
Abstract
Purpose of Review
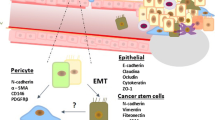
Stand out contribution of pericytes and cancer stem cells (CSC) to progression of colorectal cancer (CRC).
Recent Findings
Reported that CSC are responsible for the initiation and spread of the tumor; however, the microenvironment in the tumor plays a decisive role in prognosis; CSC and pericytes interaction are capable of contributing to neovascularization and promoting metastasis. We reviewed the contribution of cellular communication between pericytes, endothelial cells, and CSC with focus vascular remodeling, metastasis, angiogenesis, and vasculogenesis. Also, we were analyzed concise microRNAs and approved therapeutic target with the potential anti-angiogenic as treatment of CRC.
Summary
This work seeks to give an overview of pericytes in cancer and their role angiogenic with CSC in CRC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is any cancer that affects colon or rectum, it remains as a third most deadly by cancer worldwide. Actual treatment for the CRC includes surgical remove of the tumor, commonly associated with adjuvant or neo-adjuvant chemotherapy. Radiotherapy combination with surgery or chemotherapy is an option that has proven useful, but does not have a routine use in places where technical limitations are present, due to the radio-sensitivity of the rest of the bowel. Immunotherapy has been found to be useful for certain kinds of CRC, used against a wide spectrum of targets present in the malignant cells, offering a new approach of treatment. However, actual therapies for this condition are still far from being optimal. The 5-year survival rate in non-localized stages is around 60%. Metastasis and resistance treatment main cause of death.
Some theories have been structured to elucidate the main causes of failure of actual treatments, many of them indicating that the previously described heterogeneity of cells that comprise a tumor. Cancer stem cells (CSCs) has the ability of self-renew and the variety of cells that can be originated from differentiation of them; this indicates that the CSCs can initiate tumor formation and generate progenies of distinct differentiated cells, which explains the heterogeneity observed in tumor bulk. However, it is not the only cell type that participates in the tumor microenvironment, favoring its development or modifying its response to treatment. Other relevant cells that favor tumor metastasis are pericytes [1]. In this review, we purpose of remark the role of pericytes and CSC in this pathology of CRC and to describe the relationship between themselves, within the framework of this cancer. Additionally, we summarize the importance of them as target for new treatments, and we review the actual progress made in order to achieve clinical application of the knowledge that has arisen to the date.
Biology of Pericytes
Eberth, then Rouget first identified pericytes in the late nineteenth century as spatially isolated cells associated with the capillary wall and contractile properties that were localized around the endothelial cells of small blood vessels [1]. Pericytes are in close proximity to endothelial cells (20 nm apart) with a single pericyte covering several endothelial cells incompletely. The sources of pericytes are the mesoderm, the mesothelium, and the neural crest [2].
Zimmermann distinguished pericytes into three types according to their vascular location: (1) arterioles with multiple round branches; (2) capillaries with spindle-shaped, highly elongated, multiple short secondary processes, extending mainly along the long axis of blood vessels; and (3) venules where they are shorter stellate-shaped cells wrapped around endothelial components [3].
Marker pericyte expression is dynamic and varies among organs, developmental stages, activation/maturation state, the stringency of endothelial barrier properties, across individual microvascular networks as shown in Table 1 [13••]. Also, these markers are not exclusive of pericytes, and are expressed in endothelial cells, and smooth muscle cells. The presence of markers defines their properties as reported by Birbrair et al. [3] who identified two pericyte subpopulations from large blood vessels and small capillaries, named type-1 and type-2 pericytes. However, only type-2, which express PDGFRβ, CD146, and NG2 have been reported with angiogenic properties.
Pericyte Function
Pericytes function as multipotent cells with a variety of morphology and marker expression in different tissues. They are involved in the preservation of vascular stability and homeostasis including, but not restricted to, maintenance of vessel structure, regulation of blood flow, vascular permeability, and remodeling of the extracellular matrix (ECM). They are also essential for normal blood vessel development [14]. Unfortunately, in the case of abnormal pericyte function or the loss of numbers of pericytes can be associated with some pathologies such as fibrosis; ischemic organ failure; diabetes-related complications; pulmonary hypertension; Alzheimer disease; and cancer promoting tumor growth, metastasis, and angiogenesis. Some studies highlight the importance of pericytes in the progression of cancer by interaction with cancer stem cells (CSC) [15].
Cancer Stem Cells
CSC are a particular population of cancer cells that are different from those that comprise most of the tumor bulk. The presence of CSC in several solid tumors has been demonstrated in a wide variety of tissues and microenvironments, including brain tumors, melanomas, breast carcinomas, pancreatic adenocarcinomas, and gastric and colon cancer [16].
Colorectal Cancer and Cancer Stem Cells
CSCs have been associated with resistance to current therapeutic methods, and increasing knowledge about this topic suggests that development and relapse of the disease can be attributed to CSCs [17].
It is important to mention that identification of CSCs in CRC has not been particularly easy, and the techniques used include a variety of methods based on the stem cell–like properties of CSC such as spheroid formation and their ability to produce tumors that are more efficient than tumor cells when injected to immunodeficient mice. Initially, CD133 was mentioned as a reliable marker of CSC in primary human CRC, which was directly identified in xenograft tumors in immunodeficient rodents but subsequent studies have shown that expression of CD133 is also detectable in normal intestinal mucosa, in the vast majority of metastatic tumor cells; additionally, its expression is restricted to certain CSC subsets [18]. This is mentioned in order to point out that marker expression in CSCs in human CRC have been consistently difficult to identify, even when, to date, we can mention coexpression of normal stem cells. Some of these markers were CD133, and CD44, CD166, and ALDH1. An alternative to single marker identification is the evaluation of the coexpression of CD44, CD166, and EpCAM, LGR5 in the totally of tumor cells that has been reported to identify more specifically the CSC pool [19]. Also, it determines in vitro assays, the efficacy and safety of drug antineoplasic [20].
A hypothesis of carcinogenesis originated from the transformation of normal stem cells and the additional anomalous growth of epithelial cells. This hypothesis has been accumulating support due to the increasing evidence tending to corroborate it at a molecular level. Carcinogenesis in CRC from the accumulation of mutations in oncogenes and tumor suppressor genes has been demonstrated [21••]. Several pathways relevant to the carcinogenesis of CRC are as follows:
-
1)
It usually begins with an increase in expression of intracellular β-catenin in normal colon epithelial tissue. This results in the prolonged activation of the Wnt pathway, β-catenin stabilization, and C-terminal binding protein 1 (CtBP1) [22].
-
2)
APC inactivation, K-RAS activation, and β-catenin nuclear localization act synergistically to promote the progression of adenoma to carcinoma [23].
-
3)
The loss of TP53 and the loss of heterozygosity of chromosome 18q (mutations observed in advanced colorectal cancer and related to the promotion of change from adenoma to cancer) [24].
Thus, these abnormal cells changes in the colon and rectum, which form benign lesions or polyps, have the potential to develop into cancer in situ that progresses to a metastatic lesion by accumulating mutations, particularly in transforming growth factor-β receptor (TGFβR) and phosphatidylinositol 3-kinase (PIK3CA) genes, which are reported to be factors involved in tumor progression. Subsequently, additional mutation results in some clonal populations with more aggressive phenotypes that are intrinsically able to activate and regulate molecular pathways to maintain the CSC population and their particular characteristics by altering self-renewal pathways, such as Wnt/β-catenin, Notch and Hedgehog, and even interrupting the master transcriptional regulators related to sustain embryonic stem cell renewal, like NANOG, OCT-4, and SOX-2 [25, 26].
CSC are a population of cells that are obtained through dedifferentiation of epithelial cancer cells and they are a critical subset within the tumor mass involved in perpetuating tumor growth by sharing many features of intestinal CSC, such as infinite division, telomerase activity and organ-specific differentiation, high tumorigenic efficiency, chemoresistance, and promotion of recurrence and tumor metastasis. Successful metastasis is achieved through the mechanism called epithelial-to-mesenchymal transition.
Epithelial to Mesenchymal Transition
Epithelial–mesenchymal transition is defined as a dynamic process that involves changes in differentiated cells that leads them to lose their polarity, their cell-to-cell adhesion, their cell–extracellular matrix adhesion (represented by E-cadherin, claudins, and occludins), reorganize their cytoskeleton, eventually to gain invasiveness trough migratory properties; finally, become mesenchymal stem cells that are capable of differentiating into a variety of cells [27]. This process occurs in physiological situations such as embryogenesis and wound healing; however, in terms of CRC, it is widely known that this process is closely associated with the progression and metastasis of disease. It is induced by hypoxia, cytokines, and growth factors secreted by the tumor microenvironment (TME), stroma crosstalk, metabolic changes, and even with antitumor drug treatment [28].
Regulation of this process is strict, and it occurs by expression of WNT/β-catenin and TGF-β, miRNAs (miR-34 and miR-200 families), long non-coding RNAs, transcription factors such as ZEB1, ZEB2, SNAIL, SLUG, and TWIST, which produce collagen, fibronectin, vimentin, α-smooth muscle actin (α-SMA), and metalloproteases that end up degrading the extracellular matrix and facilitate the dissolution of basement membrane, destroying natural borders that contain the tumor progression, allowing migration of cancer cells through the bloodstream and finally, promoting metastasis [27].
Several findings support the idea of EMT as an important factor in the development of metastases and resistance in CRC. An important feature of these mesenchymal cells is their resistance to cellular senescence and apoptosis signaling. Additionally, evidence shows that they are subtypes of CSC:
-
1)
Those that have exclusive epithelial markers (loss of E-cadherin expression is associated with a poor prognosis in stage III CRC) [29].
-
2)
Those that have mesenchymal and epithelial markers; these ‘hybrid’ cells in partial EMT—not entirely epithelial and not entirely mesenchymal—can move as clusters of cells and, in this collective form, can gain more aggressiveness than individual cells with a fully concluded EMT phenotype [30]. Hybrid phenotypic stability factors were as GRHL2, OVOL2, ΔNp63α, and NUMB but, computational model predicts that NRF2 also permits a biphasic dynamics during EMT.
As previously mentioned, pericytes themselves can act as stem cells. For that, current opinions suggest that in tumors, pericytes come from their healthy progenitors, present in the surrounding non-cancerous tissue by stimulating TGF-β and EMT; under this stimulus, some epithelial cells are converted to tumor stem cells/ CSC and others to pericytes, which are characterized by the expression of PDGFR-β. This process, evidently different from EMT, is known as epithelial-pericyte transition (EPT) [31].
Cheng et al. reported that stem cells (glioblastoma cancer) or one fraction, between 4 and 11% of the differentiated cells, which derived from CSC under in vitro and in vivo differentiation conditions, are pericytes. This is very remarkable because if the therapeutic approach uses CSCs as a target, it would not serve to destroy the pericytes that attach to the endothelial cells to support neo-vasculature function and vascular permeability that vascularize tumor tissue and intra-tumor vasculature resulting in promotion of metastasis.
PDGF receptor β expression and PDGF-B ligand production by endothelial cells are fundamental to recruitment of pericytes into the tumor mass. Anyway, it is important to mention that the needs of local tissue/tumor microenvironment are those that rule the microvascular function of pericytes [32].
Pericytes and Cancer
The tumor microenvironment (TME) is defined as every piece of the components that surround and feed or maintain the tumor. It includes normal non-cancerous cells, molecules, and blood vessels near tumor cells [33]. Pericytes, within this framework, are a cell population that functions as stem cells, as mentioned previously, forming several cell types in a variety of pathophysiologic conditions, with self-renewal capacity and the potential to differentiate into multiple cell types such as adipocytes, chondrocytes, osteocytes, phagocytes, granulocytes, and myocytes, in culture [15]. Under pathological conditions, pericytes may differentiate into myofibroblasts, contributing to kidney fibrosis, for example. Also, they may regulate the behavior of other stem cells, such as hematopoietic stem cells or other stem cells in healthy tissues. Their tendency to reside in a particular region of the histological architecture is related to the specific microenvironment of that site, which regulates their potential for self-renewal and additionally provides maintenance through paracrine factors and direct intercellular contact that, if needed, interfere with self-renewal and differentiation pathways [34].
The TME plays a key role in tumor progression, and one of the main regulators of this are pericytes by (i) regulating oxygen flow and nutrients through the capillaries and (ii) by clearance of toxic cell products; although in the reservoir of stem cells, their self-renewal capacity and potential to differentiate into multiple cell types contributes to the regeneration of diverse tissues or even the change of different type cells that constitute the tumor [35]. Other pericyte functions are promotion or reduction of phagocytic activity, and expression of inflammatory signals involved in the immune response against the tumor. For example, low coverage of pericytes inside the tumor is a predictor of bevacizumab efficacy in metastatic colorectal cancer and in hypoxic conditions, increases lung metastasis [36]. The degree of pericyte coverage in human tumors is still controversial by participation with cancer stem cells (CSCs) that promote angiogenesis and metastasis [33, 37].
Development and survival of pericytes are regulated by several signals such as platelet-derived growth factor-β (PDGF-β), transforming growth factor-β1 (TGFβ), heparin-binding epidermal growth factor (HB-EGF); stromal-derived factor 1-a (SDF-1α); Sonic hedgehog (Shh); Jagged-1 (Jag-1), and Ephrin [38]. Cells such as macrophages and tumor-associated fibroblasts produce several of these regulatory molecules and have been implicated in the recruitment of pericytes and endothelial cells that promote intravasation, understood as a pass of cancer cells into the blood flow, or angiogenesis by increasing the expression of metalloprotease and growth factors. Promoting tumor cell intravasation, with the elevated risk of metastasis, depends on high expression of vascular endothelial growth factor (VEGF)-A and Wnt7B by macrophages that express Tie2, which mediates loosening of vascular junctions and enhances vascular permeability and matrix metallopeptidase (MMP)-9 expression by tumor-associated neutrophils [39]. Tumor-associated macrophages (TAMs) perform pro-angiogenic effects through their response to colony-stimulating factor-1 (CSF-1) and Ang2; also, tumor-associated neutrophils (TANs) contribute to tumor intravasation by inducing neovessel formation via expression of interleukin 8 (IL-8) [40]. The main molecular mediators of vascular remodeling and stability are dependent on endothelial cell–pericyte interaction as described in Table 2.
Also, inactivation of signal of TGF-β in tumor cells by dnTGFBR1 reduced the microvessel density and lumen sizes, decreasing tumor growth and producing vascular fragility and affecting the interaction of pericytes and endothelial cells. Both pericytes and endothelial cells express receptors for TGF-β and are able to produce latent forms of this growth factor, but its activation is a consequence of the interplay between these cells [46]. Therefore, the metastatic capacity on CRCs results from higher expression of TGF-β which can stimulate cancer-associated fibroblasts (CAFs) to produce IL-11, with GP130/STAT3 signaling [47]. Also, constant hypoxia can stimulate the secretion of VEGF by pericytes and TGF-β via a hypoxia inducible factor (HIF) signal [48] (Fig. 1). The relative upregulation of VEGF may promote the development of cancer vasculature and TGF-β induced GLI2 axis which inhibit the differentiation of CSC associated with tumor progression and induce the escape of chemotherapy-induced apoptosis through induction of antiapoptotic targets such as BCL-XL and XIAP [48, 49].

Damage mediated by hypoxia due to vessel compression by tumor bulk. Growth factors such as VEGF, TGF, and PDGF are released into the tumor microenvironment. Macrophages and fibroblasts near the tumor are seen. Epithelial to mesenchymal transition (EMT) is represented by its four stages, being the last one the CSC niche. Pericytes surround the external part of the vessels, while the endothelial cells remains inside the lumen, delimiting the inner vascular surface
Pericytes in Metastasis
About 25% of patients with CRC present with advance disease at the time of diagnosis and a further 35% will develop metastases during the course of the disease. Metastasis were define as a less effective process where only a minority of cancer cells dedifferentiated by EMT acquire the capacity to migrate, invade, and spread from the primary tumor to distant locations in the body by traveling through the blood or lymphatic vessels and colonizing distant organs [50]. During this process, circulating tumor cells (CTCs) in clusters and stromal cells (fibroblasts, endothelial, tumor-infiltrated myeloid cells, or pericytes) help increase the viability of tumor cells within CTC clusters by facilitating metastases formation [51]. Also, tumor progression in solid tumor de novo angiogenesis by certain subpopulations of pericytes, such as endosialin (CD248) and desmin-overexpressing pericytes, increase tumor vessel permeability [52]. Activated pericytes also produce intravasation by enhancing C–X–C motif chemokine ligand 12 (CXCL12) expression [53].
Endothelial cells in healthy established vessels remain quiescent for years. Under certain conditions such as ischemia or inflammation, they can rapidly switch to an angiogenic state and start to form new blood vessels by interaction with pericytes [15]. During metastasis tumor cells adhere to endothelial cells and then transmigrate into the extravascular stroma (platelets and myeloid cells) through paracellular migration. Multiple factors are involved in extravasation, including selectins, integrins, N-cadherin, CD44, MUC1, and intercellular cell adhesion molecule-1 (ICAM-1) [54]. The endothelial cells can transdifferentiate and acquire mesenchymal or myofibroblastic markers, such as α-smooth muscle actin (α-SMA) and type I collagen, with the stimulation of TGF-β, which leads to endothelial cytoskeleton remodeling and increased vessel permeability. MMPs (MMP-2, MMP-3, MMP-7, MMP-9, MMP-13, and MT1-MMP) degrade the extracellular matrix, which facilitates extravasation [55]. Platelets promote tumor extravasation by inducing an invasive, mesenchyme-like phenotype in tumor cells that activate the TGF-β/Smad/nuclear factor-κappa B (NF-κB) pathway [56] (Fig. 2). Extravasation occurs when the epithelial barrier is broken. Some cells that facilitate this process are platelets that interact with endothelial cells through ATP/P2Y2 pathways. Also, transdifferentiation into metastasis-associated macrophages (MAMs) and VEGF-A secretion by myeloid progenitor cells, which result in an increase in vascular permeability, thus enhancing the extravasation of cancer cells [57].

Metastasis mechanisms described in colorectal cancer. CXCL12 expression by pericytes, VEGF secretion by MAMs, metalloproteinase’s increased expression and IL-11 secretion by CAFs and TGF-β/Smad/NF-kB pathway activation by platelets are schematized. All of these end up opening a breach between endothelial cells that facilitates cancer cell intravasation
However, only a minority of disseminated tumor cells form metastasis in the secondary site because most tumor cells are cleared away by immune cells or apoptosis after extravasation. To form a metastatic tumor, tumor cells need to proliferate and colonize the target organ with the help of neovessels, which provide nutrients and oxygen for secondary tumor growth [58].
Murgai et al. reveal important pericyte function in pre-metastasic niche formation (pulmonary metastasis) [59]. They propose that the transcription factor KLF4 is essential for early tumor cell colonization and metastasis. Nevertheless, it is still unclear which molecules released by primary tumor cells act directly on pericytes, inducing pre-metastatic niche formation [59]. Also, it was recently demonstrated that CXCL12 from bone marrow arteriolar pericytes is essential for hematopoietic stem cell maintance in the bone marrow [60].
Role of Pericytes in Angiogenesis
Angiogenesis is a complex process. New vessels are formed from the interaction of pericytes and endothelial cells that supply nutrients and provide an adequate source of oxygen to the tumor, promoting the survival, and growth expansion beyond 400 μm, and they allows removal of metabolic wastes and even give an entryway to metastatic cells to the circulatory system,increasing the risk of disseminated disease [61••]. Chiaverina G using mouse models ex vivo for angiogenesis assay can studied interactions between ECs and pericytes and demostrate pericytes originate from mitotic events and can be recruited on the developing sprout by proliferation, migrate independently from endothelial cells,but also actively proliferate on the capillary-like structure.
Colorectal primary tumors with vessels greater than 100 μm in diameter provide favorable conditions for tumor intravasation, which is the most common site of metastasis in this neoplasia [62••]. Pericytes are correlated with the normal vascularization of healthy tissues through a process named vascular normalization; however, in terms of diseases like CRC, this process is affected by the cancerous cells, and paracrine signaling results in inefficient vascularization and low deliver of nutrients by structural transformations of tumor-endothelium cell contact [63].
Tumor vessels tend to be unevenly distributed and form chaotic, tortuous networks with irregular branching patterns and exhibit bidirectional blood flow and are not constantly perfused; abnormal tumor vascularization promotes the creation of new blood vessels from existing ones; this is known as angiogenesis (Fig. 3) [64••].

Role of pericytes in angiogenesis. Angiogenesis secondary to tumor hypoxia is observed; hypoxic zones decrease while angiogenesis occurs. Pericytes covering up the vessel neoformation are observed. Endothelial cells starting to cover the internal surface of mother vessels can be seen
Due to their various sizes, forms, functions, and location, distinct types of tumor vessels have been identified in tumor development: (i) mother vessels, (ii) daughter vessels (capillaries, glomeruloid microvasculature, and vascular malformations), (iii) feeder arteries, and (iv) draining veins. Mother vessels appear first and new vessels sprout from the existing ones by stimulation of microenvironment components, such as VEGF and/or tumor cells; high VEGF levels promote the formation of capillaries. A decrease in VEGF levels stimulates the formation of microvasculature and malformations in vessels [65, 66]. As a result, at the end of the angiogenesis process, feeder arteries and draining veins try to resemble the blood distribution from any other tissue, but they are generated directly upstream and downstream from the tumor.
Mosaic blood vessels have been identified in CRC tumors. Nevertheless, how does this happen? This appears to be due to a mechanism known as “vasculogenesis”, defined as the production of endothelial cells and /or pericytes from progenitor cells from bone marrow or CSC [67]. Tumor vessels comprise various stages of angiogenesis at the same time, with various different effectors, pericytes included. In relation to this, it is relevant to mention that in recent times two different pericyte populations (types 1 and 2) were proposed to be involved during angiogenesis of bone marrow. Only type 2 have been demonstrated as angiogenic in vitro and in vivo, promoting vessel maturation and stabilization at the late stages of vessel formation, finding that they make a usable option in the future as a cell therapy in cancer [34]. Another alternative that has been suggested is that endothelial cells can differentiate from CSC as reported in glioblastoma, breast carcinomas, renal carcinomas, multiple myeloma, and colon cancer [68].
Vascularization mechanisms in cancer suggest that tumors promote their own vascularization by inducing the formation of new capillary buds from preexisting capillaries of the host tissue through a transdifferentiation mechanism that cannot be controlled by VEGF expression and which permits nourishment of tumor tissues [69].
Recent studies have indicated that a novel non-angiogenesis dependent pathway exists called vasculogenic mimicry (VM), in which pericytes seem to play a role [70••]. VM makes allusion to the capacity of cancer cells to mimic certain properties of endothelial cells, such as the formation of extracellular matrix, with the consequent tumor-associated angiogenesis. This has been identified in a wide variety of tumors: glioblastoma, astrocytoma, melanoma, sarcoma, osteosarcoma, digest tract (esophageal carcinoma, gastric cancer, and colorectal cancer), breast, hepatocellular, ovarian, prostate, bladder, lung head, and neck cancers [70,71,72,73,74]. The presence of this mechanism in any neoplasia has been associated with increased aggressiveness and poor clinical outcomes.
In a strict sense, VM results in the formation of not true vessels, but in channels that imitate vessels, which come from no preexisting vascular structure; this means that these tumor cells merely mimic the function of true vessels. However, based on the previously mentioned features, periodic acid-Schiff (PAS) staining was performed by Shen et al. for the wall of channels produced by VM, and these were demonstrated positive, while tumor cells that line the external wall of the channels showed negative results for staining with endothelial markers such as CD31 or CD34 [75].
Two distinctive types of VM in aggressive malignant tumors have been identified. Type 1, known as the tubular type, is composed of non-endothelial cell-lined blood tubes, and cancer cells found lining the surface of the channels. Type 2, known as the patterned matrix type, comprises a basement membrane rich in fibronectin, collagens, and laminin, where the membrane is surrounded by tumor cells instead of endothelial cells. Fibronectin are associated with a poor prognosis in CRC, when high levels of expression are demonstrated. Furthermore, knockdown of fibronectin suppresses cancer cell proliferation via the NF-κB/p53-apoptosis signaling pathway, and keeps the cells arrested in the S-phase.
Tumor VM provides growing tumors with a new mechanism of blood perfusion and a potential dissemination route. Moreover, a recent meta-analysis concluded that tumor VM is associated with a poor prognosis in patients with gastric cancer [76, 77]. In colorectal carcinoma, VM was shown to be a strong, independent, prognostic factor of survival [78]. A meta-analysis by Cao et al. comprising 15 types of malignant tumors revealed that patients with VM-positive cancer showed a less favorable 5-year overall survival than patients with VM-negative cancer, especially in advanced stage cancer [79, 80].
Recently, **Thijssen et al. suggested that pericytes are not part of VM channels originally, but attraction and recruitment by tumor cell–derived PDGF-B makes them support the formation and stabilization of channel-vascular networks [81••]. It seems to be that tumors with VM exploit pericytes in order to provide structural support to these vascular-like networks, which are distinguished from other elements by their rich extracellular matrix depositions [81, 82]. This new knowledge suggests that more than just only being involved in channel formation at the structural level, cancer cells resemble the capacities of endothelial cells in vessel maturation stabilization.
Closely related to this findings, some therapeutics targeting pro-angiogenic factors, such as VEGF signaling, have been correlated with resistance, maybe due to VM activation in the absence of classical tumor angiogenesis, and even promotion of invasion and metastasis has been identified as a product of anti-angiogenic therapy [83, 84]. New strategies targeting VM mediators may result useful in future cancer therapeutics.
Mechanism of Regulation of Pericytes and CSC
MicroRNA (miRNA) are small non-coding RNA (18–25 bases) molecules that regulate gene expression at the post-transcriptional level through degradation or translational repression of a target mRNA. It has been reported that deregulation of miRNAs can lead to abnormal angiogenesis, which is the hallmark of cancer [85] Table 3 summarizes the main microRNAs associated with angiogenesis in endothelial cell, pericytes, and CSC.
Novel Target Anti-angiogenic and Future Treatment Approaches
Vascular normalization, understood as the restoration of normal structure and function of blood vessels, is theorized to have a series of deleterious effects in tumor ulterior development, particularly in the dynamics that follow the actual treatments as showed in Table 4 [49]. Some of them are suggested to be (1) the prevention of metastasis by the correct regulation of permeability of vessels, promoting the longevity of a localized disease, which is susceptible to be targeted with local therapies, such as surgery or radiation; (2) vessel normalization by an increase in oxygenation which secondarily enhances the effect of therapies based on oxidation processes; optimization of Starling forces that increase the pressure of interstitial fluids, making them helpful to deliver cancer cell–directed therapies; and (3) even increase adequate recognition by the host immune system.
Conclusion
Until now, treatments to block angiogenesis can resul after time in more aggressive tumors. Some mechanisms associated with progress tumors or metastasis are transdiferentiation or MV by pericytes, endothelial cells, and CSC. However, new knowledge is needed to get a comprehension of the cellular and molecular basis of cancer disease, and their effective treatment during the different cancer stages.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T. What is a pericyte? J Cereb Blood Flow Metab. 2016;36(2):451–5.
Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215.
Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O. Pericytes at the intersection between tissue regeneration and pathology. Clin Sci. 2015;128(2):81–93.
Hellström M, Kalén M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-β in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126(14):3047–55.
Ozerdem U, Stallcup WB. Early contribution of pericytes to angiogenic sprouting and tube formation. In: Angiogenesis. 2003. p. 241–9.
Jiang T, Zhuang J, Duan H, Luo Y, Zeng Q, Fan K, et al. CD146 is a coreceptor for VEGFR-2 in tumor angiogenesis. Blood. 2012;120(11):2330–9.
Berger M, Bergers G, Arnold B, Hämmerling GJ, Ganss R. Regulator of G-protein signaling-5 induction in pericytes coincides with active vessel remodeling during neovascularization. Blood. 2005;105(3):1094–101.
Hughes S, Chan-Ling T. Characterization of smooth muscle cell and pericyte differentiation in the rat retina in vivo. Investig Ophthalmol Vis Sci. 2004;45(8):2795–806.
Yamazaki T, Mukouyama Y. Tissue specific origin, development, and pathological perspectives of pericytes. Front Cardiovasc Med. 2018;27:5.
Lo HW, Zhu H, Cao X, Aldrich A, Ali-Osman F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009;69(17):6790–8.
Sena IFG, Borges IT, Lousado L, Azevedo PO, Andreotti JP, Almeida VM, et al. LepR+ cells dispute hegemony with Gli1+ cells in bone marrow fibrosis. Cell Cycle. 2017;16(21):2018–22.
Wu SP, Dong XR, Regan JN, Su C, Majesky MW. Tbx18 regulates development of the epicardium and coronary vessels. Dev Biol. 2013;383(2):307–20.
•• Harrell CR, Simovic Markovic B, Fellabaum C, Arsenijevic A, Djonov V, Volarevic V. Molecular mechanisms underlying therapeutic potential of pericytes. J Biomed Sci. 2018;25. Excellent summarises the characteristics of pericytes.
Payne LB, Zhao H, James CC, Darden J, McGuire D, Taylor S, et al. The pericyte microenvironment during vascular development. Microcirculation. 2019;26(8). Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/micc.12554. [cited 2020 Jan 21].
Thomas HM, Cowin AJ, Mills SJ. The importance of pericytes in healing: wounds and other pathologies. Int J Mol Sci. 2017;18.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11.
Phi LTH, Sari IN, Yang YG, Lee SH, Jun N, Kim KS, et al. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018;2018.
Glumac PM, LeBeau AM. The role of CD133 in cancer: a concise review. Clin Transl Med. 2018;7(1).
Fanali C, Lucchetti D, Farina M, Corbi M, Cufino V, Cittadini A, et al. Cancer stem cells in colorectal cancer from pathogenesis to therapy: controversies and perspectives. World J Gastroenterol. 2014;20(4):923–42.
Garza-Trevino EN, Rodriguez-Gonzalez MS, Delgado Gonzalez P, Alonso-Cruz YG, Alonso-Cruz YG, Soto-Dominguez A, et al. Remarkably higher efficacy and a wider safety window for nonfrontline over first-line drug combinations in the adenocarcinoma Colo 320DM cell line. J Buon. 22(5):1115–21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29135091. [cited 2020 Feb 7].
•• Nguyen LH, Goel A, Chung DC. Pathways of colorectal carcinogenesis. Gastroenterology. 2019. This paper summarises genetic changes associated with the development colorectal cancer.
Cheng X, Xu X, Chen D, Zhao F, Wang W. Therapeutic potential of targeting the Wnt/β-catenin signaling pathway in colorectal cancer. Biomed Pharmacother. 2019;110:473–81. Available from: https://www.sciencedirect.com/science/article/pii/S0753332218355744?via%3Dihub. [cited 2019 Jun 1].
Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, et al. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009;137(4):623–34.
Armaghany T, Wilson JD, Chu Q, Mills G. Genetic alterations in colorectal cancer. Gastrointest Cancer Res. 2012;5:19–27.
Pelullo M, Zema S, Nardozza F, Checquolo S, Screpanti I, Bellavia D. Wnt, Notch, and TGF-β pathways impinge on hedgehog signaling complexity: an open window on cancer. Front Genet. 2019;10.
Cathomas G. PIK3CA in colorectal cancer. Vol. 4 Front Oncol; 2014.
Vu T, Datta PK. Regulation of EMT in colorectal cancer: a culprit in metastasis, vol. 9: Cancers; 2017.
Pedrosa L, Esposito F, Thomson TM, Maurel J. The tumor microenvironment in colorectal cancer therapy. Cancers. 2019;11.
Yun JA, Kim SH, Hong HK, Yun SH, Kim HC, Chun HK, et al. Loss of E-cadherin expression is associated with a poor prognosis in stage III colorectal cancer. Oncology. 2014;86(5–6):318–28.
Bocci F, Tripathi SC, Vilchez Mercedes SA, George JT, Casabar JP, Wong PK, et al. NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr Biol (Camb). 2019;11(6):251–63.
Lu J, Shenoy AK. Epithelial-to-pericyte transition in cancer. Cancers. 2017;9.
Manzat Saplacan RM, Balacescu L, Gherman C, Chira RI, Craiu A, Mircea PA, et al. The role of PDGFs and PDGFRs in colorectal cancer. 2017;2017.
Garza Treviño EN, González PD, Valencia Salgado CI, Martinez GA. Effects of pericytes and colon cancer stem cells in the tumor microenvironment. Cancer Cell Int. 2019;19.
Ribeiro AL, Okamoto OK. Combined effects of pericytes in the tumor microenvironment. Stem Cells Int. 2015;2015:1–8.
Chen Z, Xu XH, Hu J. Role of pericytes in angiogenesis: focus on cancer angiogenesis and anti-angiogenic therapy. Neoplasma. 2016;63:173–82.
Jiang C, Huang YH, Lu JB, Yang YZ, Rao HL, Zhang B, et al. Perivascular cell coverage of intratumoral vasculature is a predictor for bevacizumab efficacy in metastatic colorectal cancer. Cancer Manag Res. 2018;10:3589–97.
Garza-Treviño EN, Said-Fernández SL, Martínez-Rodríguez HG. Understanding the colon cancer stem cells and perspectives on treatment. Cancer Cell Int. 2015;15(1):1–9.
Paiva AE, Lousado L, Guerra DAP, Azevedo PO, Sena IFG, Andreotti JP, et al. Pericytes in the premetastatic niche. Cancer Res. 2018;78(11):2779–86. Available from: http://cancerres.aacrjournals.org/lookup/doi/10.1158/0008-5472.CAN-17-3883.
Quintero-Fabián S, Arreola R, Becerril-Villanueva E, Torres-Romero JC, Arana-Argáez V, Lara-Riegos J, et al. Role of matrix metalloproteinases in angiogenesis and cancer. Front Oncol. 2019;9. Available from: https://www.frontiersin.org/article/10.3389/fonc.2019.01370/full.
Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. 2019;26.
Zonneville J, Safina A, Truskinovsky AM, Arteaga CL, Bakin AV. TGF-β signaling promotes tumor vasculature by enhancing the pericyte-endothelium association. BMC Cancer. 2018;19, 18(1).
Falcón BL, Hashizume H, Koumoutsakos P, Chou J, Bready JV, Coxon A, et al. Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels. Am J Pathol. 2009;175(5):2159–70.
Akwii RG, Sajib MS, Zahra FT, Mikelis CM. Role of angiopoietin-2 in vascular physiology and pathophysiology. Cells. 2019;8(5):471.
•• McCarty MF, Somcio RJ, Stoeltzing O, Wey J, Fan F, Liu W, et al. Overexpression of PDGF-BB decreases colorectal and pancreatic cancer growth by increasing tumor pericyte content. J Clin Invest. 2007;117(8):2114–22. Expression of PDGF-BB rendering the tumor vasculature more resistant to angiogenic therapy.
Ribatti D. Tumor refractoriness to anti-VEGF therapy. Oncotarget. 2016;7:46668–77.
Itatani Y, Kawada K, Sakai Y. Transforming growth factor-β signaling pathway in colorectal cancer and its tumor microenvironment. Int J Mol Sci. 2019;20.
Calon A, Espinet E, Palomo-Ponce S, Tauriello DVF, Iglesias M, Céspedes MV, et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22(5):571–84.
Tang YA, Chen Y feng, Bao Y, Mahara S, Yatim SMJM, Oguz G, et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci U S A 2018;115(26):E5990–E5999.
Comunanza V, Bussolino F. Therapy for cancer: strategy of combining anti-angiogenic and target therapies. Front Cell Dev Biol. 2017;5.
Pieterse Z, Sinha D, Kaur P. Pericytes in metastasis. In: Advances in experimental medicine and biology. New York: Springer; 2019. p. 125–35.
Hong Y, Fang F, Zhang Q. Circulating tumor cell clusters: what we know and what we expect (Review). Int J Oncol. 2016;49:2206–16.
Teicher BA. CD248: a therapeutic target in cancer and fibrotic diseases. Oncotarget. 2019;10:993–1009.
Kim B, Yoon J, Yoon SW, Park B. Onbaekwon suppresses colon cancer cell invasion by inhibiting expression of the CXC chemokine receptor 4. Integr Cancer Ther. 2017;16(2):244–51.
Reymond N, D’Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858–70.
Platel V, Faure S, Corre I, Clere N. Endothelial-to-mesenchymal transition (EndoMT): roles in tumorigenesis, metastatic extravasation and therapy resistance. J Oncol. 2019;2019.
Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–90.
Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell. 2013;24(1):130–7.
Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306.
Murgai M, Ju W, Eason M, Kline J, Beury DW, Kaczanowska S, et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat Med. 2017;23(10):1176–90.
Szade K, Gulati GS, Chan CKF, Kao KS, Miyanishi M, Marjon KD, et al. Where hematopoietic stem cells live: the bone marrow niche. Antioxid Redox Signal. 2018;29:191–204.
•• Chiaverina G, di Blasio L, Monica V, Accardo M, Palmiero M, Peracino B, et al. Dynamic interplay between pericytes and endothelial cells during sprouting angiogenesis. Cells. 2019;8(9):1109. This paper using mouse models ex vivo for angiogenesis assay can studied interactions between ECs and pericytes and demostrate pericytes originate from mitotic events and can be recruited on the developing sprout by proliferation, migrate independently from endothelial cells, but also actively proliferate on the capillary-like structure.
•• Li X, Li Y, Lu W, Chen M, Ye W, Zhang D. The tumor vessel targeting strategy: a double-edged sword in tumor metastasis. Cells. 2019;8(12):1602. Available from: https://www.mdpi.com/2073-4409/8/12/1602. [cited 2020 Jan 24]. This paper summarises contribution of angiogenesis to tumor metastasis.
Barlow KD, Sanders AM, Soker S, Ergun S, Metheny-Barlow LJ. Pericytes on the tumor vasculature: Jekyll or hyde? Cancer Microenviron. 2013;6:1–17.
•• Ziyad S, Iruela-Arispe ML. Molecular mechanisms of tumor angiogenesis. genes and cancer. 2011;2(12):1085–96. Excellent review highlighting the types of tumor vessels.
Nagy JA, Dvorak HF. Heterogeneity of the tumor vasculature: the need for new tumor blood vessel type-specific targets. In: Clinical and experimental metastasis; 2012. p. 657–62.
•• Bendardaf R, El-Serafi A, Syrjänen K, Collan Y, Pyrhönen S. The effect of vascular endothelial growth factor-1 expression on survival of advanced colorectal cancer patients. Libyan J Med. 2017;12(1). Relation between expression of VEGF in biopsies, development of neovascularitation and metastasis.
Chen WZ, Jiang JX, Yu XY, Xia WJ, Yu PX, Wang K, et al. Endothelial cells in colorectal cancer. World J Gastrointest Oncol. 2019;11:946–56.
Shangguan W, Fan C, Chen X, Lu R, Liu Y, Li Y, et al. Endothelium originated from colorectal cancer stem cells constitute cancer blood vessels. Cancer Sci. 2017;108(7):1357–67.
Liu Z, Qi L, Li Y, Zhao X, Sun B. VEGFR2 regulates endothelial differentiation of colon cancer cells. BMC Cancer. 2017;30:17(1).
•• Fernández-Cortés M, Delgado-Bellido D, Javier OF. Vasculogenic mimicry: become an endothelial cell “But not so much.”. Front Oncol. 2019;9. Importance of tumor microenviroment in vascular mimicry acquisition and discribe terapeutic targets.
Upile T, Jerjes W, Radhi H, Al-Khawalde M, Kafas P, Nouraei S, et al. Vascular mimicry in cultured head and neck tumour cell lines. Head Neck Oncol. 2011;3(1).
Angara K, Borin TF, Arbab AS. Vascular Mimicry: a novel neovascularization mechanism driving anti-angiogenic therapy (AAT) resistance in glioblastoma. Transl Oncol. 2017;10:650–60.
Ayala-Domínguez L, Olmedo-Nieva L, Muñoz-Bello JO, Contreras-Paredes A, Manzo-Merino J, Martínez-Ramírez I, et al. Mechanisms of vasculogenic mimicry in ovarian cancer. Front Oncol. 2019;9.
Zhang Z, Imani S, Shasaltaneh MD, Hosseinifard H, Zou L, Fan Y, et al. The role of vascular mimicry as a biomarker in malignant melanoma: a systematic review and meta-analysis. BMC Cancer. 2019;19(1):1134. Available from: https://bmccancer.biomedcentral.com/articles/10.1186/s12885-019-6350-5.
Shen Y, Quan J, Wang M, Li S, Yang J, Lv M, et al. Tumor vasculogenic mimicry formation as an unfavorable prognostic indicator in patients with breast cancer. Oncotarget. 2017;8(34):56408–16.
Guo Q, Yuan Y, Jin Z, Xu T, Gao Y, Wei H, et al. Association between tumor vasculogenic mimicry and the poor prognosis of gastric cancer in China: an updated systematic review and meta-analysis. Biomed Res Int. 2016;2016.
Li W, Ng JMK, Wong CC, Ng EKW, Yu J. Molecular alterations of cancer cell and tumour microenvironment in metastatic gastric cancer. Oncogene. 2018;37:4903–20.
Donadon M, Lleo A, Di Tommaso L, Soldani C, Franceschini B, Roncalli M, et al. The shifting paradigm of prognostic factors of colorectal liver metastases: from tumor-centered to host immune-centered factors. Front Oncol. 2018;8.
•• Cao Z, Bao M, Miele L, Sarkar FH, Wang Z, Zhou Q. Tumour vasculogenic mimicry is associated with poor prognosis of human cancer patients: a systemic review and meta-analysis. Eur J Cancer. 2013;49(18):3914–23. Vascular mimicry positive cancer patients show poor overall survival particulary in metastatic cancer.
Ge H, Luo H. Overview of advances in vasculogenic mimicry – a potential target for tumor therapy. Cancer Manag Res. 2018;10:2429–37.
•• Thijssen VLJL, Paulis YWJ, Nowak-Sliwinska P, Deumelandt KL, Hosaka K, Soetekouw PMMB, et al. Targeting PDGF-mediated recruitment of pericytes blocks vascular mimicry and tumor growth. J Pathol. 2018;246(4):447–58. Pericytes are important in vascular mimicry, facilitating sprouting, and structural support of the vascular-like networks.
Stratman AN, Davis GE. Endothelial cell-pericyte interactions stimulate basement membrane matrix assembly: influence on vascular tube remodeling, maturation, and stabilization. In: Microscopy and microanalysis. 2012. p. 68–80.
Guyot M, Hilmi C, Ambrosetti D, Merlano M, Lo Nigro C, Durivault J, et al. Targeting the pro-angiogenic forms of VEGF or inhibiting their expression as anti-cancer strategies. Oncotarget. 2017;8(6):9174–88.
Bin MM, Zaorsky NG, Deng L, Wang HH, Chao J, Zhao LJ, et al. Pericytes: a double-edged sword in cancer therapy. Future Oncol. 2015;11:169–79.
Soheilifar MH, Grusch M, Neghab HK, Amini R, Maadi H, Saidijam M, et al. Angioregulatory microRNAs in colorectal cancer. Cancers. 2020;12.
Chamorro-Jorganes A, Araldi E, Suárez Y. MicroRNAs as pharmacological targets in endothelial cell function and dysfunction. Pharmacol Res. 2013;75:15–27.
Geevarghese A, Herman IM. Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Transl Res. 2014;163:296–306.
Wang S, Olson EN. AngiomiRs-Key regulators of angiogenesis. Curr Opin Genet Dev. 2009;19:205–11.
Takahashi RU, Miyazaki H, Ochiya T. The roles of microRNAs in breast cancer. Cancers. 2015;7:598–616.
Qian X, Yu J, Yin Y, He J, Wang L, Li Q, et al. MicroRNA-143 inhibits tumor growth and angiogenesis and sensitizes chemosensitivity to oxaliplatin in colorectal cancers. Cell Cycle. 2013;12(9):1385–94.
Baraniskin A, Buchberger B, Pox C, Graeven U, Holch JW, Schmiegel W, et al. Efficacy of bevacizumab in first-line treatment of metastatic colorectal cancer: a systematic review and meta-analysis. Eur J Cancer. 2019;106:37–44.
Kanat O, Ertas H. Existing anti-angiogenic therapeutic strategies for patients with metastatic colorectal cancer progressing following first-line bevacizumab-based therapy. World J Clin Oncol. 2019;10(2):52–61. Available from: https://www.wjgnet.com/2218-4333/full/v10/i2/52.htm. [cited 2020 Jan 31].
Verdaguer H, Tabernero J, Macarulla T. Ramucirumab in metastatic colorectal cancer: evidence to date and place in therapy. Ther Adv Med Oncol. 2016;8:230–42.
Wu C-P, Lusvarghi S, Wang J-C, Hsiao S-H, Huang Y-H, Hung T-H, et al. Avapritinib: a selective inhibitor of KIT and PDGFRα that reverses ABCB1 and ABCG2-mediated multidrug resistance in cancer cell lines. Mol Pharm. 2019;16(7):3040–52. Available from: https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.9b00274. [cited 2020 Jan 31].
Hoff PM, Hochhaus A, Pestalozzi BC, Tebbutt NC, Li J, Kim TW, et al. Cediranib plus FOLFOX/CAPOX versus placebo plus FOLFOX/CAPOX in patients with previously untreated metastatic colorectal cancer: a randomized, double-blind, phase III study (HORIZON II). J Clin Oncol. 2012;30(29):3596–603.
Aljubran A, Elshenawy MA, Kandil M, Zahir MN, Shaheen A, Gad A, et al. Efficacy of regorafenib in metastatic colorectal cancer: a multi-institutional retrospective study. Clin Med Insights. 2019;13.
Tapia Rico G, Price TJ. Atezolizumab for the treatment of colorectal cancer: the latest evidence and clinical potential. Expert Opin Biol Ther. 2018;18(4):449–57.
Van Cutsem E, Yoshino T, Lenz HJ, Lonardi S, Falcone A, Limón ML, et al. Nintedanib for the treatment of patients with refractory metastatic colorectal cancer (LUME-Colon 1): a phase III, international, randomized, placebo-controlled study. Ann Oncol. 2018;29(9):1955–63.
Wang TF, Lockhart AC. Aflibercept in the treatment of metastatic colorectal cancer. Clin Med Insights. 2012;6:19–30.
•• Hidalgo M, Martinez-Garcia M, Le Tourneau C, Massard C, Garralda E, Boni V, et al. First-in-human phase i study of single-agent vanucizumab, a first-in-class bispecific anti-angiopoietin-2/anti-vegf-a antibody, in adult patients with advanced solid tumors. Clin Cancer Res. 2018;24(7):1536–45. Vancizumab impact tumor vascularity, demonstrated encouraging antitumor activity.
Andersen SE, Andersen IB, Jensen BV, Pfeiffer P, Ota T, Larsen JS. A systematic review of observational studies of trifluridine/tipiracil (TAS-102) for metastatic colorectal cancer. Acta Oncol (Madr). 2019;58(8):1149–57.
Smith KM, Desai J. Nivolumab for the treatment of colorectal cancer. Expert Rev Anticancer Ther. 2018;18(7):611–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pericytes
Rights and permissions
About this article

Cite this article
Garza Treviño, E.N., Delgado-Gonzalez, P., Valencia Salgado, C.I. et al. Pericytes Relationship with Cancer Stem Cells in the Colon. Curr. Tissue Microenviron. Rep. 1, 187–198 (2020). https://doi.org/10.1007/s43152-020-00015-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43152-020-00015-8