C18 column (5 μm, 4.6 × 250 mm). The column oven was maintained at 35 °C, and the mobile phases included solvent A [0.1% formic acid in water (v/v)] and solvent B [acetonitrile (v/v)]. The elution gradient was as follows: 0–3 min, B 10%; 3–10 min, B 10–20%; 10–20 min, B 20%; 20–30 min, B 2–35%; 30–38 min, B 35–90%; 38–39 min, B 90%; and 39–50 min, B 10%. The flow rate was 1.0 ml/min, and the injection volume was 10 μL for each run.
To analyze the content of AG in AP, the gradient eluted consisted of water with 0.1% formic acid (A) and acetonitrile (B). The mobile phase was used under binary linear gradient conditions as follows: 0–20 min, 5–100% A; 20–40 min, 100–5% A. The flow rate was 1.0 mL/min and injection volume was 10 μL. AP extract (10 mg/mL) and AG (1000 μg/mL) were dissolved methanol with appropriate dilution. Peaks were identified by comparing their retention time and UV–vis spectra with AG and were quantitated using the corresponding AG curve.
In the case of RG content analysis the gradient eluted consisted of 0.1% formic acid (A) and water with acetonitrile (B). The mobile phase was used under binary linear gradient conditions as follows: 0–47 min, 15–23% A. The flow rate was 1.0 mL/min and injection volume was 10 μL RG extract (10 mg/mL) and PGG (1000 μg/mL) were dissolved methanol with appropriate dilution. Peaks were identified by comparing their retention time and UV–vis spectra with PGG and were quantitated using the corresponding standard curve. When performed the individual 10 tests, the content of AG form AP was 14.04 ± 0.69 mg/g and PGG from RG was 230.31 ± 2.30 mg/g.
Genotoxicity study
Bacterial reverse mutation study
This study was conducted using the pre-incubation method with and without metabolic activation system (S9) in accordance with the following test guidelines: OECD Guideline for Testing of Chemicals No. 471 ‘Bacterial Reverse Mutation Test’ (Adopted 21st July 1997), Ministry of Food and Drug Safety Notice No. 2015-82 ‘Toxicity Test Standards of Medicine and Medical Supplies’ (Revised 11th November, 2015) and The standard operation procedures (SOPs) of Korea Conformity Laboratories.
Bacterial reverse mutation test was performed by using histidine-requiring strains of Salmonella typhimurium (TA98, TA100, TA1535 and TA1537) and a tryptophan-requiring strain of Escherichia coli (WP2uvrA) in the presence and absence of metabolic activation system (S9 mix). The preliminary range-finding study was performed to determine dose levels at the main study. As a result of preliminary range-finding test, precipitation was observed at 5000 μg/plate in all bacterial strains. At the main study, the test sample concentration was set at 21, 62, 185, 556, 1667 and 5000 μg/plate, with three plates for each dose. In the test, 0.05 mL of the sample, 0.1 mL of the bacterial suspension and 0.5 mL of either the S9 mix or the phosphate buffer (pH 7.4) were added to 2.0 mL of top agar which contained trace amount of histidine (for Salmonella), biotin (for Salmonella) and tryptophan (for Escherichia) maintained at 45 °C. The mixture was then poured onto a minimal glucose agar plate. The plates were inverted and incubated at 37 °C for 48 h. Dimethyl sulfoxide was used as the solvent control, and sodium azide (NaN3, TA1535 in 0.5 μg/plate), 9-aminoacridine hydrochloride hydrate (9-AA, TA1537 in 80 μg/plate), 2-(2-Furyl)-3-(5-nitro-2-furyl) acrylamide (AF-2, TA98 in 0.1 μg/plate, TA100 in 0.01 μg/plate, WP2uvrA in 0.01 μg/plate) in the absence of S9 mix, and 2-aminoanthracene (2-AA, TA98 in 0.5 μg/plate, TA100 in 1.0 μg/plate, TA1535 in 2.0 μg/plate, TA1537 in 2.0 μg/plate, WP2uvrA in 10 μg/plate) in the presence of S9 mix were used as the positive control. Additionally, 0.1 mL of S9 mix and test substance were mixed for sterility test. The test result was recorded experimental value, average value and standard deviation for the number of revertant colonies per plate. The result was judged as ‘positive’, if there is a dose-dependent increase and/or a reproducible increase at one or more concentrations in the number of revertant colonies per plate in at least one strain with or without metabolic activation system. Also, the number of revertant colony should increase more than two times than the negative control group.
Mammalian chromosome aberration study
This study was conducted in accordance with the following test guidelines: Ministry of Food and Drug Safety Notice No. 2015-82 ‘Toxicity Test Standards of Medicine and Medical Supplies’ (Revised 11th November, 2015). OECD Guidelines for the Testing of Chemicals No. 473 “In vitro mammalian chromosome aberration test” (26 September 2014). The standard operation procedures (SOPs) of Korea Conformity Laboratories (KCL/CRO).
In this study was performed the chromosome aberration test to assess the genotoxicity for APRG64 using Chinese Hamster Ovary (CHO-K1) cells in the absence (− S9 mix) and presence (+ S9 mix) of metabolic activation system. F-12 Nutrient Mixture (Gibco, Logan, UT, USA) with 10% fetal bovine serum (FBS, Hyclone, South Logan, UT, USA) and 10% Dimethyl sulfoxide (DMSO, Sigma Aldrich, St. Louis, MO, USA) were used as culture medium.
The cytotoxicity test (preliminary range-finding test) was conducted to decide the highest treatment concentration for main test, and then the main test was selected 3 concentrations with threefold as follows. Absence metabolic activation system (− S9 mix, 24 h treatment) was 2.29, 6.86, 20.58 μg/mL Absence metabolic activation system (− S9 mix, 6 h treatment and 18 h recovery) was 6.86, 20.58, 61.73 μg/mL. Presence metabolic activation system (+ S9 mix, 6 h treatment and 18 h recovery) was 6.86, 20.58, 61.73 μg/mL, respectively. Two replicate cultures were used for each concentration level.
In experiment without S9 metabolic activation, the treatment times were 4 and 24 h, respectively. 4 × 104 cells/mL were seeded in 60 mm diameter tissue culture dishes and incubated for 3 days. Before the test substance treatment, the existing culture medium was removed from the culture dish and then 4.80 mL of pre-warmed fresh medium (37 °C) and 0.20 mL of test substance solution were added to each dish which became 5.00 mL. In case of positive control, the existing culture medium was removed from the culture dish and then 4.95 mL medium (37 °C) and 0.05 mL of positive control (Mitomycin C, Sigma Aldrich) were added to each dish which became 5.00 mL. For 6 h treatment and 18 h recovery group, the culture medium was removed at 6 h after treatment and the cells were rinsed once with PBS (Ca2+ and Mg2+ free Dulbecco’s phosphate buffered saline, DPBS). Then, 5 mL of medium (37 °C) was added and the cells were incubated for additional 18 h until the harvest of metaphase-cells.
In experiment with S9 metabolic activation, the treatment time was 4 h. 4.30 mL of pre-warmed fresh medium (37 °C), 0.20 mL of test substance solution and 0.50 mL of S9 mix were added to each dish which became 5.00 mL. In case of positive control, 4.45 mL of medium (37 °C), 0.05 mL of positive control (Benzo[a]pyrene) and 0.50 mL of S9 mix were added to each dish which became 5.00 mL. The culture medium was removed at 6 h after treatment and the cells were rinsed once with PBS. Then, 5 mL of pre-warmed fresh culture medium (37 °C) was added and the cells were incubated for additional 18 h until the harvest of metaphase-cells. The chromosome slides were prepared using the harvested cells. Approximately 22 h after treatment, Colcemid (Gibco) was added to each culture plate for a final concentration of 0.2 μg/mL. The cultures were incubated for an additional 2 h. The cells were detached using 1× trypsin solution. The medium containing mitotic cells was centrifuged at 1000 rpm for 5 min. After discarded supernatant, the cell pellets were resuspended in 75 mM potassium chloride (KCl) solution and incubated at 37 °C incubator for 20 min. The cells were fixed three times with Carnoy’s fixative solution (acetic acid:methanol = 1:3 v/v) for the slide preparation. The slides were stained with 5% Giemsa (Merck, Darmstadt, Germany) solution for 20 min and observed microscopically. Two slides samples were prepared from each plate.
Mammalian erythrocyte micronucleus study
Mammalian erythrocyte micronucleus test was performed using male and female Specific Pathogenic Free (SPF) CrljOri:CD1 (ICR) mice, aged 7–8 weeks, from Orient Bio Co., Ltd. (Seongnam, Korea). Animals were acclimated for 7 days. The only healthy animals were selected for this study by observation of general symptoms during the acclimation period. They were housed in litter downed polycarbonate cage (3–4 animals per cage for preliminary range-finding study, 5 animals per cage for main study) and provided food and water ad libitum. Range of acceptable temperature and humidity for animals were 22.0 ± 0.5 °C and 44.8 ± 1.9%. According to determined dose in preliminary range-finding study, the highest dose in the main study was 2000 mg/kg/day. Test groups consisted of one negative control group (distilled water only), 3 dosing groups (500, 1000, 2000 mg/kg/day) and one positive control group [Mitomycin C (MMC) 2.0 mg/kg]. Each group consisted of five male mice. Because in the preliminary range-finding study, there was no difference in sensitivity to toxicity between male and female, so it was tested as male mice. The test substance was administrated by oral (10 mL/kg once every 24 h for 2 days) using sonde and the positive control substance was administered through intraperitoneally to test animals using disposable 1 mL syringe. After dosing, animals examined regularly for clinical signs of toxicity until sacrifice. The thigh bone of the test animal was collected with care to avoid blood contamination in autopsy room within 18–24 h after final administration. The bone marrow was collected in a centrifuge tube by flushing the thigh bone inside with in-activated FBS. The extracted bone marrow was centrifuged at 1000 rpm for 5 min and then resuspended with small aliquots of FBS after discarding the supernatant. The bone marrow was smeared on a slide glass and then dried at room temperature and fixed in methanol for 5 min. Three slides of bone marrow were prepared. The slides were stained in 4% Giemsa staining solution for the scoring of the PCE (polychromatic erythrocytes) to NCE (normochromatic erythrocytes) ratio. The observation of slides was performed with blind method and the PCE/NCE ratio was determined by scoring the number of PCEs and NCEs observed while scoring 500 erythrocytes per animal. In addition, MNPCE (micronucleated polychromatic erythrocytes) from PCEs, stained with acridine orange (40 μg/mL), was scored by analyzing 4000 PCEs per animal.
Oral toxicity study
Single dose toxicity study
Single dose oral toxicity study was performed using male and female SPF Crl:CD (Sprague–Dawley, SD) rats, aged 7 weeks, from Orient Bio Co., Ltd. Female rats were healthy young adults and they were nulliparous and non-pregnant. Animals were acclimated during 6 days after arrival. Only animals with the best appearance were selected for the test after observation during the acclimation period. All animals were housed in wire mesh cages (250 W × 350 L × 180 H mm) and provided food and water ad libitum. Range of acceptable temperature and humidity for animals were 23.5 ± 0.4 °C and 44.7 ± 1.1%. Forty SD rats were equally divided into 4 groups (5/sex/group) and once-daily oral (by gavage in the morning) of the test substance at 500 (low-dose), 1000 (middle-dose), and 2000 (high-dose) mg/kg. Dose levels were determined in accordance with the Ministry of Food and Drug Safety (MFDS) Notification [11]. A high-dose level was set as 2000 mg/kg which is selected as a limited dose generally in single dose toxicity study. Dosing volume was adjusted as 10 mL/kg body weight and vehicle used sterilized distilled water for control group.
General clinical signs or mortalities of all treated animals were carefully observed continuously during the first half-hour. After that, all animals were observed regularly at 1 h intervals until 6 h on the administration day. From the next day, animals were observed once every day up to 14 days after the administration. Also, individual animals were weighed at the date of acquisition, at grouping, dosing day prior to administration (on the administration day), 1, 4, 7, and 14 days (before necropsy) after the administration. On day 14 after the administration, all surviving animals were anesthetized with CO2 gas and terminated by exsanguination from the abdominal aorta and caudal vena cava. Complete post-mortem examinations were performed on all vital organs. This study was approved by the Institutional Animal Care and Use Committee (IACUC) (IA17-00004).
Body weight changes of all animals were analyzed by using the table and figures that were applied to the mean value and the standard deviations. Body weights among the vehicle and dosing groups were compared through the One-way ANOVA and significant difference was conceded in case of the p value is under 0.05. Statically analysis was performed in compliance with the SOP of this testing facility and SPSS for windows version 12.0 software (SPSS, Chicago, IL, USA) was used.
Rats: 4-weeks repeated dose toxicity determination study
Four-week repeated oral toxicity determination study was performed using male and female SPF SD rats, aged 5 weeks, from Orient Bio Co., Ltd. Female rats were healthy young adults and they were nulliparous and non-pregnant. Animals were acclimated during 6 days after arrival. Only animals with the best appearance were selected for the test after observation during the acclimation period. All animals were housed in wire mesh cages (250 W × 350 L × 180 H mm) and provided food and water ad libitum. Range of acceptable temperature and humidity for animals were 23.3 ± 0.3 °C and 43.0 ± 1.6%. Forty SD rats were equally divided into 4 groups (5/sex/group) and once-daily oral (by gavage in the morning) of the test substance at 500 (low-dose), 1000 (middle-dose), and 2000 (high-dose) mg/kg for 4 weeks. Dose levels were determined at the results of single dose oral toxicity study, it is considered that toxicological effects related with the test substance were not observed at the dose level of 2000 mg/kg. Dosing volume was adjusted as 10 mL/kg body weight and vehicle used sterilized distilled water for control group.
During the study period, animals were observed for mortalities, clinical signs, body weights, food and water consumption, eye examination, urinalysis, clinical pathology (hematology and serum biochemistry) and necropsy (organ weights measurement and gross findings). This study was approved by the Institutional Animal Care and Use Committee (IACUC) (IA17-00048).
Statistical analysis was performed in compliance with the SOP of this testing facility and SPSS for windows version 12.0 software (SPSS) was used. The statistical differences among the vehicle control and the all dosing groups were analyzed normally through the parametric multiple comparison procedures or non-parametric multiple comparison procedures. Statistical significance was conceded if the p value is under 0.05 and incidence rate was represented by percentage. Analysis of continuous data used the standard one-way ANOVA. If the test showed statistical significance, the data was analyzed through the parametric multiple comparison to compare the vehicle control group with the experimental groups. If the equal variance was admitted, Duncan’s test was used and if not, Dunnett’s T3 test was applied. Analysis of non-continuous data (urinalysis and urine color) was converted by scale conversion as score and then analyzed by Chi-squared test.
Rats: 13-weeks repeated dose toxicity and 4-weeks recovery study
Thirteen-week repeated oral toxicity test and 4-week recovery study was performed using male and female SPF SD rats, aged 5 weeks, from Orient Bio Co., Ltd. Female rats were healthy young adults and they were nulliparous and non-pregnant. Male and female animals were acclimated during 6 and 8 days after arrival, respectively. Only animals with the best appearance were selected for the test after observation during the acclimation period. All animals were housed in wire mesh cages (250 W × 350 L × 180 H mm) and provided food and water ad libitum. Range of acceptable temperature and humidity for animals were 23.0 ± 0.6 °C and 42.8 ± 2.9%. One hundred SD rats were divided into 4 groups (group 1, Vehicle control group, 15 male and female respectively; group 2–3, 500, 1000 mg/kg dosing group, 10 male and female respectively; group 4, 2000 mg/kg dosing group, 15 male and female respectively. Each last consecutive five animals in G1 and G4 groups are assigned to recovery groups.) once–daily oral (by gavage in the morning) of the test substance at 500 (low-dose), 1000 (middle-dose), and 2000 (high-dose) mg/kg were administered for 13 weeks. Dose levels were determined at the results of 4-week repeated oral dose range finding study, it is considered that toxicological effects related with the test substance were not observed at the dose level of 2000 mg/kg. Dosing volume was adjusted as 10 mL/kg body weight and vehicle used sterilized distilled water for control group.
During the study period, animals were observed for mortalities, clinical signs, body weights, food consumption, ophthalmological examination, clinical pathology (urinalysis, hematology, blood coagulation time and serum biochemistry), necropsy (organ weights measurement and gross findings), and histopathological examination. This study was approved by the Institutional Animal Care and Use Committee (IACUC) (IA17-00569).
Statistical analysis was performed in compliance with the SOP of this testing facility and SPSS for windows version 12.0 software (SPSS) was used. The statistical differences among the vehicle control and the all dosing groups were analyzed normally through the parametric multiple comparison procedures or non-parametric multiple comparison procedures. Statistical significance was conceded if the p value is under 0.05 and incidence rate was represented by percentage. Analysis of continuous data used the standard one-way ANOVA. If the test showed statistical significance, the data was analyzed through the parametric multiple comparison to compare the vehicle control group with the experimental groups. If the equal variance was admitted, Duncan’s test was used and if not, Dunnett’s T3-test was applied. Analysis of non-continuous data (urinalysis and urine color) was converted by scale conversion as score and then analyzed by Chi-squared test.
Beagle dogs: 4-weeks repeated dose determination study
Four-weeks repeated dose determination study was performed using five male and five female beagle dogs, aged 5 months, from Producing Beijing Marshall Biotechnology Co., Ltd. (Beijing, China). Male and female animals were acclimated during for 26 days. The healthy animals selected during the acclimation period. All animals were housed in stainless steel cages (700 W × 750 L × 750 H mm) during the experiment period. Each animal was offered a daily ration of 300 g of solid food (LAB CANINE CHOW, Cargill Agri Purina Korea Inc., Seongnam, Korea). Water was provided ad libitum for municipal tap water purified by reverse osmosis filtering system using water bottles. Range of acceptable temperature and humidity for animals were 20.0 ± 04 °C and 46.1 ± 3.1%. Male and female body weight range were 7.80–8.30 kg and 6.82–7.38 kg respectively at the administration time. Eight beagle dogs were equally divided into 4 groups (1/sex/group). once-daily oral, the test substance used in a gelatin capsule at 250 (low-dose), 500 (middle-dose), and 1000 (high-dose) mg/kg were administered for 4 weeks using hand, after opened dog’s mouth in a natural condition in cage, gelatin capsules were placed on the rear part of the tongue. Vehicle used sterilized distilled water for control group.
During the study period, animals were observed for mortalities, clinical signs, body weights, food consumption, ophthalmological examination, clinical pathology (urinalysis, hematology and serum biochemistry), necropsy (organ weights measurement and gross findings), and histopathological examination. This study was approved by the Institutional Animal Care and Use Committee (IACUC) (IA17-00740).
The statistical analysis was not performed because only one animal was used per group.
Beagle dogs: 13-weeks repeated dose toxicity and 4-weeks recovery study
Thirteen-week repeated oral dose toxicity test and 4-week recovery study was performed using male and female beagle dogs, aged 5 months, from Producing Beijing Marshall Biotechnology Co., Ltd. (Beijing, China). Male and female animals were acclimated during for 27 days. The healthy animals selected during the acclimation period. All animals were housed in stainless steel cages (700 W × 750 L × 750 H mm) during the experiment period. Each animal was offered a daily ration of 300 g of solid food (LAB CANINE CHOW, Cargill Agri Purina Korea Inc.). Water was provided ad libitum for municipal tap water purified by reverse osmosis filtering system using water bottles. Range of acceptable temperature and humidity for animals were 21.7 ± 0.5 °C and 52.8 ± 8.6%. Male and female body weight range were 6.84–8.44 kg and 5.60–6.46 kg respectively at the administration time. Sixteen beagle dogs were divided into four groups (three or five/sex/group). Once-daily oral, the test substance used in a gelatin capsule at 250 (low-dose), 500 (middle-dose), and 1000 (high-dose) mg/kg were administered for 4 weeks using hand, after opened dog’s mouth in a natural condition in cage, gelatin capsules were placed on the rear part of the tongue. Vehicle used sterilized distilled water for control group. Dose levels were determined at the results of 4-week repeated oral dose range finding study and repeated dose 13-week oral toxicity and 4-week recovery study, it is considered that abnormal clinical finding as salivation was observed in animals at 2000 mg/kg. Dosing volume was adjusted as 10 mL/kg body weight and vehicle used sterilized distilled water for control group.
During the study period, animals were observed for mortalities, clinical signs, body weights, food consumption, ophthalmological examination, clinical pathology (urinalysis, hematology, blood coagulation time and serum biochemistry), necropsy (organ weights measurement and gross findings), and histopathological examination. This study was approved by the Institutional Animal Care and Use Committee (IACUC) (IA17-00539).
Comparisons between the vehicle control group and the test substance-treated group were generally performed using parametric multiple comparison procedures or non-parametric multiple comparison procedures, and statistically significant when *p < 0.05 or **p < 0.01. The rate of occurrence was expressed as a percentage. The statistical analysis was performed according to the standard work instructions for the statistical processing of the test institute using SPSS 12.0 K program (SPSS, Chicago, IL, USA), a widely used statistical package.
Systemic safety assays
Rats: central nervous system assay
The central nervous system assay through a FOB was performed using male SPF SD rats, aged 5 weeks, from Orient Bio Co., Ltd. Animals were observed for clinical findings once daily for 6 days of the quarantine-acclimation period. Then they were acclimated to sham dosing and handling once daily for 5 days. All animals were housed in stainless wire mesh cages, (260 W × 350 D × 210 H mm) and provided food and water ad libitum for municipal tap water purified by reverse osmosis filtering system using water bottles. Range of acceptable temperature and humidity for animals were 21.7 ± 1.3 °C and 54.2 ± 14.8%. Thirty-two SD rats were equally divided into 4 groups (4/sex/group) and once-daily oral (by gavage in the morning) of the test substance at 250 (low-dose), 500 (middle-dose), and 1000 (high-dose) mg/kg. Dose levels were determined as a result of previous study that abnormal clinical finding as salivation was observed in animals at 2000 mg/kg. Thus, 250, 500 and 1000 mg/kg were selected as low, mid and high dose levels, respectively. Dosing volume was adjusted as 10 mL/kg body weight and vehicle used sterilized distilled water for control group.
During the study period, animals were observed for clinical signs and behavior, body weights, food consumption. Observation points were selected at 0 h (pre-dose) and at 0.5, 1, 3, 6 and 24 h post-dose based on the general change in the concentrations of the test substance in the plasma when dosed intravenously.
The FOB evaluations were conducted by blind method with a person for dosing and two observers. Two observers performed the blind observations, including home cage, open field, hand held and sensory-motor function observations, and the measurement of body temperature according to the “ATTACHMENT. Methods and Criteria for Evaluation of Functional Observational Battery” in the protocol. After the completion of the study, all remaining animals were euthanized in accordance with the SOP specifying the procedure of euthanasia of rodents. The FOB was performed under the condition that the animal response to external noise was reduced by generating white noise of 50 ± 10 dB. To evaluate the effects of the test substance, all parameters of the test substance groups for the FOB at each observation point were compared to those of the control group.
Statistical analysis was performed on FOB parameters using SAS Program (Ver. 9.3, SAS Institute Inc., Cary, NC, USA). Quantitative parameters were analyzed utilizing Bartlett test for homogeneity of variance (significance level: 0.05). One-way ANOVA was employed on homogeneous data (significance level: 0.05). If significant, Dunnett’s t test was employed for multiple comparisons (significance levels: 0.05 and 0.01, two-tailed). Kruskal–Wallis test was employed on heterogeneous data (significance level: 0.05). All parameters were not statistically significant. Therefore, Steel’s test was not applied for multiple comparisons. Rank value parameters were analyzed utilizing Kruskal–Wallis test (significance level: 0.05). If significant, Steel’s test was employed for multiple comparisons (significance levels: 0.05 and 0.01, two-tailed).
Rats: respiratory function assay
Respiratory function assay through whole body plethysmography system was performed using male SPF SD rats, aged 5 weeks, from Orient Bio Co., Ltd. Animals were observed for clinical findings once daily for 7 days of the quarantine-acclimation period. Then they were preconditioned to the chambers and sham dosing once daily for 3 days, and animals were placed in the chamber for a certain period of time, so as to be familiar in the administration and unrestrained chambers. All animals were housed in stainless wire mesh cages, (260 W × 350 D × 210 H mm) and provided food and water ad libitum. Range of acceptable temperature and humidity for animals were 21.9 ± 2.5 °C and 49.85 ± 4.15%. Twenty-four SD rats were equally divided into 4 groups (6/group) and once-daily oral (by gavage in the morning) of the test substance at 250 (low-dose), 500 (middle-dose), and 1000 (high-dose) mg/kg. Dose levels were determined as a result of previous study that abnormal clinical finding as salivation was observed in animals at 2000 mg/kg. Thus, 250, 500, and 1000 mg/kg were selected as low, mid and high dose levels, respectively. Dosing volume was adjusted as 10 mL/kg body weight and vehicle used sterilized distilled water for control group. All animals were observed for clinical findings including appearance, behavior and excretion once daily during the experimental period. No abnormal clinical findings were observed in any animal prior to dosing.
Animals were placed individually in the unrestrained chamber prior to dosing to acclimate the animals for approximately 30 min and pre-dose behavior was monitored until data were acquired for a total of 3 min during which their movement was stable. After administration, animals were placed in the unrestrained chamber for about 3 h. The behavior of animals was monitored for 3 min before and after each measuring point, and then transferred back to their cage. They were returned to the unrestrained chambers and acclimated from about 5–6 h post-dose and their behavior was observed for 3 min before and after each measuring point, and then transferred back to their cage. Animals were put in the unrestrained chambers within a reasonable time at 24 h post-dose. After at least 30 min of acclimation, the monitoring of animal behavior was conducted until data were acquired for a total of 3 min during which their movement was stable. After the completion of the study, all remaining animals were euthanized in accordance with the SOP specifying the procedure of euthanasia of rodents.
The monitoring of animal behavior was carried out through video footage transferred from the surveillance cameras and the video footage was recorded at the same time (SRP1650, Samsung Techwin Co., Ltd., Seoul, Korea) to be used as reference material for the interpretation of the data. When animal behavior was unstable at any measuring point, the corresponding time was recorded and the data obtained at that time were excluded from the analysis.
Measuring points of respiratory parameters were selected at 0 h (pre-dose) and at 0.5, 1, 3, 6 and 24 h post-dose based on the general change in the concentrations of the test substance in the plasma when dosed orally. For the measurement of respiratory parameters (respiratory rate, tidal volume and minute volume), each rat was placed in an unrestrained whole body plethysmography system (Buxco Electronics, Inc., Wilmington, NC, USA). Data were collected continuously for each respiratory parameter and the average values for 10 s were obtained continuously at an interval of 10 s using the FinePointe™ program (Ver. 2.3.1.9, DSI™, IWOO Scientific Corporation, Seoul, Korea).
Zero hour (0 h) (pre-dose), 24 h post-dose: The interval in which the behavior of the animal remains stable for a total of 3 min (more than 1 min in minute units). 0.5, 1, 3 and 6 h (post-dose): ± 1 min at each time point (a total of 3 min) Data obtained from unstable animals at each measurement point are excluded from the average value and if less than six data can be selected, then six data from animals stabilized close to the measurement points are selected among the data acquired before and after 3 min of each measurement point. If the number of data that can be selected for 3-min intervals before and after each measurement point is fewer than six, the average value of all the data acquired for 3 min before and after each measurement point is determined.
Statistical analysis was performed on respiratory parameters (respiratory rate, tidal volume and minute volume) using SAS Program (version 9.3, SAS Institute Inc.). Parameters were analyzed using Bartlett test for homogeneity of variance (significance level: 0.05). One-way ANOVA was employed on homogeneous data (significance level: 0.05). All parameters were not statistically significant; therefore, Dunnett’s t test was not applied for multiple comparisons. Kruskal–Wallis test was employed on heterogeneous data (significance level: 0.05). All parameters were not statistically significant. Therefore, Steel’s test was not employed for multiple comparisons.
Dogs: cardiovascular function assay
Cardiovascular function assay was performed using male, 4 beagle dogs, 14 months old, 9.20–12.22 kg. A single dose was administered to the same animal at each of four ascending dose levels (0, 250, 500, and 1000 mg/kg) in the afternoon (13:23–13:51) of each dosing day at 1-week intervals (dose volume was 5 mL/kg). The dosing formulation was administered by gastric intubation using a disposable syringe (50 mL) fitted with a catheter. Evaluation of cardiovascular parameters were blood pressure, heart rate and electrocardiogram. Measuring time points were selected on the basis of the changes of the concentration of the general drug in plasma when dosed orally at 0 h (prior to dosing), 0.5, 1, 2, 3, 4, 6, 8 and 24 h post dosing. The data of blood pressure, heart rate, body temperature and ECG were obtained from 24 h before administration to 24 h after administration using a data collecting system (Dataquest A.R.T.TM Gold Acquisition Version 4.00, Data Science Int., New Brington, MN, USA) for 20 s at 5 min intervals. The data of blood pressure, heart rate and body temperature at each measuring time point of acquisition were recorded for an average of 20 s. The data obtained before the selection range of the acquisition of data for the measuring time points prior to dosing were used as reference data for the identification and assessment of signal quality.
The data of systolic and diastolic blood pressures, heart rate and body temperature were converted to Microsoft Excel® by using a data analysis program (Dataquest A.R.T.TM Gold Analysis Version 4.00, Data Science Int.). The data of systolic and diastolic blood pressures, heart rate and body temperature at each data analysis time point were used to determine the mean value of all data recorded at 5-min intervals in the range of selection of data as described below. The data that were difficult to interpret as signal quality problem due to entry of a person or other external environmental factors were excluded from the data analysis. The mean blood pressure (MBP) of systolic and diastolic blood pressure data was calculated by the following formula:
Measuring time points | The range of data selected |
|---|---|
0 h (prior to dosing) | From approximately 85 min before dosing to approximately 30 min before dosing, data were recorded at 5-min intervals (12 collected data) |
0.5 and 1 h post dosing | Data recorded at 5-min intervals for 25 min prior to each time point (6 collected data) |
2, 3, 4, 6, 8 and 24 h post dosing | Data recorded at a 5-min intervals for 55 min prior to each time point (12 collected data) |
ECG parameters (PR, QRS, QT and QTc intervals) were analyzed (Ponemah physiology platform Version 4.60, DSI/PONEMAH, Data Science Int.) at snapshot times as described below. Five consecutive ECG waveforms with stable baseline recorded over 20 s were selected from each ‘snapshot’ time and then, averaged at each wave interval.
Snapshot: 0 h (prior to dosing) and at 0.5, 1, 2, 3, 4, 6, 8 and 24 h post dosing.
QTc interval was corrected by Fridericia’s formula.
When problems were evident at scheduled snapshot times, the suitable data having no problem of signal quality within the range of data selection at each time point were selected and analyzed.
Statistical analysis was performed using SAS Program (version 9.3, SAS Institute Inc.) on the data including actual measurement values of blood pressure, heart rate, body temperature and measurement values of ECG parameters (PR, QRS, QT and QTc intervals). Time-sequential repeated measures of ANOVA were conducted for the data of blood pressure, heart rate, body temperature and ECG parameters (significance level: 0.05) and since the effect of dose or the effect of interaction between dosage and time was not significant, Dunnett’s t-test was not applied for multiple comparisons between the control and test substance at each measuring time point.
Results
Extract
HPLC analysis
We performed high-pressure liquid chromatography (HPLC) analysis for the composition analysis of APRG64. As shown in Fig. 1, the presence of gallic acid (GA), ethyl gallate (EG), penta-galloyl glucose (PGG), and apigenin-7-O-β-glucuronide (AG) was confirmed. We selected AG as an indicator and functional ingredient of A. Pilosa and PGG as an indicator and functional ingredient of Rhus gall.

Result of HPLC analysis. a apigenin-7-O-β-glucuronide (AG) as an indicator and functional ingredient of Agrimonia Pilosa. b penta-galloyl glucose (PGG) as an indicator and functional ingredient of Rhus gall
Genotoxicity tests
Bacterial reverse mutation test
As shown in Table 1, cytotoxicity was observed at 5000 μg/plate (TA98, TA100, TA1535, TA1537 and WP2uvrA) in the absence of metabolic activation system [S9 mix(−)]. The number of revertant colonies did not show significant increase to judge positive compared with the number of revertant colonies of negative control group. In the presence of metabolic activation system [S9 mix(+)], cytotoxicity was observed at 5000 μg/plate (TA1537 and WP2uvrA) and 1667 and 5000 μg/plate (TA1535). The number of revertant colonies did not show significant increase to judge positive compared with the number of revertant colonies of negative control group.
Mammalian chromosome aberration test
As a result of main test, the number of cells with chromosome aberrations per 100 metaphase cells was 0.33, 0.67, 0.67, 1.33, and 25.67 at negative control, 2.29, 6.86, 20.58 μg/mL and positive control in the absence of metabolic activation system (24 h treatment group). The test substance did not cause statistically significant increase in the number of cells with chromosome aberration at all dose levels when compared with negative control group (Table 2). In the absence of metabolic activation system (6 h treatment and 18 h recovery group), the number of cells with chromosome aberrations per 100 metaphase cells was 0.00, 0.33, 0.67, 0.67, and 23.00 at negative control, 6.86, 20.58, 61.73 μg/mL, and positive control. The test substance did not cause statistically significant increase in the number of cells with chromosome aberration at all dose levels when compared with negative control group (Table 2). In the presence of metabolic activation system (6 h treatment and 18 h recovery group), the number of cells with chromosome aberrations per 100 metaphase cells was 0.67, 0.67, 0.67, 1.00, and 23.33 at negative control, 6.86, 20.58, 61.73 μg/mL, and positive control. The test substance did not cause statistically significant increase in the number of cells with chromosome aberration at all dose levels when compared with negative control group (Table 2). In the presence and absence of metabolic activation system, the test substance did not cause statistically significant in the number of cells with polyploidy and endoreduplication, when compared with negative control group (Table 2). However, positive controls induced statistically significant increase in the number of cells with chromosome aberration when compared with negative control group (p < 0.05) (Table 2).
Mammalian erythrocyte micronucleus test
As a result of observation, no special abnormality was observed compared to negative control group, and there was no statistically change in body weight at any group compared to negative control group (data not shown). The results of microscopic observations in the micronucleus assay for mice groups were summarized in the Table 3. There was no statistically increase in frequency of PCEs having micronuclei in administration group compared to negative control group, and clear statistically significant increase in positive control group compared with negative control group in frequency of MNPCE (p < 0.01).
Oral toxicity study
Single dose toxicity study
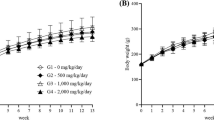
This study was conducted to investigate toxic symptoms and approximate lethal dose (LD) by single oral administration of the test substance, APRG64. There were no mortalities during the experimental period. Some animals in the male and female 2000 mg/kg dosing group showed salivation right after treating the test substance. However, they do not have toxicological significances because it is considered that the secretion of saliva was promoted temporarily. As shown in Fig. 2, in body weight, temporary decreases were observed in female of the vehicle control and the 1000 mg/kg dosing group, respectively. However, it was shown in the vehicle control group and mild change without results that support the decrease in other experimental items. Therefore, they are not considered to be affected by the test substance. As a result of the autopsy after the end of the experiment, no unusual macroscopic findings were observed in all the experimental animals.

Single dose toxicity study; body weights of male and female rats. Body weight changes of vehicle control and APRG treated group (n = 5 per each groups)
Rats: 4-weeks repeated dose toxicity determination study (DRF)
This study was conducted to investigate the no observed adverse effect level (NOAEL) by oral administration of the test substance APRG64 for 4 weeks and to set the dose of 13 weeks repeated oral toxicity study. There were no mortalities in all treated groups during the experimental period. Salivation was observed right after administration in some animals of the male and female 1000 mg/kg dosing groups intermittently and in all animals of the male and female 2000 mg/kg dosing groups consistently. Other unusual clinical signs were not observed (data not shown). And there were no statistically significant differences between the vehicle control and dosing groups during the experimental period, due to weight change, feed intake and water consumption, and ophthalmological examination results.
As shown in Table 4, bilirubin and urine color parameters shown significant differences among male and female groups (p < 0.01 or p < 0.05). In case of color, observation frequency of yellow or dark yellow color were higher in the male and female 1000 and 2000 mg/kg dosing groups compared with that of the vehicle control group (p < 0.01). This can lead to differences in metabolites by sex hormones in males and females [12] Dark yellow urine is only observed during the medication period and is probably due to urine excretion of the test substance or its metabolites. This can lead to an increase in ketone bodies due to urine pigments associated with changes in urine color [13]. In addition, female rats had significant differences among groups in parameters such as glucose, ketone body, specific gravity, occult blood and pH (p < 0.01 or p < 0.05) (Table 4). Especially specific gravity showed a tendency to increase with dose–response correlation, while pH showed a tendency to decrease with dose–response correlation. Bilirubin may produce false positive reactions due to the interference of some compounds or metabolites [14]. Except that, there were no statistically significant differences among groups in other inspection items. Also, urine sediment test on the leukocyte showed that there were no statistically significant differences among groups.
In Table 5, male rats hematological test revealed statistically significantly lower values when compared to the that of vehicle control group in the following parameters: Hb (hemoglobin) value in the male 500, 1000, and 2000 mg/kg dosing group, HCT (hematocrit) value in the male 1000 mg/kg dosing group, and MPV (mean platelet volume) value in the male 2000 mg/kg dosing group (HCT; p < 0.05, Hb, MPV; p < 0.01). Except that, there were no statistically significant differences among vehicle control and dosing groups in other inspection items.
Also, in male groups (Table 6), statistically significant increases were detected on Cl (chloride) value in the 1000 and 2000 mg/kg dosing groups and ALT (alanine aminotransferase) value in the 2000 mg/kg dosing groups when compared with that of vehicle control group (ALT; p < 0.05, Cl; p < 0.01). On the other hand, significant decreases were found on BUN (blood urea nitrogen) value in the 500, 1000, and 2000 mg/kg dosing groups, Na (sodium) value in the 1000 and 2000 mg/kg dosing groups, and GLU (glucose) and PRO (protein) values in the 2000 mg/kg dosing group (GLU, PRO; p < 0.05, Na, BUN; p < 0.01). Other statistically significant differences were not observed between the vehicle control and dosing groups in other inspection items.
At scheduled necropsy, there were no abnormal gross findings in all animals. Also, there were no statistically significant differences among vehicle control and dosing groups in absolute and relative organ weights.
Rats: 13-weeks repeated dose toxicity and 4-weeks recovery study
There were no mortalities during treatment and recovery period. Salivation was observed right after administration of the test substance in the male and female 1000 and 2000 mg/kg/day dosing groups throughout treatment period. No abnormal clinical signs were detected in other animals and experimental period (including recovery period).
In the body weight change, there were no statistically significant differences between vehicle control and dosing groups during exposure and recovery periods.
Food consumption in the female 1000 and 2000 mg/kg/day dosing group statistically significantly increased on week 5 compared to the that of vehicle control group (p < 0.05, data not shown). Except for that, there were no statistically significant differences between vehicle control and dosing groups during exposure and recovery periods. Also, there were no abnormalities detected in all animals of main and recovery groups subjected to the ophthalmological examination.
The urinalysis results (Table 7) as follows:
Main groups
In urinalysis, detection frequency of ketone body shown significant differences among female groups, which the change had increase tendency with dose–response relationship (p < 0.01, Table 7). Although there was no significant difference, increase tendency of ketone body was also found in male groups. In urine color, significant difference was observed among female groups showing dark yellow, dark brown, and dark green color urine in dosing groups (p < 0.01). Dark yellow and dark brown color urine were detected in male dosing groups (but no significant difference). Some metabolites are more significant difference in females than males because sex hormones may affect metabolic processes [12].The occurrence of dark yellow urine is only observed during the sample treatment period, which may be due to urine excretion of the test substance or its metabolites [13]. In addition, female had significant differences among groups in parameters such as occult blood (p < 0.01) and nitrite (p < 0.05). But, it concluded that trace changes occur or there is no dose-related correlation. Except that, no statistically significant differences were observed among all the male and female groups, regarding urinalysis, urine sediment test, and urine volume.
Recovery groups
In urinalysis, urine sediment test, and urine volume, there were no statistically significant differences among groups.
The hematology results (Table 8) as follows:
Main groups
Significant increases compared to the control group value were observed in following values; the LY (lymphocyte) value in the female 1000 mg/kg/day dosing group and the WBC (white blood cell), LY and MCV (mean corpuscular volume) value in the female 2000 mg/kg/day dosing group (p < 0.05). No other statistically significant differences were observed between vehicle control and dosing groups.
Recovery groups
The EO (eosinophil) value in the male 2000 mg/kg/day dosing group was significantly lower than that in the vehicle control group (p < 0.05, data not shown). No other statistically significant differences were observed between vehicle control and dosing groups. In blood coagulation test, there were no statistically significant differences between vehicle control and dosing groups in main and recovery groups.
The serum biochemistry results (Table 9) as follows.
Main groups
The Na (sodium) value in the female 500, 1000, and 2000 mg/kg/day dosing group was significantly lower when compared to that of vehicle control group (p < 0.01). There were no statistically significant differences between vehicle control and dosing groups in other inspection items.
Recovery groups
Data not shown, the Cl (chloride) value was statistically significantly decreased in the male 2000 mg/kg/day group compared with that of vehicle control group (p < 0.05) and Mg (magnesium) value was significantly increased in the female 2000 mg/kg/day group (p < 0.01). No other statistically significant differences were observed in other inspection items.
And at necropsy, there were no abnormal gross findings in all animals in main and recovery groups.
In organ weights results (Table 10) as follows:
Main groups
In absolute organ weights, right ovary of the female 1000 mg/kg/day dosing group was significantly heavier than that of vehicle control group (p < 0.05). No statistically significant differences in the absolute and relative organ weights were observed between vehicle control and treated groups in both the males and females.
Recovery groups
In absolute organ weights, significant decreases were observed in right ovary and lung weight of the female 2000 mg/kg/day group (p < 0.05, data not shown). No statistically significant differences were observed in other organ weights.
The histopathological examination results as follows:
Lung
Histopathological examination for lung in the vehicle control and 2000 mg/kg/day main and recovery groups of both sexes revealed as follows: focal/multifocal, inflammatory, cell infiltration, bronchiolalveolar was sporadically found in 1–4 animals of each main group, and 0–2 animals of each recovery group. In addition, focal foreign body with alveolar macrophage was confirmed in alveolar of one animal (animal ID G4-88) in the female 2000 mg/kg/day main group.
Other organs and tissues
Histopathological examination for the organs and tissues from the vehicle control and 2000 mg/kg/day groups were conducted. At the results, following lesions were observed solitarily in main groups; focal inflammation in kidney cortex, developmental cyst in pituitary, and unilateral focal atrophy in seminiferous. In recovery groups, multifocal vacuolization in cytoplasmic of liver and cardiomyopathy in heart were solitarily found.
Beagle dogs: 4-weeks repeated dose toxicity determination study (DRF)
This study was conducted to investigate the toxicity of the test substance APRG64 when repeatedly orally administered to non-rodent Beagle dog for 4 weeks, and to establish a capacity setting basis for the subsequent repeat toxicity test. There was no mortality during the study period. No clinical signs related to the administration of test substance were observed. Except for that, anorexia and vomiting were sporadically observed without sex and dose-dependency. As a result of body weight measurement, the decrease of body weight gain was observed only in female 500 and 1000 mg/kg/day administration group. However, this is considered to be a change not within the normal weight range and not related to the administration of the test substance. There were no food consumptions changes related to the administration of test substance were observed. And no abnormal signs related to the administration of test substance were observed in ophthalmoscopy.
The urinalysis results that there were no significant changes related to the administration of test substance compared with the control group and before administration (data not shown).
In the hematology results (Table 11), increasing tendency of LYP (percent of lymphocyte) values was observed in males at dosing group compared with before administration. Decreasing tendency of retic (reticulocyte) values was observed in females at dosing group compared with before administration. Except for that, there were no significant changes in other items.
And serum biochemical results (Table 12) that increasing tendency of K (potassium) values in males, GLU (glucose) and A/G ratio (albumin/globulin ratio) values in females were observed at dosing group compared with before administration. Decreasing tendency of CHO (total cholesterol) values in males and Ca (calcium) in females was observed at dosing group compared with before administration. Except for that, there were no significant changes in other items.
When necropsy, in male at 1000 mg/kg/day group, small testis (bilateral), epididymis (bilateral) and prostate were observed (data not shown).
In male at 1000 mg/kg/day group, decrease tendency in absolute and relative weights of testis, epididymis and prostate were observed. In females, increasing tendency in absolute and relative weights of thyroid (bilateral) at dosing group were observed compared with control group. Except for that, there were no significant changes in other inspection organs. In histopathological examination, in male at 1000 mg/kg/day group, small testis (bilateral), epididymis (bilateral) and prostate were considered to immature lesion (data not shown).
Beagle dogs: 13-weeks repeated dose toxicity and 4-weeks recovery study
The test substance, APRG64, was orally administered to non-rodent beagle dog for 13 weeks, and the toxicity of the substance was investigated and the recovery was evaluated by 4 weeks of the recovery period. There was no mortality during the study period. Salivation was observed in 250 mg/kg/day treated male, 500 and 1000 mg/kg/day treated male and female groups. In addition, compound-colored feces were observed in 500 and 1,000 mg/kg/day treated male and female dogs, and anorexia was observed in male and female dogs treated with 250, 500, and 1000 mg/kg/day. As a result, there was a tendency to decrease feed consumption in the dosing group, and a tendency to lose weight and decrease weight gain was observed. And no abnormal signs related to the administration of test substance were observed in ophthalmoscopy. No changes related to the administration of the test substance were observed as a result of ophthalmological examination, electrocardiogram, and hematological test.
The urinalysis results (Table 13) as follows:
Main groups
There was a statistically significant increase (p < 0.05) in ketone body and protein in the dosing group males. Urinary proteins are generally found in trace amounts in normal condition, and are known to be more prominent in rats and dogs. Males are known to be more affected than females, and many reports have shown similar trends [15,16,17,18,19,20,21].
Recovery groups
No changes associated with the administration of the test substance were observed. A significant increase (p < 0.05) was observed in the occult blood of the female dosing group compared to the vehicle control group. However, changes not observed in the main groups are not considered to be related to the transfer of test substances.
In the hematology results (Table 14),
Main groups
In the blood test at the 4th week of administration, there was a significant decrease in MCH (Mean corpuscular hemoglobin) levels and female LYP (Percent of lymphocyte) values in males treated with 1000 mg/kg/day as compared with the vehicle control group (p < 0.05). NEP (percent of neutrophil) and NE (Neutrophil) levels of females treated with 1000 mg/kg/day were significantly increased compared to the vehicle control group (p < 0.05). At the 13th week of administration, there was a significant decrease in male HGB (hemoglobin, p < 0.05), HCT (hematocrit, p < 0.05), MCV (Mean corpuscular volume, p < 0.01), MCH (p < 0.01) and females MCHC (Mean corpuscular hemoglobin concentration, p < 0.01) levels of compared to the vehicle control group at 1000 mg/kg/day. Also, Retic (%, reticulocyte) values of male 500 and 1000 mg/kg/day treated group were significantly increased compared with the vehicle control group (p < 0.01).
Recovery groups
No abnormal signs related to the administration of test substance were observed in hematology test.
The serum biochemical test results (Table 15) were as follows,
Main groups
At the 13th week of administration, a significant increase was observed in male AST (Aspartate aminotransferase) and ALT (Alanine aminotransferase) treated with 1000 mg/kg/day compared to the vehicle control group (p < 0.01). And, AST and ALT in female group of 1000 mg/kg/day showed a tendency to increase compared to the vehicle control group. Therefore, histopathological examination was implemented in both females and males to confirm the above results. Except, a significant increase was observed in CRE (creatinine) from the male 250 mg/kg/day group at the 13th week of administration compared to the vehicle control group. However, it was concluded that there was no dose-related correlation.
As a result of blood coagulation time test. (Table 16),
Main groups
APTT (active partial thromboplastin time) was significantly increased in 1000 mg/kg/day of male and female compared to vehicle control (p < 0.01).
Recovery groups
APTT increase tendency was observed in male and female 1000 mg/kg/day.
And at necropsy, black-gray colored mottled was observed in the duodenum of the male 1000 mg/kg/day group and gray colored degeneration was observed in the duodenum of the female 1000 mg/kg/day group, respectively one. These changes are estimated to be due to the test substance. However, there was no toxicological significance because the degree was mild and there was no continuous change in the recovery group.
In absolute organ weights, no statistically significant changes were observed in either the main groups or recovery groups.
Finally, histopathological examination was as follows,
Main groups
In both sex animals of 1000 mg/kg/day groups, we observed the central lobular ballooning degeneration in the liver (three male and one female, respectively). Also, in male 500 mg/kg/day group, we observed the liver central lobular rarefaction (one of the three).
Recovery groups
In the liver of 1000 mg/kg/day group, the central lobular ballooning degeneration was observed in two male and two female. Thus, NOAEL in beagle dog of the test substance is judged to be 500 mg/k/day for both male and female, and the target organ is judged to be liver.
Systemic safety assays
Central nervous system assay
This study evaluated the effects on the central nervous system by performing a single oral administration of the test substance to male SD rats at 6 weeks of age and performing functional observational battery. No statistically significant or biologically relevant differences were noted in any parameter in animals at 250 and 500 mg/kg when compared to the control group at any measuring point. Therefore, it is considered that there was no effect of the test substance on the central nervous system. A statistically significant increase in the number of urine pools was noted in animals at 1000 mg/kg when compared to the control group at 1 h post-dose. However, it is considered not to be a test substance-related effect because there was no biologically relevant difference in comparison to the measured values prior to dosing (0 h) and no significant differences in the related parameters such as the number of fecal pellets. No statistically significant or biologically relevant differences were noted in the other parameters when compared to the control group.
In Home Cage Observations (Table 17), no statistically significant or biologically relevant differences in body posture, behavior or ease of removal from cage were noted in any animal at 250, 500, and 1000 mg/kg when compared to the control group prior to dosing (0 h) and at 0.5, 1, 3, 6 and 24 h post-dose.
In Open Field Observations (Table 18), no statistically significant or biologically relevant differences in the number of unit areas crossed, rearing counts, respiration rate, stereotypy, unusual behavior, tremor, convulsion, gait, arousal level or number of fecal pellets and urine pools were noted in any animal at 250 and 500 mg/kg when compared to the control group prior to dosing (0 h) and at 0.5, 1, 3, 6, and 24 h post-dose. A statistically significant increase (p < 0.05) in the number of urine pools was noted in animals at 1000 mg/kg at 1 h post-dose when compared to the control group, and no significant difference was noted at other observation time points. No statistically significant or biologically relevant differences were noted in any animal at 1000 mg/kg when compared to the control group prior to dosing (0 h) and at 0.5, 1, 3, 6, and 24 h post-dose.
In Hand Held Observations, no statistically significant or biologically relevant differences in fur and skin appearance, piloerection, contaminated abdominal region, lacrimation, salivation, palpebral closure, pupil size, pupillary reflex, muscle tone or extensor-thrust reflex were noted in any animal at 250, 500, and 1000 mg/kg when compared to the control group prior to dosing (0 h) and at 0.5, 1, 3, 6 and 24 h post-dose (data not shown).
In sensori-motor function observations (data not shown), no statistically significant or biologically relevant differences in the visual, touch, click and tail pinch responses, aerial righting reflex, hindlimb landing foot splay, forelimb grip strength or hindlimb grip strength were noted in any animal at 250, 500, and 1000 mg/kg when compared to the control group prior to dosing (0 h) and at 0.5, 1, 3, 6, and 24 h post-dose.
Also, in determined of body temperature (data not shown), no statistically significant or biologically relevant difference in rectal temperature was noted in any animal at 250, 500, and 1000 mg/kg when compared to the control group prior to dosing (0 h) and at 0.5, 1, 3, 6 and 24 h post-dose.
No change in the test substance 250, 500, and 1000 mg/kg administration groups was observed in all parameters of FOB. From the above results, it can be concluded that the test substance had no effect on the central nervous system at doses of 250, 500, and 1000 mg/kg in a single oral administration using the rats under the test conditions.
Respiratory function assay
This study evaluated respiratory effects by measuring the respiratory rate, tidal volume, and minute volume under no anesthesia and no restraint conditions after single oral administration of the test substance, APRG64, to male SD rats at 6 weeks of age. No statistically significant or biologically relevant differences in the respiratory rate, tidal volume or minute volume were noted in the animals at 250, 500, and 1000 mg/kg when compared to the control group at any measuring point. Therefore, it is considered that there were no effects of the test substance after the administration.
In the respiratory rate (Fig. 3a), no statistically significant or biologically relevant differences in the respiratory rate were noted in any animal at 250, 500, and 1000 mg/kg when compared to the control group at 0 h (pre-dose) and at 0.5, 1, 3, 6, and 24 h post-dose.

Respiratory function. a respiratory rate, b tidal volume, c minute volume. No statistically significant or biologically relevant differences were noted in any animal at 250, 500, and 1000 mg/kg when compared to the control group at 0 h (pre-dose) and at 0.5, 1, 3, 6, and 24 h post-dose
Tidal volume results (Fig. 3b) that no statistically significant or biologically relevant differences in the tidal volume were noted in any animal at 250, 500, and 1000 mg/kg when compared to the control group at 0 h (pre-dose) and at 0.5, 1, 3, 6, and 24 h post-dose.
In minute volume (Fig. 3c), no statistically significant or biologically relevant differences in the minute volume were noted in any animal at 250, 500, and 1000 mg/kg when compared to the control group at 0 h (pre-dose) and at 0.5, 1, 3, 6, and 24 h post-dose.
From the above results, it was concluded that APRG64, which is a test substance, has no effect on the respiratory system at doses of 250, 500, and 1000 mg/kg in a single oral administration using the rats under the test conditions.
Cardiovascular function assay
This test was conducted to evaluate the effects on the cardiovascular system by blood pressure, heart rate and electrocardiogram after oral administration of the test substance to four male beagle dogs with no anesthesia and no restraint and inserted with a remote transmitter. In cardiovascular parameters, no statistically significant differences or biologically relevant differences in the blood pressure (systolic, diastolic and mean blood pressures), heart rate, and electrocardiogram parameters (PR, QRS, QT and QTc intervals) were noted in animals dosed at 250, 500, and 1000 mg/kg at any measuring time point when compared to the control values. Therefore, it is considered that there were no effects of the test substance on the blood pressure (systolic, diastolic and mean blood pressures), heart rate and electrocardiogram parameters (PR, QRS, QT and QTc intervals) after administration.
In blood pressure and heart rate results (Fig. 4) were follow as: No statistically significant differences or biologically relevant differences in the blood pressure (systolic, diastolic and mean blood pressures) and heart rate were noted in animals dosed at 250, 500, and 1000 mg/kg at any measuring time point when compared to the control values.

Blood pressure and heart rate. a Systolic blood pressure, b diastolic blood pressure, c mean blood pressure, d heart rate. No statistically significant differences or biologically relevant differences in the blood pressure (systolic, diastolic and mean blood pressures), heart rate, and electrocardiogram parameters were noted in animals dosed at 250, 500, and 1000 mg/kg at any measuring time point when compared to the control values
The result of electrocardiogram (Fig. 5) were follow as: No statistically significant differences or biologically relevant differences in the electrocardiogram parameters (PR, QRS, QT and QTc intervals) were noted in animals dosed at 250, 500, and 1000 mg/kg at any measuring time point when compared to the control values.

Electrocardiogram. a PR interval, b QRS duration, c QT interval, d QTc interval. No statistically significant differences or biologically relevant differences in the electrocardiogram parameters (PR, QRS, QT and QTc intervals) were noted in animals dosed at 250, 500, and 1000 mg/kg at any measuring time point when compared to the control values
Discussion
We conducted a genotoxic and general toxicity test to determine the safety of APRG64, which we intend to develop as a new natural product medicine. First, the results of the three genotoxicity tests are as follows. As a result of the bacterial reverse mutation test (Ames test) using the direct method and the metabolic activation method, it was judged that APRG64 did not induce a reverse mutation under the test conditions set in this study. As a result of chromosome aberration test using CHO-K1 cell derived from Chinese hamster, APRG64 did not induce chromosome aberration under the test conditions set in this study. In addition, an erythrocyte micronucleus induction test using an ICR mouse was carried out, and it was confirmed that the micronucleus was not induced in the bone marrow cells of the mice under the conditions of this test.
General toxicity studies consist of a single dose with rodent, 4-week repeated oral dose toxicity determination study (DRF), and 13-week repeated oral dose toxicity and 4-week recovery study with rodent and non-rodent. In addition, the central nervous system, respiratory system, and cardiovascular function safety study was conducted. The rats were dosed once orally to determine the approximate lethal dose (LD). No toxic symptoms were observed and the approximate LD was > 2000 mg/kg. Based on this result, the high-dose was set at 2000 mg/kg/day and the 4-week repeated dose toxicity determination study (DRF) was performed. No toxicological changes were observed by APRG64. Thereafter, the NOAEL was assumed to be 2000 mg/kg/day, and conducted to 13-week repeated dose toxicity and 4-week recovery study. In conclusion, the NOAEL of APRG64 for rodents was confirmed to be 2000 mg/kg/day, and no toxic target organ was observed. At the same time, DRF was performed on non-rodents beagle dogs. Since no toxicity 1000 mg/kg/day of APRG64 was observed when repeated oral administration for 4 weeks, the groups of 13-week repeated dose toxicity and 4-week recovery studies was set at concentrations of 250, 500, 1000 mg/kg/day. Under this study condition, it was confirmed that there is no effect in the central nervous system, respiratory system and cardiovascular test conditions. However, we confirmed the central lobular balloon degeneration of the liver at 1000 mg/kg. Thus, the NOAEL of APRG64 for beagle dogs was 500 mg/kg/day for both males and females, and the toxic target organ was judged to be liver.
The results of this safety study will be of great help in the subsequent pharmacological use and development of APRG64.
Abbreviations
- AG:
-
Apigenin-7-O-β-glucuronide
- AP:
-
Agrimonia Pilosa
- APRG64:
-
AP and RG of each 50% ethanolic extracts mixed 64
- CHO-K1:
-
Chinese hamster ovary
- EG:
-
Ethyl gallate
- FOB:
-
The functional observational battery
- GA:
-
Gallic acid
- HPLC:
-
High performance liquid chromatography
- ICR:
-
CrljOriCD1
- KCL/CRO:
-
Korea Conformity Laboratories
- MBP:
-
The mean blood pressure
- MFDS:
-
The Ministry of Food and Drug Safety
- MMC:
-
Mitomycin C
- MNPCE:
-
Micronucleated polychromatic erythrocytes
- NCE:
-
Normochromatic erythrocytes
- PCE:
-
Polychromatic erythrocytes
- PGG:
-
Penta-galloyl glucose
- RG:
-
Rhus javanica gall
- SOPs:
-
Standard operation procedures
- SPF:
-
Specific pathogenic free
References
Wyk AV, Prinsloo G (2018) Medicinal plant harvesting, sustainability and cultivation in South Africa. Elsevier 277:335–342
Raynor DK, Dickinson R, Knapp P, Long AF, Nicolson DJ (2011) Buyer beware? Does the information provided with herbal products available over the counter enable safe use? BMC Med 9:94
Boas GRV, Santos ACD, Souza RIC, Araújo FHSD, Traesel GK, Marcelino JM, Silveira APSD, Farinelli BCF, Cardoso CAL, Lacerda RBD et al (2018) Preclinical safety evaluation of the ethanolic extract from guavira fruits (Campomanesia pubescens (D.C.) O. BERG) in experimental models of acute and short-term toxicity in rats. Elsevier 118:1–12
Nguyen DH, Seo UM, Zhao BT, Le DD, Seong SH, Choi JS, Min BS, Woo MH (2017) Ellagitannin and flavonoid constituents from Agrimonia pilosa L. with their protein tyrosine phosphatase and acetylcholinesterase inhibitory activities. Bioorg Chem 72:293–300
Min BS, Kim YH, Tomiyama M, Nakamura N, Miyashiro H, Otake T, Hattori M (2001) Inhibitory effects of Korean plants on HIV-1 activities. Phytother Res 15:481–486
Shin WJ, Lee KH, Park MH, Seong BL (2010) Broad-spectrum antiviral effect of Agrimonia pilosa extract on influenza viruses. Microbiol Immunol 54:11–19
Zhou RJ, Ye H, Wang F, Wang JL, Xie ML (2017) Apigenin inhibits d-galactosamine/LPS-induced liver injury through upregulation of hepatic Nrf-2 and PPARgamma expressions in mice. Biochem Biophys Res Commun 493:625–630
Lee HA, Hong SH, Han SJ, Kim OJ (2011) Antimicrobial effects of the extract of Galla rhois on the long-term swine clinical trial. J Vet Clin 28:1–6
Kim SH, Park HH, Lee S, Jun CD, Choi BJ, Kim SY, Kim SH, Kim DK, Park JS, Chae BS et al (2005) The anti-anaphylactic effect of the gall of Rhus javanica is mediated through inhibition of histamine release and inflammatory cytokine secretion. Int Immunopharmacol 5:1820–1829
Behrendt P, Perin P, Menzel N, Banda D, Pfaender S, Alves MP, Thiel V, Meuleman P, Colpitts CC, Schang LM et al (2017) Pentagalloylglucose, a highly bioavailable polyphenolic compound present in Cortex moutan, efficiently blocks hepatitis C virus entry. Antiviral Res 147:19–28
MFDS (2015) Toxicity test standards of medicine and medical supplies. Ministry of Food and Drug Safety (MFDS), No. 2015-82
Fan S, Yeon A, Shahid M, Anger JT, Eilber KS, Fiehn O, Kim J (2018) Sex-associated differences in baseline urinary metabolites of healthy adults. Sci Rep 8:11883
Shin SH, Koo KH, Bae JS, Cha SB, Kang IS, Kang MS, Kim HS, Heo HS, Park MS, Gil GH et al (2013) Single and 90-day repeated oral dose toxicity studies of fermented Rhus verniciflua stem bark extract in Sprague–Dawley rats. Food Chem Toxicol 55:617–626
Baader E, Bickel M, Damm D, Donaubauer HH, Fehlhaber HW, Grotsch H, Gunzler V, Teetz V, Volz M (1994) Interference in clinical laboratory tests, with special regard to the bilirubin assay: effects of a metabolite of the new prolyl 4-hydroxylase inhibitor, Lufironil. Eur J Clin Chem Clin Biochem 32:515–520
Zhong-Ze Han H-DX, Kim Kwang-Ho, Ahn Tae-Hwan, Bae Jin-Sook, Lee Ji-Young, Gil Ki-Hyun, Lee Joo-Young, Woo Su-Jung, Yoo Hyun-Jung, Lee Hyun-Kul, Kim Kap-Ho, Park Chan-Koo, Zhang Hu-Song, Song Si-Whan (2010) Reference data of the main physiological parameters in control sprague–dawley rats from pre-clinical toxicity studies. Lab Anim Res 26:153–164
Snell KC (1967) Pathology of laboratory rats and mice. Blackwell, Oxford, pp 104–145
Bolton WK, Benton FR, Maclay JG, Sturgill BC (1976) Spontaneous glomerular sclerosis in aging Sprague–Dawley rats. I. Lesions associated with mesangial IgM deposits. Am J Pathol 85:277–302
Alt JM, Deerberg F, Hackbarth HJ, Stolte H (1980) The study of urinary protein excretion in male rats as compared with human proteinuria. Contrib Nephrol 19:79–87
Verhagen AM, Attia DM, Koomans HA, Joles JA (2000) Male gender increases sensitivity to proteinuria induced by mild NOS inhibition in rats: role of sex hormones. Am J Physiol Renal Physiol 279:F664–F670
Kohn DF (2002) Laboratory animal medicine. Academic Press, New York, pp 121–165
Baylis C (2005) Changes in renal hemodynamics and structure in the aging kidney; sexual dimorphism and the nitric oxide system. Exp Gerontol 40:271–278
Acknowledgements
This research was supported by the Ministry of Trade, Industry & Energy (MOTIE), Korea Institute for Advancement of Technology (KIAT) through the Global collaborate R&D program and the MSIT (Ministry of Science and ICT), Korea, under the ICT Consilience Creative program (IITP-2019-2015-0-00742) supervised by the IITP (Institute for Information and Communications Technology Planning and Evaluation).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Rights and permissions
About this article

Cite this article
Park, JH., Ra, JS., Kwon, J.E. et al. Evaluation of genetic toxicity, acute and sub-chronic oral toxicity and systemic safety of Agrimonia pilosa and Rhus gall 50% ethanolic extract mixture (APRG64) in vitro and in vivo (rodent and non-rodent animal models). Toxicol Res. 36, 367–406 (2020). https://doi.org/10.1007/s43188-020-00042-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43188-020-00042-5