
Abstract
To find an asphaltene thermodynamic model that can predict phase behavior and precipitation at reservoir and surface conditions and can consider mixing and titration with intermediate hydrocarbons is a challenge for design of petroleum production systems. In addition, regression of equation of state (EOS) to describe the precipitated solid can lead to non-physical and inconsistent parameters during model preparation. In this study, Flory–Huggins (FH) solution theory is combined with two EOSs, Peng–Robinson (PR) and perturbed chain statistical associating fluid theory (PC-SAFT) for fluid phase behavior modeling and asphaltene precipitation prediction. In this modeling, pseudo-liquid asphaltene-rich phase is considered and based on the different natures of asphaltene molecules, polydisperse (PD) approach is implemented for characterization to provide a physically consistent model in addition to a monodisperse (MD) approach. For evaluation of the suggested modeling approach, two samples of laboratory data published by Buenrostro‐Gonzalez et al. (AIChE J 50:2552–2570, 2004) are studied. Several combinations of models are used including PC-SAFT + FH + MD, PC-SAFT + FH + PD, PR + FH + MD and PR + FH + PD for these samples. The accuracy of the predictions for fluid C1 and Y3 are 2.7, 5.2 (PC-SAFT + FH + MD, bubble pressure), 3.4, 2.6 (PC-SAFT + FH + MD, titration), 0.08, 6.3 (PC-SAFT + FH + MD, onset pressure), 2.4, 6.3 (PR + FH + MD, bubble pressure), 3.4, 2.5 (PR + FH + MD, titration), 1.2, 5.7 (PR + FH + MD, onset pressure) respectively compared to 4.53, 6.3 (bubble pressure), 7.39, 6.9 (titration), 4.57, 7.008 (onset pressure) for SAFT-VR from the literature. In addition, the deviations are 2.7, 5.2 (PC-SAFT + FH + PD, bubble pressure), 2.3, 1.8 (PC-SAFT + FH + PD, titration), 0.4, 0.8 (PC-SAFT + FH + PD, onset pressure), 2.4, 6.2 (PR + FH + PD, bubble pressure), 2.6, 2.3 (PR + FH + PD, titration), 1.3, 2.4(PR + FH + PD, onset pressure).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Crude oil is a mixture of different organic compounds with different boiling points. During the crude oil production process, from porous media to production facilities, the hydrodynamic and thermodynamic conditions change. These changes in some cases cause problems in various sectors of the oil industry. One of the most well-known and important problems is asphaltene precipitation, which imposes excessive costs during the crude oil production process. Asphaltene are the heaviest and polar component of crude oil, which are separated by addition of non-polar solvents such as normal alkanes to oil and are soluble in aromatic compounds such as toluene. Among crude oil compounds, asphaltene have the highest molecular weight, which varies from several hundred to several thousand depending on the type of oil and its conditions (Ancheyta et al. 2002). Asphaltene contain substances with different chemical and physical structures; they include aromatic and saturated rings, aliphatic, some heteroatoms, as well as some metals such as iron, nickel, and vanadium (Ashtari et al. 2011). Many factors are effective including pressure change, temperature change and flow regime change. It is therefore vital to know “when”, “how much” and “under what conditions” these heavy compounds precipitate. Asphaltene precipitation causes many problems in the oil recovery, transport, and production industries, such as precipitation and clogging of the porous surfaces of the reservoir formations, clogging of well equipment, transmission lines and oil production equipment. Prediction of formation conditions and amount of precipitated asphaltene, is necessary to prevent and reduce damage (Neuhaus et al. 2019; Afra et al. 2020). There are two well-known structures for asphaltene structure: the island and the archipelago model. In the island model, there is an aromatic core to which 7–10 alkyl chains are attached. These molecules are of distinct sizes and different natures even in one sample of crude oil. The main indication of this is different amount/nature of precipitated asphaltenes from a crude sample by titration. In the archipelago model, there are several cores connected by branches (Schulze et al. 2015; Chacón-Patiño et al. 2017). Asphaltene precipitation models can be classified into two broad categories: reversible and irreversible. Irreversible models are in fact the same colloidal model based on the assumption of stability of asphaltene with resin and were first used by Leontaritis and Mansoori (1987). These reversible models are based on thermodynamic equilibrium, which are the largest group of models, have attracted more attention (and are more physically consistent with reversible precipitation of asphaltene) (Hirschberg et al. 1984).
The reversible models can be divided into three categories considering the tools employed to model the precipitated phase: activity coefficients’ models (mostly regular solution theory) solid (solid solution and multisolid approaches) models and EOS-based models (including cubic and SAFT families). The latter assumes asphaltene phase is a liquid like mixture and calculates compressibility factor for it. For example, if a model uses PC-SAFT for liquid hydrocarbon and solid solution for asphaltene phase, then it lies within second category.
Solubility models
Hildebrand and Scott (1962) first defined the solubility parameter as the square root of the ratio of the cohesion energy to the volume of pure liquid. Asphaltene has the highest solubility parameter among crude oil components and its value is between 19 and 24 \((\text{MPa}^{0.5})\). Any change in temperature, pressure and crude oil composition changes the value of the solubility parameter and causes asphaltene precipitation. Flory (1942) and Huggins (1941) later developed the Gibbs free energy equation for polymers, and then Scott and Magat (1945) based on this theory, developed their own theory for heterogeneous polymer structures. In this theory, asphaltene is considered as a combination of polymers that have different molecular weights. Hirschberg et al. (1984) used Flory–Huggins theory and SRK equation to model the vapor–liquid-asphaltene three-phase system. For convenience, they first calculated vapor–liquid equilibrium and then assumed that the asphaltene precipitation did not affect the vapor–liquid equilibrium. They also investigated temperature and pressure effect on asphaltene flocculation. However, this model overestimated the asphaltene solubility at very high dilution ratios. Similarly, the model predicted that asphaltenes are in dissolved state at much lower pressures than the experimental onset pressures. Anyway, this model was used as a basis for the some later published models. De Boer et al. (1995) used Hirschberg’s thermodynamic model of asphaltene solubility to come up with a simple method to identify crude oils which have a tendency to cause asphalt (asphaltene + resins) precipitation problems. Cimino et al. (1995) developed a model by considering the assumption that solvent phase is pure (despite of Hirschberg’s model). Their model relies on cloud point measurement for investigate the phase behavior. Coutinho et al. (1995) used an activity coefficient model to predict solid–liquid equilibrium in mixture of hydrocarbons with large size difference among the mixture species. An important shortcoming of these models is considering asphaltene as a single component. To improve this, Akbarzadeh et al. (2005) divided asphaltene into fractions of different molar mass based on the gamma molar mass distribution. They also successfully used the regular solution theory and assumed liquid–liquid equilibrium for Asphaltene precipitation modeling with various normal alkanes at different temperatures. Later, Pazuki and Nikookar (2006) to achieve a higher accuracy, added an interaction term to the Flory–Huggins equation and predict phase behavior of asphaltene precipitation by solvent titration. Soroush et al. (2007) investigated the solubility and molar volume of four pseudo-components obtained from SARA analysis using PR and FH.
Solid models
The modeling studies in the second category consider asphaltene as a solid phase and without addressing any compressibility calculation, they calculate asphaltene fugacity using a solid model. Thomas et al. (1992) developed three solid–liquid precipitation models based on the model of Won (1986). The first was a pure component solid phase fugacity correlation, the second was a multicomponent regular solution theory model and the third was the same multicomponent solid model that was modified for pressure and temperature influences. Yarranton and Masliyah (1996) considered asphaltene as a mixture of sub-fractions with different densities and molar masses. The asphaltene were fractionated by solvent extraction technique and molar mass distribution of asphaltene was obtained using surface tension and vapor pressure measurements. The solubility was modeled using solid–liquid equilibrium calculation with the equilibrium ratios derived from Flory–Huggins entropy of mixing. Finally, they successfully predicted both precipitation point and amount of asphaltene precipitation. Lira‐Galeana et al. (1996) developed a thermodynamic framework base on the multi-solid model, wherein each solid phase considered a pure component that cannot be dissolved in other solid phase. Nghiem and Coombe (1997) presented a thermodynamic model for asphaltene precipitation that captured the behavior of asphaltene precipitation during primary depletion. They modeled asphaltene precipitation, using three-phase flash calculation. They divided the crude oil into two pseudo-components, precipitating and non-precipitating and considered asphaltene as a pure solid phase. Abouie et al. (2017) compared the solid model and PC-SAFT in both static and dynamic asphaltene modeling. According to their work, the PC-SAFT model is superior to the solid model in term of the extrapolation accuracy when the experimental data are not available, they showed that a tuned solid model can also reproduce the experimental data with an acceptable accuracy. Dehaghani et al. (2018) studied the poly-dispersity behavior of asphaltene, divided it into three pseudo-components based on normal alkanes. They used a combination of solid model and PC-SAFT equation. The association properties of asphaltene are often overlooked in modeling for simplicity. Their modeling approach was extended in a later study. They replaced multi-solid approach with a solid solution approach, using a UNIQUAC activity coefficient model to describe solid phase. However, for liquid phase, PC-SAFT and PR were used. The results showed improvement when a solid-solution method was used instead of a multi-solid approach (Dehaghani et al. 2020).
EOS for asphaltene phase
Studies including EOS tool for description of precipitated phase are the majorities of recent works. Sabbagh et al. (2006) investigated the precipitation of asphaltene in a mixture of toluene and normal alkane using the PR and association properties of asphaltene. Abutaqiya et al. (2020) used PR for modeling asphaltene onset pressure with hydrocarbon and non-hydrocarbon gas injection to show that cubic EOS are at least as good as PC-SAFT. Their model was tested against high-pressure and high-temperature data from four wells from the Middle East. Kontogeorgis et al. (1996) used the CPA equation (including a SAFT based association term) for asphaltene precipitation. This equation is based on a combination of a physical part and an association term. Through the past decades, CPA equation of state has been successfully applied to several complex mixtures of water, alcohols, glycols, organic acids, and hydrocarbons. Li and Firoozabadi (2010) applied CPA equation to study asphaltene precipitation in two categories: light oil samples and heavy oil/bitumen samples. In the first category, they considered liquid–liquid equilibrium for upper onset pressure, bubble pressure, and gas–liquid–liquid equilibrium between lower onset and bubble pressure. In the second category, the asphaltene was modeled as liquid–liquid equilibrium over a range of temperatures, pressures and compositions. Shirani et al. (2012) used CPA for three live oil samples and investigated effect of CO2 injection. This model was expressed in two terms: the physical interaction between molecules in the mixture was considered in the physical part, and the chemical interaction and hydrogen bonding between some molecules in the mixture was included in the association part. The results of this model were compared to experimental data of three live oils.
SAFT equations were first proposed by Chapman et al. (1990) for mixtures. SAFT-based equations, define the residual Helmholtz free energy using statistical thermodynamics, and have several versions, the most important is the PC-SAFT proposed by Gross and Sadowski (2001). PC-SAFT considers the hard-chain fluid instead of the hard-sphere reference fluid used in the original SAFT for perturbation theory. The PC-SAFT has been widely used to predict asphaltene precipitation under different scenarios including natural depletion and gas injection. In this model, in contrast to the van der Waals family of cubic equations of state, the chain length, which represents molecular shape is taken into account by the developed reference term. Ting (2003) investigated thermodynamic stability and phase behavior of asphaltene in oil and other highly asymmetric mixtures. He proposed a characterization approach for PC-SAFT model and determined the model’s parameters through regression against experimental data. Gonzalez et al. (2005) demonstrated a simple and recombined live oil models using PC-SAFT to predict the onset pressure of asphaltene by investigating the effect of injection gases and pressure depletion. They injected different concentrations of C1, C2, CO2 and N2, and compared them with each other. In a later work, the effect of drilling mud and reinjection of associated gas on asphaltene precipitation was examined (Gonzalez et al. 2007). Also, in case of gas injection, asphaltene was treated as both monodisperse and polydisperse. Using an extension of the monodisperse SAFT asphaltene parameter fitting procedure, they were able to assign a set of SAFT parameters to represent each of the asphaltene subfractions. Behbahani et al. (2011) applied PC-SAFT, solid model and Flory–Huggins model to correlate onset pressures and solvent ratios at the onset point of precipitation for different solvents. PC-SAFT showed better performance and Flory–Huggins model accuracy was between PC-SAFT model and solid model ones. Panuganti et al. (2012) divided the crude oil composition into nine sections. In their work, the petroleum fractions heavier than C4 were considered as pseudo-component, and the plus-fraction was divided according to SARA analysis. They modeled different injection gases with PC-SAFT by considering asphaltene as a single component. Punnapala and Vargas (2013) continued this research to improve the work of Panuganti et al. (2012) by reducing the adjustable parameters to molecular weight and aromaticity. Their model was able to predict effect of gas injection on asphaltene stability and properties of crude oil with a constant set of simulation parameters. Tavakkoli et al. (2014), focused on modeling of the polydisperse asphaltene’s onset pressure and amount of precipitation from solvent-diluted crude oil using PC-SAFT over a wide range of crude oil density. Their work was compared to a previous model (Ting 2003) that had fewer adjustable parameters. Zúñiga-Hinojosa et al. (2014) applied PC-SAFT to model asphaltene precipitation from heavy oils and bitumen by n-alkane dilution. Liquid–liquid equilibrium was assumed between a dense liquid phase (asphaltene-rich phase) and a light liquid phase. The liquid–liquid equilibrium calculation (with asphaltene as the only precipitating component) was considered through a Michelsen stability approach. In their work, asphaltene was split into sub-fractions of different molecular weights by a gamma distribution function. In another study, PC-SAFT and solid model were used to predict the effects of aromatic solvents on the onset concentration and asphaltene precipitation weight percent. They showed that, by changing the type of solvent and titration ratio, the accuracy of the solid model decreases, while this does not affect the accuracy of the PC-SAFT. However, both models predicted an almost linear relationship between UOP (upper onset pressure) and solvent concentration. This was in accordance with the experimental results (Ebrahimi et al. 2016). To better understand the effect of association properties of asphaltene in modeling, Arya et al. (2016) compared asphaltene precipitation results obtained from different modeling approaches based on CPA, PC-SAFT with association and PC-SAFT without association models, and found that the CPA was more reliable compared to the other two approaches. Both versions of PC-SAFT provided good results for different fluids; even though in some case PC-SAFT with association has offered a better prediction for upper onset pressure. However, in most recent studies PC-SAFT without association has been used.
A comparative study of PC-SAFT and CPA to estimate the UOP and stability has been presented in a recent study. The authors investigated cross-association between resin and asphaltene molecules and self-association between asphaltene molecules. The estimated cross-association parameters of the PC-SAFT exhibited a better linear effect on the onset temperature than those of CPA. This could be a clue that the PC-SAFT could be more reliable for UOP (upper onset pressure) estimation outside the experimental temperature range. They showed that PC-SAFT predicts an increase in the stability of the oil sample at higher temperatures while the CPA model estimates no considerable changes in the asphaltene stability at such conditions (Nascimento et al. 2019).
In another work, the authors investigated the role of asphaltene characterization in the accuracy of PC-SAFT to predict asphaltenes precipitation behavior. They used an alternative characterization procedure where the molecular weight of asphaltenes is assumed as a temperature dependent parameter (AlHammadi and AlBlooshi 2019). A research group introduced a modified Barker and Henderson’s second-order perturbation term to the PC-SAFT (a modified PC-SAFT) to predict UOP. Even though predicting UOPs at lower temperatures, using this method could reduce or eliminate the crossover temperature (additional parameters for modified PC-SAFT) when there is CO2 enrichment of the petroleum system (Cañas-Marín et al. 2019). Masoudi et al. (2020) proposed modified PC-SAFT characterization technique for modeling asphaltene crude oil phase behavior. They assumed asphaltene as a combination of poly-nuclear aromatics, benzene derivatives, and saturates, and a set of weighting factors is also used based on uncertainties in experimental data. To achieve the best match between measured and model predictions, a trust region-based optimization method was applied. Nazari and Assareh (2021) used a new characterization to expand the crude oil composition to improve the work of Panuganti et al. (2012). They also presented a new adjustment scenario for the parameters to calculate onset pressure, bubble pressure and density.
A research group provided a critical review of the PC-SAFT in asphaltene precipitation modeling. According to their studies the main investigated aspects in recent years include the effect of asphaltene polydispersity, pressure–temperature-composition changes, and binary interaction coefficients (BICs). Based on this study, the polydisperse models increase the accuracy of calculation and monodisperse modeling overestimates the precipitated asphaltene weight percent. There is still no reliable method for generating BICs for such complex systems as reservoir fluid with asphaltene. PC-SAFT has been used more frequently in recent years than other EOSs while CPA can demonstrate comparable results with the PC-SAFT (Seitmaganbetov et al. 2021). A recent research work presented a thermodynamic model of asphaltene precipitation for depletion and gas injection at elevated pressures and temperatures by PC-SAFT. One of the main novelties of this work was the simultaneous adjustment of regression parameters including aromaticity of aromatics + resins and the aromaticity of asphaltene species (Ghasemi et al. 2022). The PC-SAFT has been used without considering the association term during gas injection and mixing processes to predict asphaltene precipitation behavior. The authors suggested a new method based on material balance concept for characterization when the SARA (saturates-aromatics-resins-asphaltenes) analysis is not reported (Moghaddam and Jamshidi 2022). In recent years, generally, the researchers have increasingly used a combination of existing models to achieve a more efficient model.
Contributions of this study
In earlier studies, mostly, the asphaltene fraction has been regarded as one single pseudo-component (monodisperse). The main drawback of the monodisperse models is that they do not take into account different natures of asphaltene molecules (Andersen and Speight 1999). The molecular weight of asphaltene has been debated for years and no unique value has been considered for it. On the other hand, the complex structure of asphaltene includes aromatic and saturated rings, aliphatic, some heteroatoms, as well as some metals such as iron, nickel, and vanadium (Groenzin and Mullins 2000). Therefore, it is necessary to use a multi-component approach of asphaltene to predict its performance in reservoir and surface conditions. Recent studies have been conducted based on the nature of asphaltene. For example, Tavakkoli et al. (2014) compared the graph of asphaltene precipitation in two cases, single component and multi-component approach. However, representative precipitation modeling approach for pressure depletion and mixing process is still a challenge. FH is a simple method with satisfactory results for predicting asphaltene precipitation. However, FH theories cannot be applied independently to live crude oil below the bubble pressure (when gas is released). Although the results of previous studies have shown that PC-SAFT alone has performed well in asphaltene precipitation modeling, in this research, to avoid non-physical-tuning the equation of state parameters, PC-SAFT was combined with Flory–Huggins and only one adjustable parameter (asphaltene solubility) was considered. Since PC-SAFT has not been developed for solid phase, its regression is highly sensitive specially in binary interaction coefficients and asphaltene parameters. This means that small changes in the interaction parameters can diverge equilibrium calculations. Moreover, the light and intermediate component are also present in the solid phase as mentioned in the work of Punnapala and Vargas (2013). FH can help to avoid such problems with asphaltene phase description.
Contrary to cubic equations that are developed for simple molecules, due to the inclusion of interaction details in the SAFT equation, this equation is effective for a wide range of pure compounds and mixtures in a wide range of pressure and temperature. A range that includes simple molecules as well as complex fluids such as electrolytes, polar solvents, covalent fluids, polymers, liquid crystals, and plasma. In this work, in PC-SAFT, the asphaltene is assumed to be macromolecules with pre-aggregation properties, but the association properties of these macromolecules with each other and with other components are ignored. Therefore, in this study, we tried to create a model using the combination of PC-SAFT/PR equations + FH, considering heterogeneous nature of asphaltene by PD, which can predict the asphaltene precipitation due to different diluents. This is in addition to predicting bubble pressure and onset pressure at different temperatures.
Methodology
In this study, the liquid phase is modeled using PC-SAFT while the asphaltene phase is modeled using FH solution theory. In the following sections, at first a brief description of FH implementation is presented. Afterward, the essentials of PC-SAFT are given. Later, the equilibrium calculations with PC-SAFT + FH are explained.
Hirschberg et al. (1984) used the Flory–Huggins solution theory to examine the asphaltene-maltene equilibrium. By dividing the crude oil into two components, dilute (without asphaltene) and concentrated (asphaltene) and assuming that the concentrated phase is pure, they used the chemical potential equality of the two phases, which is also basis of this study.
where \({\varphi }_{a}\), \({V}_{a}\) and \({V}_{m}\) are volume fraction of asphaltene, molar volume of asphaltene and maltene. \({\delta }_{a}\) and \({\delta }_{m}\) are solubility of asphaltene and maltene respectively that most important parameters of the Flory–Huggins solution theory. According to Alboudwarej et al. (2003) the equilibrium ratio is defined as follows:
The asphaltenes are treated as a mixture of pseudo-components of different molar mass as well. Since the dense phase consists only of asphaltenes, it is assumed to be a mixture of these asphaltene components. Therefore, the activity coefficients in the dense phase are unity and only the activity coefficients in the light phase are calculated:
And equilibrium ratio can be written as Eq. 4 that use in equilibrium calculation in this work:
The solubility of asphaltene is an adjustable parameter, but the solubility of the remaining liquid (maltene) is calculated using the Hildebrand and Scott (1962) equation:
where \({U}^{res}\) is residual internal energy that calculated by equation of state. PC-SAFT cannot be used directly to calculate \({U}^{res}\). In PC-SAFT, at first the residual Helmholtz energy is calculated and then the residual internal energy of the liquid is derived using the following steps and formulations:
The residual Helmholtz free energy consists of a hard chain and a dispersion term:
where \({g}_{ii}^{hs}\) is radial distribution function, \({I}_{1}\) and \({I}_{2}\) calculated by simple power series in density, \({C}_{1}\) is an abbreviation for the compressibility factor. \({m}_{i}\) is the segment length and \(m\) is the average segment length of the phase. Therefore, the derivative with respect to temperature becomes:
The hard-chain term of this derivative should be calculated as
\(\frac{\partial {A}^{hs}}{\partial T}\) is the hard sphere term which is:
The derivative of radial distribution function with respect to temperature has the following form:
And finally, the dispersion contribution to this temperature derivative is:
where \({\zeta }_{n}\) (1, 2, and 3) are dimensionless densities, and \({d}_{i}\) is temperature-dependent segment diameter. After substitution we can calculate \({U}^{res}\).
In this study, PR and PC-SAFT equations for the liquid part and Flory–Huggins solution theory for the asphaltene part (which is considered as pseudo-liquid) are used. The inputs of the PR equation have been studied in detail in literature (Peng and Robinson 1976). The PC-SAFT equation has five input parameters; Of course, considering that the association property of asphaltene is not considered in this work, only three input parameters are needed, which include the temperature independent diameter (σ), the number of segment (m) and the segment dispersion energy (\(\varepsilon /k\)). This compressibility equation was defined by Gross and Sadowski (2001) as follows:
The coefficients for fugacity are linked to the residual chemical potentials based on:
In which:
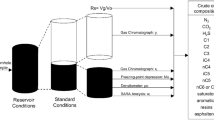
Dead oil is needed for titration, therefore, first, the initial feed (live oil) composition is flashed under standard temperature and pressure conditions. Characterization of crude oil based on SARA analysis, limitations, and challenges in asphaltene characterization and especially multi-component asphaltene are explained in detail in the next section.
The process of calculating the density and bubble pressure as a flowchart is shown in Fig. 1; this workflow explains the fitting process, fitting parameters, and regression loop in the optimization. The density and bubble pressure are calculated at same time, just by one objective function. Adjustable parameters, for the PR, include volume shift (\({C}_{shift}\)) for plus fraction and binary interaction coefficients (\({k}_{ij}\)), and for the PC-SAFT include m, \(\varepsilon /k\) for Aromatic-saturates and \({k}_{ij}\). The allowable change intervals during optimization of these parameters are within \(\pm 10\%\) of original values. The alpha parameter is adjusted during the regression process and only the BIC of the plus fraction, aromatic-saturate, resin and asphaltene are used.

Summary of fitting procedure for the tuning variables for density and bubble pressure
Titration is done for three normal alkanes in monodisperse (MD) and polydisperse (PD) model. According to Fig. 2, a Rachford–Rice (RR) equation is used to obtain the precipitated solid moles as follows (\(l\) stands for liquid and \(s\) stands for solid, \(x\) is mol fraction, \(K\) is equilibrium ratio, \({N}_{c}\) is the number of components and \({N}_{a}\) is the number of asphaltene pseudo-components and \({n}_{s}\) moles of solid):

Flow diagram of fitting procedure and calculation of titration
For those components that do not precipitate \({x}_{i}^{s}\) and \({K}_{i}\) equal to zero and this can be written for the components that do not precipitate (RR1) as:
In the above relations, RRP1, is the derivative with respect to \(ns\). Also, by inserting Eqs. 19 and 20, in Eq. 21 this equation for the components that precipitate (RR2) is as follows:
Which \(RRP2\) is a derivative of the Rachford–Rice function and finally, the sum of the equations mentioned forms the final Rachford–Rice equation. To calculate the precipitated solid moles, an initial estimation of the equilibrium ratio (\({K}_{i}\)) is required, which uses Eq. 4 that depends on asphaltene solubility (\({\delta }_{a}\)). To check the correctness of the \({K}_{i}\) value, the equilibrium function is defined as Eq. 26, \({g}_{equil}\) in a Newton–Raphson iteration cycle close to zero and it confirms that the \({K}_{i}\) are converged and the mole fraction of asphaltene in the dilute phase can be calculated. By calculating the weight percentage of asphaltene and comparing it with the laboratory value, we can figure out if a new estimate for solubility still necessary.
In calculating the (upper) onset pressure, no precipitant has formed yet, and the predominant phase is liquid. In fact, the compound is on the verge of biphasic. In addition, since the onset pressure is always above the bubble pressure, there is no gas phase. Figure 3 shows the modeling process performed at this stage.

Flow diagram of fitting procedure and calculation of onset pressure
As it is shown, two equilibrium functions are used, one to study the composition at the onset pressure (Eq. 26) and the other to study the pressure changes and the desired onset pressure (Eq. 27). In this loop, the pressure is updated each iteration, in this equation \({g}_{onsetp}\) is derivative with respect to \(\ln\left(P\right)\). Finally, the calculated onset pressure is compared with the experimental value, and if there is a large difference, a new value for the solubility is guessed.
Case studies and characterization
In this study, two Mexican fluids named C1 and Y3 are used. The compositional data for these two fluids are given in the Table 1. The characterization of SARA analysis of these two fluids is from the work of Buenrostro‐Gonzalez et al. (2004) and is in accordance with the Table 2. Studies on asphaltene onset pressure, bubble pressure and titration with three alkanes, normal-pentane, normal-heptane, normal-nonane have been performed by Buenrostro‐Gonzalez et al. (2004). The experimental values for titration using the mentioned three normal alkanes in standard conditions are given in the Table 3. According to Table 3, different normal alkane precipitates different amounts of asphaltene (which is compatible with polydisperse nature of asphaltenes). Therefore, the crude oil composition is extended from C7+ to C12+ for titration with three different alkanes. This is done by Pedersen et al. (2006) correlations:
In this correlation, \(A\) and \(B\) are constants for splitting functions and \(Mw\) is the molecular weight of single carbon number fractions.
C12+ should be split to characterize asphaltene. For this purpose, according to SARA analysis in Table 2, the plus fraction can be divided into three pseudo-components. This, later, enables us to split the asphaltene physically into three meaningful pseudo-components.
To reduce the adjustable parameters, the aromatics and saturates parts are considered as one pseudo-component. Weights of light saturates (like C6, C7, C8, …, C11) and aromatic (like cyclo-C5, cyclo-C6…) is calculated and subtracted from the weight of Saturates-Aromatic. The results of this analysis are shown in the Table 4. The critical properties and acentric factor of the aromatic-saturated fractions are estimated with the Riazi and Al-Sahhaf (1996) correlations and for resin and asphaltene the Avaullee et al. (1997) correlations are used. To determine the parameters of the PC-SAFT equation of state for the aromatic-saturate pseudo-component, the Assareh et al. (2016) correlations are used (by molecular weight, MW and specific gravity, Sg):
for the resin and asphaltene sections, the Gonzalez et al. (2007) equations are used (by molecular weight, MW and aromaticity, \(\gamma\)):
The binary interaction coefficients (BICs) are also obtained from Chueh and Prausnitz (1967) correlations.
In this equation, the value of \(\alpha\) is found by regression.
Result and discussion
Buenrostro‐Gonzalez et al. (2004) used the SAFT-VR equation, assuming asphaltene to be a single pseudo-component (mono-disperse). We make use of this as a reference for comparison. This research is divided into two parts; mono-disperse and poly-disperse asphaltene. Both are described in the following sections. To better compare the results of these two parts with each other’s, the mean of absolute percentage error is used (\({\Omega }^{exp}\) is experimental value and \({\Omega }^{calc}\) is the calculated ones):
Mono-disperse modeling
In this section, asphaltene is separated from the plus fraction as a homogeneous component; its molecular weight and density are constant and the values in the work of Buenrostro‐Gonzalez et al. (2004) are used. To improve the modeling results, a series of adjustable parameters are considered, and the best value is calculated using the genetic algorithm, a regression technique, as a powerful optimization tool. The objective function (OF) in the regression process is defined as follows:
As mentioned earlier, the density and bubble pressure were adjusted at same time. Therefore, to achieve better results, adjustable parameters should be selected in such a way that they can have the greatest impact with the least change. For this purpose, sensitivity analysis was performed on the parameters of both equations of state. In the PR equation, the parameter \({C}_{shift}\) and in the PC-SAFT equation, the parameter \(m\) and \(\frac{\varepsilon }{k}\) had a pronounced effect on the density, and the parameter BIC was effective on the bubble pressure.
Table 5 shows the adjustable parameters before and after optimization. Figure 4 also shows the results of this modeling for bubble pressure at different temperatures compared to laboratory values. In fluid C1 the PR equation results are slightly better than PC-SAFT, but in fluid Y3 the PC-SAFT equation performed more accurately. Figure 12 (left) shows that Buenrostro‐Gonzalez et al. (2004) obtained results for Y3 fluid compared to this study.

(Top) Chart for calculation bubble point pressure in mono-disperse with PR (left) and PC-SAFT (right) for fluid C1. (Bottom) Chart for calculation bubble point pressure in mono-disperse with PR (left) and PC-SAFT (right) for fluid Y3. Dashed lines and bullet represent this study and experimental data (Buenrostro‐Gonzalez et al. 2004)
In mono-disperse titration, to find the precipitated asphaltene due to the injection of each normal alkane, there is no need to divide asphaltene into fractions. The aim is to perform titrations for three distinct types of asphaltenes. Titration is performed for all three normal alkanes (pentane-heptane and Nonane) while the only adjustable parameter is asphaltene solubility (Fig. 2). Although in this model, the solubility of different asphaltenes is not adjusted together, it is considered that the solubility of asphaltene increases with the increase in molecular weight. The predicted values for solubility are listed in Table 6. Figure 5 show the weight percentage obtained for different volumes of normal alkanes for C1 and Fig. 6 for Y3. The results of both fluid samples show that the PC-AFT equation performs better than PR. Figure 12 also shows the Buenrostro‐Gonzalez et al. (2004) calculation results compared to this study.

The amount of precipitated asphaltene in mono-disperse approach with PR (left) and with PC-SAFT (right) for fluid C1. Dashed lines and bullet represent this study and experimental data (Buenrostro‐Gonzalez et al. 2004)

The amount of precipitated asphaltene in mono-disperse with PR (left) and with PC-SAFT (right) for fluid Y3. Dashed lines and bullet represent this study and experimental data (Buenrostro‐Gonzalez et al. 2004)
Fortunately, there was no obstacle to adjust in the onset pressure calculation. The use of only one adjustable parameter (\({\delta }_{a}\)) not only created a simple model, but also due to the high influence of solubility on onset pressure, the adjustment process was not complicated. According to the results of earlier studies, there is a linear relationship between the solubility of asphaltene and temperature that use in this study:
Constants A and B are considered in the adjustable regression process. To calculate these two parameters, the linear equation between two points (two onset pressures at two different temperatures) must be written, for which two laboratory onset pressures are used. The final values of these two parameters are shown in the Table 7.
The final amount of asphaltene solubility for both equations and both fluid samples are shown in the Table 8. Figure 7 shows the results of onset pressures in the mono-disperse for the two equations of PR and PC-SAFT. In fluid C1, the PC-SAFT equation works better, but in the case of fluid Y3, the opposite is true. Figure 12 shows Buenrostro‐Gonzalez et al. (2004) is calculated results in addition.

(Top) Chart for calculation onset point pressure in mono-disperse with PR (left) and PC-SAFT (right) for fluid C1. (Bottom) Chart for calculation onset point pressure in mono-disperse with PR (left) and PC-SAFT (right) for fluid Y3. Dashed lines and bullet represent this study and experimental data (Buenrostro‐Gonzalez et al. 2004)
Poly-disperse modeling
Asphaltene is a heterogeneous compound, and ignoring this property, while simplifying modeling, is at odds with the nature and physics of asphaltene. For this purpose, based on the normal alkanes used in the titration section, asphaltene is divided into three components (NC5-Asp, NC7-Asp, and NC9-Asp). To find the molecular weight and density, the mixtures roles was used as (\({wt}_{i}\) are the weight percent):
The properties of each of the asphaltene pseudo-component are listed in the Table 9. According to Table 9, the aromaticity parameter was considered the same for all three types of asphaltene; this is similar to the work of Tavakkoli et al. (2014) with the aim of reducing adjustable parameters. Specific gravity and molecular weight are calculated from the mixing rules as mentioned before. It should be noted that the molecular weight of any specific part of asphaltene must be guessed initially. The higher the normal alkane number, the higher the molecular weight of asphaltene. A first guess must be made for the last two asphaltene sub-components and this causes uncertainty in the results and provides leverage for regression.
Undoubtedly, the division of asphaltene into several pseudo-components affects the results of modeling; In calculating the bubble pressure with the PC-SAFT equation, two parameters related to aromatic-saturate (m, \(\varepsilon /k\)) were adjustable. There are three adjustable parameters, including the \({k}_{ij}\) parameter. The PC-SAFT equation showed more sensitivity to the \(\varepsilon /k\). In the case of the PR equation, there are only two adjustable parameters (\({C}_{shift}\), \({k}_{ij}\)). Figure 8 show bubble pressure in poly-disperse model; according to the percentage of error calculated in the Table 10 for fluid C1, the results obtained from the PR equation are slightly better than PC-SAFT, but for fluid Y3 the result is the opposite. The results of the poly-disperse model are also slightly better than the mono-disperse.

(Top) Bubble point pressure in poly-disperse with PR (left) and PC-SAFT (right) for fluid C1. (Bottom) Chart for calculation bubble point pressure in poly-disperse with PR (left) and PC-SAFT (right) for fluid Y3. Dashed lines and bullet represent this study and experimental data (Buenrostro‐Gonzalez et al. 2004)
In poly-disperse titration, by adding normal alkane, since not all the available asphaltene precipitates at once, by assigning a specific solubility to each asphaltene component, the amount of precipitation of each component can be calculated. For example, by adding normal heptane to the oil composition, the part of normal heptane and all subsequent parts (normal nonane) are precipitated; then, the total sediment obtained from these two parts gives the amount of asphaltene sediment due to normal heptane injection. To determine the solubility parameter, the titration process starts from the smallest component (nonane) and its solubility is calculated by regression; then, the nC7-asphaltene is considered as a single component and its solubility is calculated. Then it is divided two parts. The properties of nC9-asphaltene obtained in the earlier step will substitute one of these two parts. Once again, the regression process is performed on these two parts to calculate the best solubility. This procedure is also performed to calculate the normal pentane titration. Table 6 shows the solubility of asphaltene after regression. According to the Figs. 9 and 10, the PC-SAFT equation performs better than the PR equation, and also the poly-disperse model except in one case (normal heptane with PR equation) is better than the mono-disperse model. For low amount of normal alkane, the adjustment is difficult, but with the increase of the amount of normal alkane, the graph becomes smoother, and a better match is achieved. The parameters adjusted in the previous section to calculate the density and bubble pressure are selected after the sensitivity analysis to have the least effect on the titration and onset pressure because the purpose of this work is to investigate the effect of asphaltene solubility.

The amount of precipitated asphaltene in poly-disperse with PR (left) and with PC-SAFT (right) for fluid C1. Dashed lines and bullet represent this study and experimental data (Buenrostro‐Gonzalez et al. 2004)

The amount of precipitated asphaltene in poly-disperse with PR (left) and with PC-SAFT (right) for fluid Y3. Dashed lines and bullet represent this study and experimental data (Buenrostro‐Gonzalez et al. 2004)
The onset pressure is highly sensitive to the solubility of asphaltene, a linear equation is used to calculate the solubility parameters, A and B, which are calculated in the mono-disperse step, are unchanged in this step as well, but Eq. 43 is used to calculate the solubility of each of the asphaltene sub-components. This relationship shows the volume fraction of each component in terms of the solubility of that component.
The solubility parameter should be estimated by considering two points for each asphaltene component: first, according to the literature (Yarranton and Masliyah 1996), the solubility increases with the increase in molecular weight. Second, solubility has an inverse relationship with temperature.
The values for the solubility of each component at the reservoir temperature are in Table 8. The results for onset pressure at reservoir temperature are shown in Fig. 11. For fluid C1, the results of the PC-SAFT equation are much better than the PR equation, and the poly-disperse model has less error than the mono-disperse model.

(Top) Onset point pressure in poly-disperse with PR (left) and PC-SAFT (right) for fluid C1. (Bottom) Chart for calculation onset point pressure in poly-disperse with PR (left) and PC-SAFT (right) for fluid Y3. Dashed lines and bullet represent this study and experimental data (Buenrostro‐Gonzalez et al. 2004)
The modeling results of Buenrostro‐Gonzalez et al. (2004) was done only for single-component (mono-disperse) asphaltene, and in Table 9, the results of this work in single-component model are compared with the Buenrostro‐Gonzalez et al. (2004) model. As can be seen, the chart on the left shows a comparison of the bubble pressure, only in fluid Y3 for Buenrostro‐Gonzalez et al. (2004) results. The other two charts, which are related to titration and bubble pressure respectively, show the drastic difference between these two models (Fig. 12).

(Left) Chart to compare the results of this work and Buenrostro‐Gonzalez et al. (2004) for: (left) bubble pressure, (middle) titration, (right) onset pressure
Table 10 shows minimum absolute deviation percent error for bubble pressure, titration, and onset pressure for both of C1 and Y3. In the comparison of two equations of state PR and PC-SAFT, in few cases the PR equation of state has performed better than PC-SAFT and in some cases, result of monodisperse better than polydisperse.
Conclusion
In this study, a new method based on combination of equation of state and Flory–Huggins theory was developed for modeling improvement of asphaltene precipitation and onset pressure predictions. The calculation of bubble point, onset pressure and precipitation in monodisperse and polydisperse asphaltene using PR and PC-SAFT equation, presented improved results compared to the work of Buenrostro‐Gonzalez et al. (2004). Using the PC-SAFT equation for solid phase and adjusting its parameters in the regression process is a challenging task. This problem was addressed by combining PC-SAFT with FH in this work. FH is a simple equation and has inaccuracies, however combining it with PR and PC-SAFT equations gave satisfactory results and removed the necessity of extensive parameters’ regression. To obtain a better match between experimental result and calculated values, while improving physical description of mixing processes, asphaltene was broken to three pseudo-components based on precipitating solvent. In this work, the solubility parameter played an effective role to improve the calculation of onset pressure and titration with various normal alkanes. Considering a linear function in term of temperature proved to be an effective way to estimate asphaltene solubility for surface and underground conditions. In some cases, the single-component model performed better, but it is important to note that the multi-component model is a better representation of the nature of asphaltene.
Data availability
All the data used for comparision and the modeling results can be provided upon request.
Abbreviations
- \(A\) :
-
Helmholtz free energy
- \(a\) :
-
Parameter in Peng–Robinson
- \(b\) :
-
Parameter in Peng–Robinson
- \({C}_{shift}\) :
-
Volume shift in Peng–Robinson
- \({C}_{1}\) :
-
Abbreviation
- \(d\) :
-
Temperature independent segment diameter
- \(g\) :
-
Equilibrium function
- \({g}_{ii}^{hs}\) :
-
Average radial distribution function of hard segment
- \({I}_{1}\) :
-
Simple power series in density
- \({I}_{2}\) :
-
Simple power series in density
- \(K\) :
-
Equilibrium ratio
- \({k}_{ij}\) :
-
Binary interaction coefficient
- \(k\) :
-
Boltzmann constant
- \(m\) :
-
Number of segments per chain
- \(N\) :
-
Number of components
- \(n\) :
-
Mol
- \(P\) :
-
Pressure
- \(R\) :
-
Gas universal constant
- \(T\) :
-
Temperature
- \(U\) :
-
Internal energy
- \(V\) :
-
Molar volume
- \(Z\) :
-
Compressibility factor
- \(z\) :
-
Initial feed
- \(w\) :
-
Weighting factor
- \(x\) :
-
Mole fraction
- \(\varphi\) :
-
Volume fraction
- \(\delta\) :
-
Solubility
- \(\omega\) :
-
Acentric factor
- \(\varepsilon\) :
-
Segment–segment dispersion energy
- σ:
-
Segment diameter
- γ:
-
Aromaticity factor
- α:
-
Prausnitz equation multiplier for interaction coefficients
- β:
-
Prausnitz equation power for interaction coefficients
- \(\mu\) :
-
Chemical potential
- \(\rho\) :
-
Total number density of molecules
- \(\varsigma\) :
-
Abbreviation
- \(SARA\) :
-
Saturates, aromatics, resins and asphaltene
- \(PC\_SAFT\) :
-
Perturbed chain from of the statistical associating fluid theory
- \(PR\) :
-
Peng–Robinson
- \(SAFT\_VR\) :
-
Statistical associating fluid theory for potential of variable attractive range
- \(EOS\) :
-
Equation of state
- \(PD\) :
-
Polydisperse model
- \(MD\) :
-
Monodisperse model
- \(Asph\) :
-
Asphaltene
- \(Res\) :
-
Resin
- \(Sat\_Aro\) :
-
Saturates + aromatics
- \(Sto\) :
-
Stock tank
- \(MAPE\) :
-
Mean absolute percentage error
- \(BIC\) :
-
Binary interaction coefficient
- \(Mw\) :
-
Molecular weight
- \(SG\) :
-
Specific gravity
- \(RR\) :
-
Rachford–Rice
- \(wt\) :
-
Weight
- \(a\) :
-
Asphaltene
- \(m\) :
-
Maltene
- \(mix\) :
-
Mixture
- \(c\) :
-
Component
- \(i,j\) :
-
Component
- \(equil\) :
-
Equilibrium
- \(tot\) :
-
Total
- \(dens\) :
-
Density
- \(sat\) :
-
Saturation
- \(coh\) :
-
Cohesion
- \(res\) :
-
Residual
- \(l\) :
-
Liquid
- \(s\) :
-
Solid
- \(id\) :
-
Ideal
- \(hc\) :
-
Hard chain
- \(disp\) :
-
Dispersion
- \(exp\) :
-
Experimental
- \(calc\) :
-
Calculation
References
Abouie A, Darabi H, Sepehrnoori K (2017) Data-driven comparison between solid model and PC-SAFT for modeling asphaltene precipitation. J Nat Gas Sci Eng 45:325–337
Abutaqiya MI, Sisco CJ, Khemka Y, Safa MA, Ghloum EF, Rashed AM, Gharbi R, Santhanagopalan S, Al-Qahtani M, Al-Kandari E (2020) Accurate modeling of asphaltene onset pressure in crude oils under gas injection using Peng–Robinson equation of state. Energy Fuels 34:4055–4070
Afra S, Samouei H, Golshahi N, Nasr-El-Din H (2020) Alterations of asphaltenes chemical structure due to carbon dioxide injection. Fuel 272:117708
Akbarzadeh K, Alboudwarej H, Svrcek WY, Yarranton HW (2005) A generalized regular solution model for asphaltene precipitation from n-alkane diluted heavy oils and bitumens. Fluid Phase Equilib 232:159–170
Alboudwarej H, Akbarzadeh K, Beck J, Svrcek WY, Yarranton HW (2003) Regular solution model for asphaltene precipitation from bitumens and solvents. AIChE J 49:2948–2956
Alhammadi AA, Alblooshi AM (2019) Role of characterization in the accuracy of PC-SAFT equation of state modeling of asphaltenes phase behavior. Ind Eng Chem Res 58:18345–18354
Ancheyta J, Centeno G, Trejo F, Marroquin G, García J, Tenorio E, Torres A (2002) Extraction and characterization of asphaltenes from different crude oils and solvents. Energy Fuels 16:1121–1127
Andersen SI, Speight JG (1999) Thermodynamic models for asphaltene solubility and precipitation. J Petrol Sci Eng 22:53–66
Arya A, Liang X, Von Solms N, Kontogeorgis GM (2016) Modeling of asphaltene onset precipitation conditions with cubic plus association (CPA) and perturbed chain statistical associating fluid theory (PC-SAFT) equations of state. Energy Fuels 30:6835–6852
Ashtari M, Bayat M, Sattarin M (2011) Investigation on asphaltene and heavy metal removal from crude oil using a thermal effect. Energy Fuels 25:300–306
Assareh M, Ghotbi C, Tavakkoli M, Bashiri G (2016) PC-SAFT modeling of petroleum reservoir fluid phase behavior using new correlations for petroleum cuts and plus fractions. Fluid Phase Equilib 408:273–283
Avaullee L, Trassy L, Neau E, Jaubert JN (1997) Thermodynamic modeling for petroleum fluids I. Equation of state and group contribution for the estimation of thermodynamic parameters of heavy hydrocarbons. Fluid Phase Equilib 139:155–170
Behbahani TJ, Ghotbi C, Taghikhani V, Shahrabadi A (2011) Experimental investigation and thermodynamic modeling of asphaltene precipitation. Sci Iran 18:1384–1390
Buenrostro-Gonzalez E, Lira-Galeana C, Gil-Villegas A, Wu J (2004) Asphaltene precipitation in crude oils: theory and experiments. AIChE J 50:2552–2570
Cañas-Marín WA, González DL, Hoyos BA (2019) A theoretically modified PC-SAFT equation of state for predicting asphaltene onset pressures at low temperatures. Fluid Phase Equilib 495:1–11
Chacón-Patiño ML, Rowland SM, Rodgers RP (2017) Advances in asphaltene petroleomics. Part 1: asphaltenes are composed of abundant island and archipelago structural motifs. Energy Fuels 31:13509–13518
Chapman WG, Gubbins KE, Jackson G, Radosz M (1990) New reference equation of state for associating liquids. Ind Eng Chem Res 29:1709–1721
Chueh P, Prausnitz J (1967) Vapor-liquid equilibria at high pressures: calculation of partial molar volumes in nonpolar liquid mixtures. AIChE J 13:1099–1107
Cimino R, Correra S, Bianco AD, Lockhart TP (1995) Solubility and phase behavior of asphaltenes in hydrocarbon media. In: Asphaltenes Fundamentals and Applications. Springer US, Boston, pp 97–130. https://doi.org/10.1007/978-1-4757-9293-5_3
Coutinho JA, Andersen SI, Stenby EH (1995) Evaluation of activity coefficient models in prediction of alkane solid-liquid equilibria. Fluid Phase Equilib 103:23–39
De Boer R, Leerlooyer K, Eigner M, Van Bergen A (1995) Screening of crude oils for asphalt precipitation: theory, practice, and the selection of inhibitors. SPE Prod Facil 10:55–61
Dehaghani YH, Assareh M, Feyzi F (2018) Asphaltene precipitation modeling with PR and PC-SAFT equations of state based on normal alkanes titration data in a multisolid approach. Fluid Phase Equilib 470:212–220
Dehaghani YH, Ahmadinezhad H, Feyzi F, Assareh M (2020) Modeling of precipitation considering multi-component form of Asphaltene using a solid solution framework. Fuel 263:116766
Ebrahimi M, Mousavi-Dehghani S, Dabir B, Shahrabadi A (2016) The effect of aromatic solvents on the onset and amount of asphaltene precipitation at reservoir conditions: experimental and modeling studies. J Mol Liq 223:119–127
Flory PJ (1942) Thermodynamics of high polymer solutions. J Chem Phys 10:51–61
Ghasemi S, Behbahani TJ, Mohammadi M, Ehsani MR, Khaz’ali AR (2022) Experimental investigation and thermodynamic modeling of asphaltene precipitation during pressure depletion and gas injection at HPHT conditions in live oil using PC-SAFT EoS. Fluid Phase Equilib 562:113549
Gonzalez DL, Ting PD, Hirasaki GJ, Chapman WG (2005) Prediction of asphaltene instability under gas injection with the PC-SAFT equation of state. Energy Fuels 19:1230–1234
Gonzalez DL, Hirasaki GJ, Creek J, Chapman WG (2007) Modeling of asphaltene precipitation due to changes in composition using the perturbed chain statistical associating fluid theory equation of state. Energy Fuels 21:1231–1242
Groenzin H, Mullins OC (2000) Molecular size and structure of asphaltenes from various sources. Energy Fuels 14:677–684
Gross J, Sadowski G (2001) Perturbed-chain SAFT: an equation of state based on a perturbation theory for chain molecules. Ind Eng Chem Res 40:1244–1260
Hildebrand JH, Scott RL (1962) Regular solutions. Prentice-Hall, Hoboken
Hirschberg A, Dejong LN, Schipper B, Meijer J (1984) Influence of temperature and pressure on asphaltene flocculation. Soc Petrol Eng J 24:283–293
Huggins ML (1941) Solutions of long chain compounds. J Chem Phys 9:440–440
Kontogeorgis GM, Voutsas EC, Yakoumis IV, Tassios DP (1996) An equation of state for associating fluids. Ind Eng Chem Res 35:4310–4318
Leontaritis KJ, Mansoori GA (1987) Asphaltene flocculation during oil production and processing: a thermodynamic colloidal model. In: SPE international symposium on oilfield chemistry, Texas, San Antonio, OnePetro
Li Z, Firoozabadi A (2010) Cubic-plus-association equation of state for asphaltene precipitation in live oils. Energy Fuels 24:2956–2963
Lira-Galeana C, Firoozabadi A, Prausnitz JM (1996) Thermodynamics of wax precipitation in petroleum mixtures. AIChE J 42:239–248
Masoudi M, Miri R, Hellevang H, Kord S (2020) Modified PC-SAFT characterization technique for modeling asphaltenic crude oil phase behavior. Fluid Phase Equilib 513:112545
Moghaddam AK, Jamshidi S (2022) Performance evaluation and improvement of PC-SAFT equation of state for the asphaltene precipitation modeling during mixing with various fluid types. Fluid Phase Equilib 554:113340
Nascimento FP, Costa GM, De Melo SAV (2019) A comparative study of CPA and PC-SAFT equations of state to calculate the asphaltene onset pressure and phase envelope. Fluid Phase Equilib 494:74–92
Nazari F, Assareh M (2021) An effective asphaltene precipitation modeling approach using PC-SAFT with detailed fluid descriptions for gas injection conditions. Fluid Phase Equilib 532:112937
Neuhaus N, Nascimento PT, Moreira I, Scheer AP, Santos AF, Corazza ML (2019) Thermodynamic analysis and modeling of Brazilian crude oil and asphaltene systems: an experimental measurement and a PC-SAFT application. Braz J Chem Eng 36:557–571
Nghiem LX, Coombe DA (1997) Modelling asphaltene precipitation during primary depletion. SPE J 2:170–176
Panuganti SR, Vargas FM, Gonzalez DL, Kurup AS, Chapman WG (2012) PC-SAFT characterization of crude oils and modeling of asphaltene phase behavior. Fuel 93:658–669
Pazuki G, Nikookar M (2006) A modified Flory–Huggins model for prediction of asphaltenes precipitation in crude oil. Fuel 85:1083–1086
Pedersen KS, Christensen PL, Shaikh JA, Christensen PL (2006) Phase behavior of petroleum reservoir fluids. CRC Press, Boca Raton
Peng D-Y, Robinson DB (1976) A new two-constant equation of state. Ind Eng Chem Fundam 15:59–64
Punnapala S, Vargas FM (2013) Revisiting the PC-SAFT characterization procedure for an improved asphaltene precipitation prediction. Fuel 108:417–429
Riazi MR, Al-Sahhaf TA (1996) Physical properties of heavy petroleum fractions and crude oils. Fluid Phase Equilib 117:217–224
Sabbagh O, Akbarzadeh K, Badamchi-Zadeh A, Svrcek WY, Yarranton HW (2006) Applying the PR-EoS to asphaltene precipitation from n-alkane diluted heavy oils and bitumens. Energy Fuels 20:625–634
Schulze M, Lechner MP, Stryker JM, Tykwinski RR (2015) Aggregation of asphaltene model compounds using a porphyrin tethered to a carboxylic acid. Org Biomol Chem 13:6984–6991
Scott RL, Magat M (1945) The thermodynamics of high-polymer solutions: I. The free energy of mixing of solvents and polymers of heterogeneous distribution. J Chem Phys 13:172–177
Seitmaganbetov N, Rezaei N, Shafiei A (2021) Characterization of crude oils and asphaltenes using the PC-SAFT EoS: a systematic review. Fuel 291:120180
Shirani B, Nikazar M, Mousavi-Dehghani SA (2012) Prediction of asphaltene phase behavior in live oil with CPA equation of state. Fuel 97:89–96
Soroush S, Vafaei SM, Masoudi R (2007) Applying the PR-EOS to predict the onset of asphaltene precipitation from n-alkane diluted bitumens. Iran J Chem Chem Eng 26(3):111–119
Tavakkoli M, Panuganti SR, Taghikhani V, Pishvaie MR, Chapman WG (2014) Understanding the polydisperse behavior of asphaltenes during precipitation. Fuel 117:206–217
Thomas F, Bennion D, Bennion D, Hunter B (1992) Experimental and theoretical studies of solids precipitation from reservoir fluid. J Can Pet Technol 31(1):23–31
Ting P-LD (2003) Thermodynamic stability and phase behavior of asphaltenes in oil and of other highly asymmetric mixtures. Rice University, Houston
Won K (1986) Thermodynamics for solid solution-liquid-vapor equilibria: wax phase formation from heavy hydrocarbon mixtures. Fluid Phase Equilib 30:265–279
Yarranton HW, Masliyah JH (1996) Molar mass distribution and solubility modeling of asphaltenes. AIChE J 42:3533–3543
Zúñiga-Hinojosa MA, Justo-García DN, Aquino-Olivos MA, Román-Ramírez LA, García-Sánchez F (2014) Modeling of asphaltene precipitation from n-alkane diluted heavy oils and bitumens using the PC-SAFT equation of state. Fluid Phase Equilib 376:210–224
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mousavian, M., Assareh, M. Asphaltene precipitation modeling using PC-SAFT and Flory–Huggins theory considering polydisperse asphaltene description. Braz. J. Chem. Eng. 41, 543–567 (2024). https://doi.org/10.1007/s43153-023-00335-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43153-023-00335-w