
Abstract
Purpose of the Review
Intratumoral pericytes (PC) do not share the same tumor niche as peritumoral PC. Furthermore, glioblastoma multiforme (GB) cells do not seem to affect them equally. Therefore, for a better understanding of the effects of GB on PC, in this chapter, we will classify them according to whether they are intratumoral or peritumoral PC, focusing mainly on peritumoral effects, which seem to have better future prospects for finding effective therapies in GB cancer.
Recent Findings
Recently, it has been shown that PC could be the main target of the tumor infiltration front and have a fundamental role in the proliferation, expansion, and survival of the tumor, as well as in the regulation of anti-tumor immune responses. Modulation of the immune function of PC through molecular mechanisms such as chaperone-mediated autophagy (CMA) seems to be essential to prevent an immunosuppressive microenviroment that facilitates tumor growth.
Summary
GB is the most frequent and aggressive brain tumor. In the last years, PC have been gaining special attention due to their role in GB progression. GB cells infiltrate away from the tumor core more often and faster when they are associated with perivascular cells. However, to find targeted therapies against PC to promote their brain defense function and improve anti-tumor immune responses requires a better understanding of the heterogeneity, markers, and distribution of PC at origin.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma Multiforme
Glioblastoma multiforme (GB) is the most frequent and aggressive brain tumor. Patients survive 15 months on average and less than 5% survive 5 years [1]. The cellular origin of GB is unknown, but some studies suggest that it could be generated from astrocytes, oligodendrocyte progenitor cells, and neural stem cells [2, 3]. The limits of the tumor are diffuse everywhere and the name “multiform” refers to the variability of the histological morphology of GB, which has rounded and spindle-shaped cells, small or very large [4]. GB is characterized by a highly variable appearance due to the presence of small areas of necrotizing tissue that are surrounded by cells with poor cellular differentiation and tumor stem cells infiltrated by blood vessels (Fig. 1). GBs are frequently formed in the subcortical white matter of the cerebral hemispheres and, during their development, tumor cells infiltrate and invade the brain parenchyma interacting with cells from perivascular areas. GB tumor cells establish a functional network of microtubes with the cells surrounding the tumor to ensure intercommunication and supply of organelles and nutrients required for tumor survival [5, 6••, 7, 8].

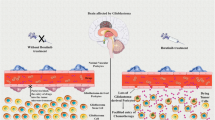
PC in the intratumoral and peritumoral areas. An original drawing of the brain with GB is shown at the top. Examples of intratumoral and peritumoral areas of GB are indicated with red boxes. Below is a schematic drawing of the PC characteristics of each environment. In the intratumoral area, PC derived from GB stem cells predominate, which mediate the effects for tumor survival, such as angiogenesis or immune evasion. In contrast, in the peritumoral area, GB interacts with preexistent PC by altering them to cause effects such as vascular co-optation or immune evasion that allow survival and growth of the tumor infiltration front
Clinically, the disease is characterized by progressive neurological deficits whose nature depends on the location of the tumor. Recently, it has been shown that GB cells also affect neurons with the network of tumor microtubes (TMs) causing neurodegeneration in a process defined as “vampirization.” GB cells establish a positive feedback loop through cJun N-terminal kinase (JNK)/matrix metalloproteinase (MMP) signaling with vampirized neurons to promote their expansion [9•].
Several mechanisms that allow the progression of GB have been reported [10,11,12,13,14]. The high heterogeneity of GB cells prevents that the applied treatments to eliminate the cancer progression (resection, radiotherapy, and chemotherapy with temozolomide) can be successful. Furthermore, GB evades the immune system by preventing anti-tumor immune responses and supporting malignant growth, which is a major obstacle to successful immunotherapy [6••, 15,16,17,18].
Immune Evasion of GB
The immune system is highly specialized in the recognition of foreign antigens and unhealthy cells, including tumor cells. However, cancers have developed different strategies to suppress the anti-tumor immune response, escaping recognition of the immune system.
Tumor cells induce several mechanisms in the tumor microenvironment (TME) that alter innate and adaptive anti-tumor immune responses. These mechanisms include downregulation of the major histocompatibility complex (MHC) expression and polarization of macrophages to an M2 immunosuppressive phenotype, decreased T cell activation and proliferation, induction of anergic T cells, and differentiation of T regulatory cells (Tregs) [10, 19, 20••]. GB cells are able to evade anti-tumor T cell responses by several mechanisms, such as increased infiltration of Tregs and T cells with high expression of PD-1 (programmed cell death protein 1) and CTLA-4 (cytotoxic T lymphocyte-associated protein 4), by preventing the processing and presentation of antigen on MHC-class I and upregulating the PDL1 expression, the PD-1 receptor ligand, and negative regulator of the T cell activation [10, 21, 22].
It is well-known that the microenvironment of the brain tumor is characterized by the secretion of a variety of anti-inflammatory molecules, not only by tumor cells themselves but also by other peritumoral cells previously conditioned by the tumor. These peritumoral cells are peripheral immune cells (i.e., tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSC), Tregs, and T cells), and various specialized organ-resident cells including astrocytes, microglial cells, and pericytes (PC) [6••, 15, 17].
Immune Function of PC
In the last years, PC have been gaining special attention due to their role in GB progression. In the brain, PC are located on the abluminal wall of microvessels and venules, forming part of the blood–brain barrier (BBB) together with astrocytic endfeet and endothelial cells. PC are heterogeneous in their morphology, distribution, molecular markers, origin, and function [23]. At least two PC subsets have been described in brain microvessels: type-1 and type-2 PC, which are distinguished by Nestin gene expression [24, 25].
The BBB forms a neurovascular unit that regulates homeostasis of the central nervous system [26,27,28]. PC contribute to the maintenance of the BBB integrity through the elimination of toxic products, the regulation of vesicle trafficking, and the control of the expression of endothelial tight junction and adherens proteins [26, 28,29,30,31].
PC are contractile cells that regulate the tone and morphology of the vessels [29, 32] and are involved in new vessel formation during angiogenesis [33]. PC have stem cell-like properties [34,35,36] and release pro-regenerative growth factors and extracellular vesicles [31, 37, 38].
PC show immune properties, both innate and adaptive, and represent an immune defense in the brain modulating neuroinflammation [39,40,41,42]. They can respond to pro-inflammatory stimuli and are able to sense different types of danger due to the expression of functional pattern recognition receptors (PPRs), contributing to the onset of innate immune response. PC show interconvertible properties with macrophages [43] and have the ability to promote inflammation in response to brain damage [27, 38], enhancing BBB disruption through paracrine secretion of several vasculotoxic molecules and reactive oxygen species (ROS) [27, 31, 38]. In this context, PC not only secrete a variety of chemokines and cytokines but also overexpress adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), which guide innate immune cells after transendothelial migration across vessel walls, facilitating their co-stimulation [44]. It has been reported that in response to interferon-gamma cytokine (IFN-γ), PC upregulate the expression of ICAM-1 and MHC class I and II molecules, acquiring the capacity to present antigen to T cells, whereas under exposure to tumor necrosis factor-alpha (TNF-α), PC upregulate levels of VCAM-1 [39]. In addition to a role in innate immune responses, PC are also involved in adaptive immune responses [38, 43]. PC seem to be able to anergize T cells, at least in part, through the expression of PDL-1 and are able to modulate Tregs creating an immunosuppressive microenvironment [44].
Effects of GB on PC
It is currently known that the neurovascular unit represents the perivascular niche, a privileged environment for tumor support, expansion, and growth, where cells with stem cell properties are found [45]. GB cells infiltrate and invade the brain parenchyma, interacting with cells of the perivascular space and brain microvessels, which are composed of endothelial cells and PC [8, 29, 46], establishing a functional network of microtubes from GB [5,6,7] that leads to its progression. Importantly, PC do not act as a defense against the tumor progression, but their immune function fails to contribute to the elimination of GB [6••, 20••, 41, 47••].
An overall analysis of the functional properties of PC reveals that these are multifaceted cells with the ability to significantly influence tumor development. As a component of the TME, PC may actively contribute to some classic cancer hallmarks such as induction of angiogenesis, sustained tumor growth, metastasis, and evasion of immune system.
But 90% of recurrences in GB occur in the peritumoral area [48]. The tumor infiltration front [49], classically little studied and without repercussion in the grade of the tumor stage and therapeutic decision-making, seems to be a key place to understand GB growth and find targets for new immunological therapies [50]. Therefore, there is a growing interest in studying more precisely the peritumoral tissue [51] in order to improve the individualized therapeutic management of each patient.
It has been previously reported that glioma cells use different routes of invasion such as intraparenchymal invasion along white matter tracks [52, 53] or invasion along blood vessels [52]. GB cells infiltrate away from the tumor core more often and faster when they are associated with perivascular cells [54•].
Recently, it has been shown that PC could be the main target of the tumor infiltration front and have a fundamental role in tumor proliferation, expansion, and survival, as well as in the regulation of the immune system [6••, 41, 47••].
Therefore, for a better understanding of the effects of GB on PC, in this chapter we will classify them according to whether they are intratumoral or peritumoral PC, focusing mainly on peritumoral effects which seem to have better future prospects for finding effective therapies in GB cancer (Fig. 1, Table 1).
Effects of GB on Intratumoral PC
In pathophysiology, PC have been implicated as mediators of processes associated with cancer, including angiogenesis and metastasis [55]. Glioma stem cells (GSC) promote tumor angiogenesis by increasing VEGF expression [56], which might include VEGF release from PC [37, 57]. Type-2 PC have been described to participate in tumor angiogenesis [58] and seem to be generated from GSC during angiogenesis process to allow the development of blood vessels and tumor growth [13].
GB is one of the most angiogenic tumors, being essential the hypoxia conditions to trigger tumor angiogenesis. It is known that hypoxia in TME may recruit PC. Under hypoxia conditions, GB cells upregulate their production of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF). VEGF and bFGF induce the recruitment of PC. VEGF increases the secretion of platelet-derived growth factor-BB (PDGF-BB) by endothelial cells, and FGF-2 increases the expression of PDGF receptor in PC. Activation of PDGF receptor by PDGF-BB leads to activation of PC. These PC then solidify immature vasculatures, making them less permeable (Fig. 1, Table 1) [57].
Interestingly, under hypoxia conditions such as what occurs in GB, brain PC are able to differentiate into neural and vascular lineage cells as well as activated microglial cells [59, 60]. Microglial cells have been demonstrated to be recruited and activated by the TME, contributing to glioma growth [61, 62]. Thus, these studies support an important role of PC, as precursor cells of other stromal cells that might be facilitating tumor growth as a consequence of the TME.
On the other hand, intratumoral PC also modulate T cell responses. Bose et al. (2013) showed for the first time that GB-derived PC induce dysfunction or anergy of CD4+ T cells, which contributes to the GB immune evasion that favors progressive tumor growth [40]. This study revealed that the immunoregulatory phenotype/function associated with PC in the tumor niche appears to be reinforced by the cytokine IL-6 and requires of intrinsic expression of regulator of G protein signaling 5 (RGS5). Significant overexpression of RGS5 on GB-derived PC was detected, along with upregulation of the co-stimulatory molecule CD80 and the expression of the negative regulator of the anti-tumor T cell responses, programmed death ligand 1 (PDL-1). Although IL-6 promoted the expression of MHC-II, it also promoted a higher co-inhibitory/co-stimulatory molecules ratio in GB-derived PC than in control PC. RGS5 expression was shown to correlate with induced T cell anergy by tumor-derived PC. In addition, a significant amount of adhesion molecules, such as ICAM-1 and VCAM-1, were found in GB-derived PC, being identified ICAM-1 as responsible of T cell anergy induction through interaction with LFA-1 of CD4+ T cells [40].
Effects of GB on Peritumoral PC
GB Switches PC Function from Being Tumor Suppressor to Tumor Promoter
Initially, according to Folkman (1971), it was considered that tumor growth was only “dependent on angiogenesis,” which provides nutrients and oxygen to the tumor through blood flow. But it is currently known that GB can progress without angiogenesis [63]. Instead, GB can perform vascular co-optation, a mechanism by which GB interacts with preexisting blood vessels by kidnapping them to promote infiltration and tumor progression. This effect might explain why anti-angiogenic treatments have a lower effect than expected [64].
In fact, Caspani et al. (2014) show that GB malignancy proceeds via specific, Cdc42-dependent, and previously unknown interactions of tumor cells, through actin-based cytoplasmic microtubes, with brain PC. This interaction results in the co-option of modified preexisting blood vessels that support the expansion of the tumor margin.
In addition, GB cells induce immunomodulatory changes in PC in a dependent manner on cellular interaction that changes the function of PC from being tumor suppressor to tumor promoter (Fig. 1, Table 1) [41]. As a result of their interaction with GB, PC acquire an immunosuppressive function that helps to evade the anti-tumor immune response and, consequently, participate in the promotion of tumor growth [6••, 41, 47••, 65].
GB Upregulates Chaperone-Mediated Autophagy Aberrantly in PC
The GB-induced phenotypic switch of PC from being tumor suppressor to tumor promoter likely requires major changes in PC proteome. Proteins are regulated through changes in transcriptional programs as well as through active degradation of proteins to ensure that specific functions in cells act properly or are finished. Selective degradation of intracellular proteins occurs mainly via the ubiquitin proteasome system or through selective forms of autophagy.
Chaperone-mediated autophagy (CMA) is a selective process of lysosomal degradation of soluble cytosolic proteins with KFERQ-like motifs. These motifs are recognized by the chaperone heat shock cognate 71 kDa protein (Hsc70) on the substrate to carry it to the lysosome with the help of other co-chaperones. The protein substrate binds to the CMA receptor at the lysosomal membrane; the limiting component of this pathway known as lysosome-associated membrane protein type 2A (LAMP2A). The protein is unfolded and translocated into the lysosome to be degraded with the help of a resident chaperone Hsc70 in the lysosomal lumen [66, 67].
Regulation of CMA activity is key for proper cell function and homeostasis, since its downregulation increases the intracellular accumulation of damaged proteins, defective regulation of many cell functions, and defective responses to different stresses, such as oxidative stress, toxic exposure, or nutrient deprivation [68,69,70]. CMA is a degradation pathway that modulates the function of some immune cells [71, 72], including PC [47••].
Studies in our lab on CMA on PC have let us demonstrate that GB interaction with PC in peritumoral areas promotes a ROS burst in GB cells that leads to an upregulation of the CMA receptor expression (LAMP-2A) in PC (Fig. 2). Then, LAMP2-A is delivered to the lysosomal membrane to upregulate CMA activity aberrantly in PC [47••]. In this way, the aberrant upregulation of GB-induced CMA in PC degrades or interferes with proteins that are required to prevent the establishment of cell–cell interactions and the specific release of cytotoxic products that impair growth and survival of GB. Furthermore, GB-induced CMA changes the immune function of PC, impairing the generation of anti-tumor immune responses (Fig. 2) and, thus, switching PC function from being tumor suppressor to tumor promoter, as described below [47••].

Model of the effects of GB-induced CMA on the PC. An original drawing shows the GB interaction with PC promoting a ROS burst that induces an aberrant upregulation of the CMA in PC through the expression of LAMP-2A (big red arrow). The Hsc70 chaperone binds to KFERQ motifs in CMA substrate proteins and, in a complex with other co-chaperones, delivers them to the lysosomal membrane where they interact with LAMP-2A. Then, they are translocated to the lysosomal lumen assisted by a lysosomal Hsc70 to be degraded. As a consequence of the induction of CMA by GB, the degradation of specific proteins modulates the change of the PC function from tumor-suppressor to tumor-promoter. GB-induced CMA promotes an immunosuppressive function in PC that fails to support T cell responses GB (blue arrows). Modulation of the PC proteome eventually results in changes in programs of gene expression that include upregulation of the anti-inflammatory cytokines TGF-β and IL-10, which exert a negative regulation on T cells and antigen-presenting cells. T cell activation and proliferation are affected and regulatory T cells are generated (blue arrows). The absence of co-stimulatory molecules, such as CD80 and CD86, and the expression of the negative regulator PDL-1 also facilitate an immunosuppressive function in PC. GB-induced CMA in PC also inhibits the secretion of proteins that may have anti-tumor activity, stabilizes GB-PC interaction to facilitate tumor growth, and modulates their MSC-like properties such as increasing in the secretion of vesicles with pro-regenerative factors that assist tumor proliferation (white arrows)
GB Ablates the Immune Function of PC
GB has been shown to induce an anti-inflammatory phenotype on brain PC [6••, 47••]. Studies from our lab on the immune function of PC have revealed high levels of anti-inflammatory cytokine expression in vitro and in vivo in brain PC that interact with GB cells. PC secrete high levels of anti-inflammatory cytokines IL-10 and TGF-β when contacting with GB cells and, conversely, produce hardly or much lower levels of pro-inflammatory cytokines such as IL-1, IL-23, IL-12, or TNF-α compared to control PC [6••]. Incubation of PC with supernatants from GB culture media is not enough to lead to an anti-inflammatory phenotype, which supports that cell–cell interaction between GB and PC is essential for the acquisition of an immunomodulatory phenotype acquired in PC in response to GB [6••].
GB prevents anti-tumor inflammatory responses in PC as a result of the aberrant upregulation of CMA in PC. The anti-inflammatory phenotype of PC contacting with GB is prevented in CMA-deficient LAMP-2A knockout PC (KO PC), which show reduced mRNA level expression of Tgf-β and Il-10 gene and higher levels of the pro-inflammatory cytokine TNF-α compared to WT PC [47••]. Therefore, GB-induced CMA is the molecular mechanism responsible for this immune change in PC (Fig.1).
On the other hand, GB cells also promote expression of surface membrane molecules that are needed for an acquired immunosuppressive function in PC that favors tumor growth [6••, 47••]. Activated PC have been reported to present macrophage-like properties acquiring phagocytic activity and the ability to present antigens to T cells [73,74,75]. PC gain some of the immunosuppressive properties of tumor-associated macrophages, in response to their interaction with GB cells [6••]. In vitro, PC that are interacting with GB cells express high levels of IL-4RA, IL-1RN, and other membrane molecules implicated in the inhibition of anti-tumor responses [6••], [76, 77]. The immunosuppressive ligand of PD-1, PDL-1, which is a negative regulator of T cell activation and has been associated with glioblastoma progression [10, 78, 79], is expressed in PC under resting conditions and its level of expression is maintained upon GB cell interaction in vitro and in vivo [6••]. However, the expression of the co-stimulatory molecules for promotion of effective anti-tumor T cell responses such as CD80 and CD86 is significantly reduced in GB-conditioned PC compared to control PC and dependent on GB-induced CMA [47••]. Furthermore, the ability of PC to present antigen to T cells is also affected by GB cell interaction since surface expression of major histocompatibility complex class II molecules (MHC-II) is drastically reduced in PC co-cultured with GB cells compared to control PC [6••].
Activated PC by inflammatory challenge have the ability to present antigen on MHC molecules to T cells, regulating the activity of different T cell populations [39, 40, 80]. But, the ability of PC to activate T cell responses is suppressed by the interaction with GB cells through GB-induced CMA (Fig. 2) [6••, 47••].
Then, the acquired anti-inflammatory phenotype and the expression of an immunosuppressive molecule pattern in GB-conditioned PC interacting with GB cells in peritumoral area result in a failed antigen presentation to activate T cells. In vitro studies from our lab have shown that this effect is mediated by GB-induced CMA [47••]. In fact, CD4+ T cells are defective in proliferation and IL-2 cytokine production when co-cultured with antigen-loaded antigen-presenting cells (APCs) in the presence of GB-conditioned PC. By contrast, CD4+ T cells are activated properly, producing high levels of IL-2 and proliferating in response to APCs and in presence of control PC [6••]. This implies that, when PC are in contact with GB cells, they not only show a reduced ability to cross-present tumor antigens but also hinder the function of APCs. Furthermore, GB-conditioned PC secrete anti-inflammatory cytokines that seem to be enough to affect T cell responses, which might explain the observed effect on the failed activation of T cells in presence of APCs. Upon T cell receptor and co-stimulation engagement, following activation with anti-CD3 and anti-CD28 antibodies, CD4+ T cells show reduced proliferation and IL-2 production in the presence of culture medium from PC co-cultured with GB, compared to activated CD4+ T cells in the presence of medium from PC alone [6••].
To sum up, peritumoral PC reduce T cell responses through the induction of an anti-inflammatory response and the development of an immunosuppressive function in response to interaction with GB cells. These effects in PC are consequences of the ablation of their immune function by GB cells through aberrant upregulation of CMA (Table 1, Fig. 2). By contrast, LAMP2-A KO PC with impaired CMA activity do not allow to GB cells to form stable interactions with them, impairing to be sensibilized by the tumor cells. LAMP2-A KO PC, in vitro, maintain co-stimulatory molecules such as CD-80 and are able to present antigen to T cells to activate them. In vivo studies demonstrate that T cells from mice grafted with PC and GB present higher levels of T cell inhibitor receptors, PD-1 and cytotoxic T lymphocyte-associated protein-4 (CTLA-4), while T cell from mice grafted with KO PC and GB do not. Those T cells are found in the meningeal brain space and perivascular areas co-localizing with peritumoral GB cells that are favored to proliferate, leading to tumor growth. By contrast, mice grafted with CMA-deficient PC are able to develop tumor antigen-specific T cells that promote tumor elimination [47••].
GB Promotes the Establishment of Stable Interactions with PC
GB cells induce stable interactions with PC to modulate their functions and ensure tumor growth. All GB changes on PC seem to indicate that GB cells need upregulated CMA in PC to stabilize GB-PC interactions and, consequently, to allow GB tumor cell growth and survival. By contrast, GB cells are not able to interact properly with CMA-deficient PC, indicating that GB cells promote the establishment of stable interactions with PC through aberrant protein degradation by CMA (Fig. 3a). In fact, studies with a GB mouse model revealed that infiltrated GB cells in perivascular areas, where PC are interacting with the tumor cells, proliferate considerably more than in GB mice that were grafted with CMA-deficient PC, in which only residual number of GB cells can be found (Fig. 3b) [47••].

GB needs stable interactions with PC to survive (reproduced from Valdor et al., Glioblastoma ablates pericytes anti-tumor immune function through aberrant upregulation of chaperone-mediated autophagy. Proc Natl Acad Sci U S A, 2019. 116(41): pp. 20655–20665. This open-access article is distributed under Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND). a Interaction of WT PC and KO PC (for LAMP-2A) with GB cells. Images obtained by scanning electron microscopy are representative of at least 3 independent experiments. Arrows indicate interaction through nanotubes (yellow arrow) and cytosol fusion (red arrow) after 72 h of co-culture (scale bar, 10 μm). b GB tumor growth in mice that were xenografted with co-cultures of WT PC + GB cells (GB/WT PC) and compared to mice xenografted with KO PC + GB cells (GB/KO PC) (scale bars of left, 250 μm)
GB cells interact with PC through microtube-like ultralong protrusions towards the PC surface, while PC project small nanotubes to maintain stable interaction with GB cells. GB-interacting PC reduce the expression of the interaction protein occludin through GB-induced CMA (Fig. 2). PC without GB-induced CMA fail to promote stable interactions and upregulate the expression of occludin, which might indicate, in part, that the defective cell–cell interaction may be due to the deregulation of adhesion proteins. Interestingly, occludin forms part of the tight junctions that provide stability to the BBB, and PC seem to regulate the formation of tight junctions by the interaction with endothelial cells [81]. The BBB is disrupted during tumor progression and is then, known as the blood–tumor barrier (BTB). BTB is highly heterogeneous and characterized by a non-uniform permeability and active efflux of molecules [82,83,84,85]. Thus, it seems that the decreased expression of occludin in PC through GB-induced CMA might be responsible, at least in part, for the rupture of BBB during tumor progression.
On the other hand, occludin in PC also promotes expression and activation of AMPK and transcription factors such as NFkB and SP1. These transcription factors regulate expression of glucose transporters and mediate the rapid response to metabolic stress, including inflammation, through the production of inflammatory molecules such as TNF-α and interleukins [86]. Therefore, the reduced expression of occludin in PC might be also contributing to the hard detection of pro-inflammatory cytokine expression in GB-conditioned PC.
GB Prevents the Anti-tumor Protein Secretory Function of PC
Proliferation of GB cells is not affected by the presence of PC, and even more, GB cells need to interact with PC to support optimal proliferation and survival through nutrients providing and growth factor exchange. But, GB proliferation is significantly reduced and cell death is increased when tumor cells try to interact stably with CMA-deficient PC, which demonstrates that GB-induced CMA effect on PC is essential for the following effects on several PC functions that facilitate tumor survival and growth.
GB cells induce proteome changes in PC through CMA, also preventing their defense secretory function against the tumor. CMA-deficient PC have been shown to release a significant secretion of toxic proteins for the tumor that reduce tumor cell survival and prevent PC-GB interactions. GB cells in presence of CMA-deficient PC release danger signals such as ROS and granulocyte–macrophage colony-stimulating factor (GM-CSF), correlating with a high increase of tumor cell death. In addition, these danger signals can be reproduced in GB cells when are treated only with culture media from co-cultures of GB cells with CMA-deficient PC, which contain toxic proteins for the tumor cells. Thus, the toxic protein secretion of CMA-deficient PC is enough to induce GB cell death and negatively impact tumor survival, supporting that GB cells avoid the toxic secretion of proteins by PC upon cell–cell interaction [47••].
GB Modulates Mesenchymal Stem Cell-Like Properties in PC
The secretome of PC consists of a broad variety of functional molecules including inflammatory modulators, angiogenic, trophic factors, and extracellular matrix proteins [31]. GB-induced CMA in PC alters the expression of markers/properties associated with mesenchymal stem cells (MSC) and contributes to regulating interactions with GB cells. The expression of several angiogenic factors such as VEGF, angiotensin I, and cytokine IL-6, which are associated to altered PC proliferation and regenerative properties [31, 37, 38], is clearly increased in PC when contacting with GB cells [6••, 47••]. However, these GB-induced changes in PC remain unaffected in the same conditions in CMA-deficient PC [47••], which demonstrates that those changes are dependent of GB-induced CMA in PC. By contrast, anti-angiogenic factors associated with tumor regression, such as anti-thrombin and osteonectin (SPARC) [87,88,89,90], are secreted by CMA-deficient PC contacting with GB cells [47••]. Interestingly, the release of these proteins might be part of the PC secretion that is toxic for the tumor, but this needs further studies to be elucidated.
GB cells are also able to modulate MSC-like markers in PC such as Sca-1, CD105, and CD90 through aberrant upregulation of CMA activity [47••]. In addition, GB cells promote increased vesicles secretion in PC, containing cytokines and other pro-regenerative factors [31, 37]. Scanning microscopy analyses from our lab showed that GB cells interacting with PC lead to accumulation of secretory vesicles in the sites of contact between PC and GB, whereas they are rarely observed in CMA-deficient PC. Interestingly, only secreted vesicles from CMA-deficient PC that are contacting with GB cells were found to inhibit GB proliferation, which may be due to qualitative changes in vesicle composition/content rather than just mere changes in their quantity [47••].
In conclusion, GB cells induce several changes in PC for their own benefit through the proteome modulation of PC by abnormal upregulation of the selective form of autophagy, CMA. Those proteome changes lead to subvert PC’s anti-tumor responses through the degradation of proteins that may participate in the expression of inflammatory mediators and proper induction of T cell responses, regulate MSC-like properties, and strengthen PC-GB interaction, all together contributing to maintain tumor survival and expansion (Fig. 2).
Conclusions and Future Directions
GB is often resistant to standard treatments, which include resection, radiotherapy, and chemotherapy with temozolomide [91]. Numerous studies are focused on new molecular targets to treat GB [92, 93]; however, none of them seem to be effective to eliminate the tumor progression definitely. The main reasons for this elusive behavior of GB are the rapid transformation of GB cells and the heterogeneity of biochemical characteristics in these tumors, which hinder the effectiveness of targeted therapies and also favor the evasion of the brain immune responses. Immune evasion within the TME supports malignant growth and is also a major obstacle for successful immunotherapy. Many drugs are not able to cross the BBB to act on the tumor. Therefore, immunotherapy will only be successful if it is targeted and can be successfully delivered to the brain to deal effectively with cellular heterogeneity [94].
Intratumoral PC seem to come from GSC, allowing the development of blood vessels of the tumor [13]. Therefore, targeted therapy against specific markers of GSC-derived PC might block and, probably, reverse GB tumor growth, but not prevent the GB spreading in peritumoral areas where not GSC-derived PC interact and facilitate tumor progression [47••]. Then, a better understanding of the molecular differences between the GSC-derived PC and not GSC-derived PC is needed.
On the other hand, the molecular mechanisms underlying specific interactions between GB and PC remain poorly characterized. Deciphering these mechanisms may allow finding new specific therapeutic targets capable of inhibiting the development of GB in the host brain microenvironment [47••].
In vitro and in vivo data support that GB-PC interaction in the peritumoral area switches PC function from being tumor suppressor to tumor promoter by changes dependent on GB-induced CMA. Therefore, preventing the GB-induced upregulation of CMA in PC that interact with GB cells may represent a targeting strategy for the development of new therapies against GB [47••]. Even more, the immunosuppressive function that PC acquire in the presence of GB can be reversed with the modulation of CMA activity, and therefore, effective anti-tumor immune responses can be generated [47••]. Excitingly, when the aberrant activation of CMA in PC is avoided, a greater number of memory T cells is detected, which correlates with an efficient regression of the tumor. These T cells can represent cells that have been activated by tumor antigens and could generate an efficient anti-tumor response [47••].
In conclusion, an in-depth study of the changes in the proteome of WT or LAMP-2A-deficient PC in presence of GB cells is essential to find CMA substrates, as possible specific markers that might be implicated in the regulation and consequences of GB-induced CMA in PC and might let us eliminate GB cancer.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ostrom QT, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro Oncol. 2019;21(Supplement_5):v1–v100.
Zong H, Verhaak RG, Canoll P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev Mol Diagn. 2012;12(4):383–94.
Zong, H., L.F. Parada, and S.J. Baker, Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb Perspect Biol, 2015. 7(5).
Pintus D, Marruchella G, Masia M, Maestrale C, Cancedda MG, Contu C, et al. Glioblastoma with oligodendroglioma component in a ewe. J Vet Diagn Investig. 2016;28(4):449–54.
Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528(7580):93–8.
•• Valdor R, et al. Glioblastoma progression is assisted by induction of immunosuppressive function of pericytes through interaction with tumor cells. Oncotarget. 2017;8(40):68614–26 This paper shows that glioblastoma cells promote an immune function on pericytes that facilitates tumor progression.
Farin A, Suzuki SO, Weiker M, Goldman JE, Bruce JN, Canoll P. Transplanted glioma cells migrate and proliferate on host brain vasculature: a dynamic analysis. Glia. 2006;53(8):799–808.
Hjelmeland AB, Lathia JD, Sathornsumetee S, Rich JN. Twisted tango: brain tumor neurovascular interactions. Nat Neurosci. 2011;14(11):1375–81.
• Portela M, et al. Glioblastoma cells vampirize WNT from neurons and trigger a JNK/MMP signaling loop that enhances glioblastoma progression and neurodegeneration. PLoS Biol. 2019;17(12):e3000545 This article describes a molecular mechanism for tumor microtube production, infiltration, and maintenance that can explain both neuron-dependent tumor progression and GB-associated neural decay.
Nduom, E.K., M. Weller, and A.B. Heimberger, Immunosuppressive mechanisms in glioblastoma. Neuro Oncol, 2015. 17 Suppl 7: p. vii9-vii14.
Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol. 2015;11(9):504–14.
Kast RE, Hill QA, Wion D, Mellstedt H, Focosi D, Karpel-Massler G, et al. Glioblastoma-synthesized G-CSF and GM-CSF contribute to growth and immunosuppression: potential therapeutic benefit from dapsone, fenofibrate, and ribavirin. Tumour Biol. 2017;39(5):1010428317699797.
Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153(1):139–52.
Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17(2):170–82.
Otvos B, Silver DJ, Mulkearns-Hubert EE, Alvarado AG, Turaga SM, Sorensen MD, et al. Cancer stem cell-secreted macrophage migration inhibitory factor stimulates myeloid derived suppressor cell function and facilitates glioblastoma immune evasion. Stem Cells. 2016;34(8):2026–39.
Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32(2):253–67 e5.
Henrik Heiland D, Ravi VM, Behringer SP, Frenking JH, Wurm J, Joseph K, et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat Commun. 2019;10(1):2541.
Arrieta VA, et al. The possibility of cancer immune editing in gliomas. A critical review. Oncoimmunology. 2018;7(7):e1445458.
Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31(3):326–41.
•• Molina ML, et al. Autophagy in the immunosuppressive perivascular microenvironment of glioblastoma. Cancers (Basel). 2019;12(1) This work describes different forms of autophagy in the glioblastoma perivascular niche as possible therapeutic targets against tumor proliferation and survival.
Jacobs JF, et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro-Oncology. 2009;11(4):394–402.
Reardon DA, Freeman G, Wu C, Chiocca EA, Wucherpfennig KW, Wen PY, et al. Immunotherapy advances for glioblastoma. Neuro-Oncology. 2014;16(11):1441–58.
Dias Moura Prazeres PH, et al. Pericytes are heterogeneous in their origin within the same tissue. Dev Biol. 2017;427(1):6–11.
Birbrair A, Wang ZM, Messi ML, Enikolopov GN, Delbono O. Nestin-GFP transgene reveals neural precursor cells in adult skeletal muscle. PLoS One. 2011;6(2):e16816.
Birbrair A, Zhang T, Files D, Mannava S, Smith T, Wang ZM, et al. Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res Ther. 2014;5(6):122.
Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68(3):409–27.
Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14(11):1398–405.
Cheng J, Korte N, Nortley R, Sethi H, Tang Y, Attwell D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018;136(4):507–23.
Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443(7112):700–4.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–6.
Gaceb A, Barbariga M, Özen I, Paul G. The pericyte secretome: potential impact on regeneration. Biochimie. 2018;155:16–25.
Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508(7494):55–60.
Ding Y, Song N, Luo Y. Role of bone marrow-derived cells in angiogenesis: focus on macrophages and pericytes. Cancer Microenviron. 2012;5(3):225–36.
Appaix F, Nissou MF, van der Sanden B, Dreyfus M, Berger F, Issartel JP, et al. Brain mesenchymal stem cells: the other stem cells of the brain? World J Stem Cells. 2014;6(2):134–43.
Dore-Duffy P. Pericytes: pluripotent cells of the blood brain barrier. Curr Pharm Des. 2008;14(16):1581–93.
Corselli M, et al. Identification of perivascular mesenchymal stromal/stem cells by flow cytometry. Cytometry A. 2013;83(8):714–20.
Gaceb A, Özen I, Padel T, Barbariga M, Paul G. Pericytes secrete pro-regenerative molecules in response to platelet-derived growth factor-BB. J Cereb Blood Flow Metab. 2018;38(1):45–57.
Rustenhoven J, Jansson D, Smyth LC, Dragunow M. Brain pericytes as mediators of neuroinflammation. Trends Pharmacol Sci. 2017;38(3):291–304.
Balabanov R, Beaumont T, Dore-Duffy P. Role of central nervous system microvascular pericytes in activation of antigen-primed splenic T-lymphocytes. J Neurosci Res. 1999;55(5):578–87.
Bose A, Barik S, Banerjee S, Ghosh T, Mallick A, Bhattacharyya Majumdar S, et al. Tumor-derived vascular pericytes anergize Th cells. J Immunol. 2013;191(2):971–81.
Caspani EM, Crossley PH, Redondo-Garcia C, Martinez S. Glioblastoma: a pathogenic crosstalk between tumor cells and pericytes. PLoS One. 2014;9(7):e101402.
Pieper C, Marek JJ, Unterberg M, Schwerdtle T, Galla HJ. Brain capillary pericytes contribute to the immune defense in response to cytokines or LPS in vitro. Brain Res. 2014;1550:1–8.
Thomas WE. Brain macrophages: on the role of pericytes and perivascular cells. Brain Res Brain Res Rev. 1999;31(1):42–57.
Navarro R, et al. Immune regulation by pericytes: modulating innate and adaptive immunity. Front Immunol. 2016;7:480.
Brooks MD, Sengupta R, Snyder SC, Rubin JB. Hitting them where they live: targeting the glioblastoma perivascular stem cell niche. Curr Pathobiol Rep. 2013;1(2):101–10.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307.
•• Valdor R, et al. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2019;116(41):20655–65 This work shows the effects of glioblastoma on perivascular pericytes in its own benefit.
Lemee JM, Clavreul A, Menei P. Intratumoral heterogeneity in glioblastoma: don’t forget the peritumoral brain zone. Neuro-Oncology. 2015;17(10):1322–32.
Engelhorn T, Savaskan NE, Schwarz MA, Kreutzer J, Meyer EP, Hahnen E, et al. Cellular characterization of the peritumoral edema zone in malignant brain tumors. Cancer Sci. 2009;100(10):1856–62.
Osta WA, Chen Y, Mikhitarian K, Mitas M, Salem M, Hannun YA, et al. EpCAM is overexpressed in breast cancer and is a potential target for breast cancer gene therapy. Cancer Res. 2004;64(16):5818–24.
Aubry M, de Tayrac M, Etcheverry A, Clavreul A, Saikali S, Menei P, et al. From the core to beyond the margin: a genomic picture of glioblastoma intratumor heterogeneity. Oncotarget. 2015;6(14):12094–109.
Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117.
Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neuro-Oncol. 2004;70(2):217–28.
• Alieva M, et al. Intravital imaging of glioma border morphology reveals distinctive cellular dynamics and contribution to tumor cell invasion. Sci Rep. 2019;9(1):2054 This work shows the different parts of the tumor niche and identifies the perivascular space as the invasive margin of the glioblastoma tumor.
Raza A, Franklin MJ, Dudek AZ. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am J Hematol. 2010;85(8):593–8.
Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66(16):7843–8.
Wong ET, Brem S. Antiangiogenesis treatment for glioblastoma multiforme: challenges and opportunities. J Natl Compr Cancer Netw. 2008;6(5):515–22.
Birbrair A, Zhang T, Wang ZM, Messi ML, Olson JD, Mintz A, et al. Type-2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol. 2014;307(1):C25–38.
Ozen I, et al. Brain pericytes acquire a microglial phenotype after stroke. Acta Neuropathol. 2014;128(3):381–96.
Nakagomi T, Kubo S, Nakano-Doi A, Sakuma R, Lu S, Narita A, et al. Brain vascular pericytes following ischemia have multipotential stem cell activity to differentiate into neural and vascular lineage cells. Stem Cells. 2015;33(6):1962–74.
Daginakatte GC, Gianino SM, Zhao NW, Parsadanian AS, Gutmann DH. Increased c-Jun-NH2-kinase signaling in neurofibromatosis-1 heterozygous microglia drives microglia activation and promotes optic glioma proliferation. Cancer Res. 2008;68(24):10358–66.
Ellert-Miklaszewska A, Dabrowski M, Lipko M, Sliwa M, Maleszewska M, Kaminska B. Molecular definition of the pro-tumorigenic phenotype of glioma-activated microglia. Glia. 2013;61(7):1178–90.
Wesseling P, van der Laak JAWM, de Leeuw H, Ruiter DJ, Burger PC. Quantitative immunohistological analysis of the microvasculature in untreated human glioblastoma multiforme. Computer-assisted image analysis of whole-tumor sections. J Neurosurg. 1994;81(6):902–9.
Donnem T, Hu J, Ferguson M, Adighibe O, Snell C, Harris AL, et al. Vessel co-option in primary human tumors and metastases: an obstacle to effective anti-angiogenic treatment? Cancer Med. 2013;2(4):427–36.
Ochs K, Sahm F, Opitz CA, Lanz TV, Oezen I, Couraud PO, et al. Immature mesenchymal stem cell-like pericytes as mediators of immunosuppression in human malignant glioma. J Neuroimmunol. 2013;265(1–2):106–16.
Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15(8):305–9.
Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19(6):365–81.
Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273(5274):501–3.
Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15(11):4829–40.
Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2006;103(15):5805–10.
Valdor R, Macian F. Autophagy and the regulation of the immune response. Pharmacol Res. 2012;66(6):475–83.
Valdor R, Mocholi E, Botbol Y, Guerrero-Ros I, Chandra D, Koga H, et al. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol. 2014;15(11):1046–54.
Matsumoto J, Takata F, Machida T, Takahashi H, Soejima Y, Funakoshi M, et al. Tumor necrosis factor-alpha-stimulated brain pericytes possess a unique cytokine and chemokine release profile and enhance microglial activation. Neurosci Lett. 2014;578:133–8.
Guijarro-Munoz I, et al. Lipopolysaccharide activates toll-like receptor 4 (TLR4)-mediated NF-kappaB signaling pathway and proinflammatory response in human pericytes. J Biol Chem. 2014;289(4):2457–68.
Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75(3):388–97.
Komohara Y, Jinushi M, Takeya M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014;105(1):1–8.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–96.
Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology. 2016;18(2):195–205.
Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wöhrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncology. 2015;17(8):1064–75.
Domev H, Milkov I, Itskovitz-Eldor J, Dar A. Immunoevasive pericytes from human pluripotent stem cells preferentially modulate induction of allogeneic regulatory T cells. Stem Cells Transl Med. 2014;3(10):1169–81.
Jo DH, Kim JH, Heo JI, Kim JH, Cho CH. Interaction between pericytes and endothelial cells leads to formation of tight junction in hyaloid vessels. Mol Cells. 2013;36(5):465–71.
Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95(8):4607–12.
Monsky WL, Mouta Carreira C, Tsuzuki Y, Gohongi T, Fukumura D, Jain RK. Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: mammary fat pad versus cranial tumors. Clin Cancer Res. 2002;8(4):1008–13.
Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology. 2018;20(2):184–91.
Arvanitis CD, Ferraro GB, Jain RK. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat Rev Cancer. 2020;20(1):26–41.
Castro V, Skowronska M, Lombardi J, He J, Seth N, Velichkovska M, et al. Occludin regulates glucose uptake and ATP production in pericytes by influencing AMP-activated protein kinase activity. J Cereb Blood Flow Metab. 2018;38(2):317–32.
Luengo-Gil G, Calvo MI, Martín-Villar E, Águila S, Bohdan N, Antón AI, et al. Antithrombin controls tumor migration, invasion and angiogenesis by inhibition of enteropeptidase. Sci Rep. 2016;6:27544.
Isaka T, Yoshimine T, Maruno M, Kuroda R, Ishii H, Hayakawa T. Altered expression of antithrombotic molecules in human glioma vessels. Acta Neuropathol. 1994;87(1):81–5.
Rivera LB, Brekken RA. SPARC promotes pericyte recruitment via inhibition of endoglin-dependent TGF-beta1 activity. J Cell Biol. 2011;193(7):1305–19.
Chlenski A, Liu S, Guerrero LJ, Yang Q, Tian Y, Salwen HR, et al. SPARC expression is associated with impaired tumor growth, inhibited angiogenesis and changes in the extracellular matrix. Int J Cancer. 2006;118(2):310–6.
Messaoudi K, Clavreul A, Lagarce F. Toward an effective strategy in glioblastoma treatment. Part I: resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov Today. 2015;20(7):899–905.
Zhu Y, Bassoff N, Reinshagen C, Bhere D, Nowicki MO, Lawler SE, et al. Bi-specific molecule against EGFR and death receptors simultaneously targets proliferation and death pathways in tumors. Sci Rep. 2017;7(1):2602.
Osuka S, Van Meir EG. Overcoming therapeutic resistance in glioblastoma: the way forward. J Clin Invest. 2017;127(2):415–26.
Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. 2020;20(1):12–25.
Funding
Dr. Valdor reports grants, personal fees and non-financial support from Ministerio de Economia y Competitividad de España (MINECO)-SAF2015-73923-JIN, Agencia estatal de investigación (AEI/FEDER/UE), and grants and non-financial support from Fundacion Seneca, agencia de ciencia y tecnologia de la region de Murcia, project-PI 20840/PI/18, during the conduct of the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pericytes
Rights and permissions
About this article

Cite this article
Molina, M.L., Valdor, R. The Effect of Glioblastoma on Pericytes. Curr. Tissue Microenviron. Rep. 1, 171–181 (2020). https://doi.org/10.1007/s43152-020-00016-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43152-020-00016-7