
Abstract
Polycystic ovary syndrome (PCOS) is one of the most common endocrine disorders, affecting approximately 5–20% of women of reproductive age. PCOS is a multifactorial, complex, and heterogeneous disease, characterized by hyperandrogenism, ovulatory dysfunction, and polycystic ovaries, which may lead to impaired fertility. Besides the reproductive outcomes, multiple comorbidities, such as metabolic disturbances, insulin resistance, obesity, diabetes, and cardiovascular disease, are associated with PCOS. In addition to the clear genetic basis, epigenetic alterations may also play a central role in PCOS outcomes, as environmental and hormonal alterations directly affect clinical manifestations and PCOS development. Here, we highlighted the epigenetic modifications in the multiplicity of clinical manifestations, as well as environmental epigenetic disruptors, as intrauterine hormonal and metabolic alterations affecting embryo development and the adulthood lifestyle, which may contribute to PCOS development. Additionally, we also discussed the new approaches for future studies and potential epigenetic biomarkers for the treatment of associated comorbidities and improvement in quality of life of women with PCOS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
History and Pathophysiology of PCOS
The first report of polycystic ovary syndrome (PCOS) was noted more than 2000 years ago when Hippocrates (460 AC-377 AC) related menstrual disorders in “robust” healthy women with “masculine characteristics” [1]. In 1721, Vallisneri described a case of an infertile woman with bright white surface ovaries of the size of pigeon eggs [2]; in the following years, many reports of women with symptoms similar to PCOS emerged, but it was not then systematized. In 1935, Stein and Leventhal described for the first time the most similar phenotype related to PCOS, which included women with menstrual disorder, hirsutism, and enlarged ovaries with small follicles, later referred to as the Stein–Leventhal syndrome [3, 4].
Despite the advances in the understanding of the pathophysiology and treatment of PCOS-related symptoms, the diagnosis criteria were first established in a conference at the National Institutes of Health (NIH) in 1990. PCOS was classified as chronic anovulation or ovulatory dysfunction due to excessive presence of androgens, in the absence of any other disorders with similar symptoms [5]. In 2004, this classification was revised in the Rotterdam consensus, with inclusion of the size and morphology of the ovaries in the diagnosis. According to this classification system, PCOS must present at least two of the following three characteristics: (1) hyperandrogenism (clinical and/or biochemical), (2) ovulatory dysfunction, and (3) polycystic ovaries, with 12 or more cysts in one ovary and/or ovarian volume of >10 mL [6].
The Rotterdam criteria are well accepted and widely used, although in 2006 the Androgen Excess Society issued a statement in an attempt to establish hyperandrogenism as a key condition for PCOS diagnosis, excluding cases in which only ovulatory dysfunction and polycystic ovaries are the main features underlying the pathophysiology of chronic anovulation and infertility observed in PCOS women [7]. Insulin resistance (IR) [8] and metabolic alterations, such as obesity, dyslipidemia, arterial hypertension, abnormal glucose metabolism, and chronic inflammation are common disorders associated with the pathophysiology of PCOS, which increases the risk of developing type 2 diabetes mellitus (T2DM) and cardiovascular disease (CVD) [9]. The prevalence of PCOS may vary depending on the diagnosis criteria; it affects 5–20% of women worldwide, thereby being the main cause of anovulatory infertility [10]. The large variation in the number of affected women reflects the heterogeneous nature of PCOS phenotypes and associated comorbidities, making it difficult to understand the etiology of this syndrome [11]. This suggests a complex and multifactorial origin of PCOS, in which genetic alterations, environmental factors, and epigenetic modifications contribute to its development or the worsening of its clinical manifestations [12].
Genetic Basis of PCOS
The large number of clinical phenotypes is mainly due to hormonal and metabolic disturbances that directly affect the reproductive outcomes in women with PCOS. Familial studies strongly support the genetic origin of PCOS; most of them suggest autosomal dominant inheritance with incomplete penetrance and variable expressivity due to a combination of multiple genetic components and environmental interactions [13]. The genetic origin of PCOS is confirmed by a hereditary factor observed in first-degree relatives, since mothers or sisters with PCOS are at an increased risk (35–40%) of developing the disease [14]. In addition, the susceptibility of the disease in monozygotic twins is at least twice as high as in dizygotic twins [15].
Despite the clear genetic influence, it is unlikely that a single gene is responsible for the onset of the disease. Genome-wide association studies (GWAS) have identified a large number of variants involved in crucial pathways related to endocrine disturbances and hyperandrogenism observed in PCOS pathophysiology [16,17,18,19,20]. A intricate network involved in the regulation of cell proliferation, insulin metabolism, follicular growth and maturation, hormonal regulation, and diabetes susceptibility are required for ovarian steroidogenesis and oocyte maturation and PCOS development [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40]. Single nucleotide polymorphisms (SNPs) and repetitive DNA sequences have been associated with several aspects and increased risk of PCOS development. Among the main molecular markers reported in GWAS, most are involved in androgen biosynthesis and metabolism, insulin resistance, oxidative stress, and T2DM. The genetic variants were mainly located in the LHCGR, FSHR, CYP11A, THADA, ERBB4, GATA4, HSD17B5, FSHB, HMGA2, RAB5B/SUOX, INSR, and H6PD genes that play an important role in PCOS susceptibility (Table 1). Genes involved in cellular growth and apoptosis are related to the reproductive outcomes of PCOS, including ovarian dysfunction, folliculogenesis, and infertility (Table 1).
Repetitive DNA sequences are related to transcriptional regulation of gene expression and the maintenance of genomic stability [41]. Triplet repeat sequences are associated with several human diseases, including reproductive disorders. The repetitive (CAG)n polymorphism in the androgen receptor gene (AR) has a physiological role in ovarian function and folliculogenesis and may also be a risk factor for PCOS development. The increased number of CAG repeats negatively correlates with AR activity, which affects androgen action in women with PCOS [42]. Alterations in the repetitive telomeric region are also related to PCOS; however, the results are contradictory, as they are directly affected by clinical characteristics [43,44,45]. Metabolic disturbances observed in PCOS may contribute to the progressive telomeric loss, leading to cellular senescence, reproductive aging, and chronic anovulation. However, the excess androgen may stimulate telomerase activity and protect against telomere erosion [46].
Although the genetic origin of PCOS is widely supported, the underlying molecular mechanisms and pathways remain unexplored. The genetic contribution to PCOS pathophysiology involves a complex network of interactions; various genes and pathways are associated with these interactions. However, the common variants reported have a small effect on disease development and do not explain the variety of clinical alterations observed in PCOS [47]. The identification of rare genetic variants using high-throughput genetic strategies and the regulation of gene expression may contribute to a better understanding of PCOS pathophysiology. Indeed, environmental changes during the lifetime, whether intrauterine or after birth through adulthood, contribute to the heterogenic phenotype of PCOS, suggesting an epigenetic component as the main cause of its development, as well as the associated endocrine and hormonal disturbances.
Epigenetic Fetal Origin of PCOS
Epigenetic marks, such as DNA methylation, histone modifications, and noncoding RNA regulation, modify the structure of chromatin, conferring a differential program of gene expression, irrespective of changes in the DNA sequence [48]. Epigenetic modifications control gene expression dynamically and reversibly, as the epigenome of a cell has great plasticity and can be reprogrammed. Epigenetic reprogramming changes cell fate throughout development and adulthood, and environmental factors play a crucial role in the maintenance and establishment of epigenetic marks [49]. Disruptions in the epigenetic machinery are related to the etiology of various neurodevelopmental disorders, such as the Beckwith–Wiedemann, Silver–Russell, and Fragile X syndrome [50, 51], as well as complex and multifactorial diseases such as cancer [52], T2DM, and metabolic alterations [53].
Owing to the multifactorial and complex etiology of PCOS, epigenetic disturbances may be the main alteration underlying the development and severity of this disorder. Environmentally induced epigenetic changes caused by an intrauterine or postnatal adverse environment may trigger PCOS-like symptoms or its associated clinical alterations after birth [54, 55]. Metabolic and hormonal dysfunctions during fetal development may promote aberrant epigenetic reprogramming, leading to physiological changes in the fetus and PCOS emergence in adult life. This hypothesis may also explain the familial genetic basis and susceptibility to acquiring PCOS in first-degree relatives [56].
Hyperandrogenism seems to be the main factor underlying the fetal origin of PCOS, as the intrauterine androgen excess appears to predispose to PCOS in adulthood via epigenetic mutations. This has been observed in various studies, wherein the embryo exposed to the hyperandrogenic environment during prenatal life presented PCOS-like alterations, such as elevated levels of testosterone, progesterone, estradiol, and luteinizing hormone (LH), irregular estrous cycles, reduced preovulatory follicles, and increased preantral follicles [57,58,59,60]. In addition, Rhesus monkeys exposed to high levels of androgens during fetal development reportedly presented a PCOS-like phenotype after birth [61].
Altered DNA methylation is the main reported epigenetic marker in developmental epigenetic reprogramming of PCOS. These modifications lead to gene silencing or reduction of its activity and regulation of important developmental processes, including genomic imprinting [62] and X-chromosome inactivation [63], as well as the transcriptional and posttranscriptional regulation of gene expression and chromatin remodeling [64]. In a previous study, the hyperandrogenic intrauterine environment altered the DNA methylation patterns of prenatally androgenized Rhesus monkeys, with 163 genes in infants and 325 in adults, especially those linked to the antiproliferative transcription factor TOB in T cells and the transforming growth factor-ß (TGF-β) signaling pathways, such as TGFBR1, KRAS, BMP2, TFE3, RUNX3, and HOXC8 [61]. Previous studies have also implicated members of the TGF-ß superfamily, such as fibrillin-3 and inhibin B, in the pathophysiology of PCOS [40].
Androgen excess during prenatal life lead to DNA hypomethylation in androgenized rats at specific CpG sites from the promoter region of GATA6 (-520) and STAR (-822) genes involved in steroidogenesis and steroid biosynthesis [65]. In addition to the influence of androgens, other external factors may alter the epigenetic landscape, thus leading to PCOS. Rats exposed to the agricultural fungicide vinclozolin and the insecticide dichloro-diphenyl-trichloroethane (DDT), reportedly presented changes in the transcriptome and epigenome of ovarian granulosa cells in the F3 generation, suggesting an epigenetic transgenerational inheritance of ovarian pathology [66].
Assuming that hyperandrogenism during development play a role in the emergence of PCOS in adulthood, the information regarding the time of development and the endocrine disruptors that can lead to PCOS phenotype and/or ovary dysfunction, is crucial for a better understanding of the mechanisms underlying this syndrome. One hypothesis is that the major influence of androgen levels occurs during mid-gestation when intrauterine hyperandrogenism can affect fetal development, including ovarian morphological alterations and sex steroid production [67,68,69]. Ewes prenatally treated with testosterone showed growth retardation, reduced ovarian reserve, and increased ovarian follicular recruitment [70]. However, the origin of PCOS due to excess androgen may occur earlier in the development process depending on the species. Zebrafish embryos exposed to androgens in early development presented changes in global DNA methylation levels in ovaries, but these alterations were not observed during late androgen exposure. Early embryonic androgen exposure seems to be transferred to offspring with transgenerational changes in the ovarian epigenome and glucose homeostasis [71].
Echiburú and colleagues (2020) analyzed the DNA methylation profile in 368 CpG sites in the promoter regions of seven reproductive and metabolic genes (LEP, ADIPOQ, AMH, LEPR, ADIPOR1, ADIPOR2, and AR) in children born to women with PCOS treated with metformin during pregnancy. Daughters of the PCOS women showed differences in one CpG site of LEPR (Chr1-65419664 site, hypermethylation) and ADIPOR2 (Chr12-1690290, hypermethylation) genes and two CpG sites in the AR gene (ChrX-67543969 and ChrX-67544981 sites, hypomethylated) when compared to the non-PCOS controls. In sons, five CpG sites in LEP, three in AMH, and nine in AR genes showed differences in methylation levels between the groups. Moreover, the Chr7-128240906 and Chr7-128241078 sites in LEP exhibited higher methylation levels in the infants from PCOS women treated with metformin. The authors suggested that an intrauterine PCOS environment predisposes a sex-dependent DNA methylation pattern [72].
Epidemiological studies support the adverse intrauterine environment linked to health problems in adult life as developing PCOS. Birth weight studies suggest that hyperandrogenism is more prevalent in infants small for gestational age, and the prevalence of PCOS is twice as high as in the infants appropriate for gestational age [73]. Increased levels of testosterone during prenatal life are related to other developmental disorders, as women with PCOS present autistic characteristics and a greater predisposition to having a child with autism [74]. Alterations in the androgen receptor (AR) gene and non-random (skewed) X chromosome inactivation (XCI) may play a role in PCOS development in early gestational age. The repeat trinucleotide sequence is differentially methylated in the active and inactive X-chromosomes, which is usually a random process [75]. Although random XCI seems to be present in PCOS, the skewed XCI is related to shorter CAG repeats in the AR gene [76,77,78,79]. In addition, the AR CAG repeats and the nonrandom XCI appear to exert considerable effects on LH and FSH levels [80].
Despite the growing evidence of developmental PCOS and aberrant epigenetic reprogramming during embryonic and fetal life, the related pathways and the mechanisms underlying the inherited PCOS remain unclear. However, the hyperandrogenic intrauterine environment and other endocrine disruptors induce reproductive and metabolic dysfunctions in adulthood via epigenetic mutations [55]. The relationship between hyperandrogenism and metabolic intrauterine exposure during development provides new insights regarding familial inheritance and the environmental influence on PCOS development and its related comorbidities; this information also contributes to the development of intrauterine preventive treatments in the offspring.
PCOS Comorbidities and Epigenetic Markers in Adulthood
The genome of an individual responds in a coordinated way to environmental changes during life [49]. Besides mutational events, disruption in the epigenetic control of gene function also alters many biological processes and is related to the pathophysiology of human diseases. The multifactorial nature of PCOS and the diversity of clinical features observed suggest that genetic predisposition and environmental changes alter the epigenome and may be the main alteration behind its etiology. Aberrant epigenetic reprogramming through endocrine disruptors or lifestyle changes may lead to PCOS development and play a role in ovarian alterations and reproductive dysfunctions related to this disorder [81]. The epigenetic biomarkers, as well as their modifications, diagnostic potential, and tissues analyzed in PCOS, are summarized in Table 2.
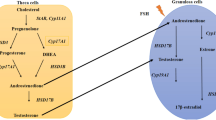
The altered epigenetic biomarkers in PCOS seem to participate in an intricate network of biological interactions, as shown in Fig. 1. Based on the biomarkers suggested in Table 2, a functional network among PCOS-interacting proteins was predicted using the STRING database (https://string-db.org, version 11.0) [105]. The network interactions were built with a score of 0.4 or greater, using multiple proteins, selecting Homo sapiens as an organism and an initial input of 28 proteins; the scores obtained for each interaction are summarized in Supplementary Table S1. This network had significantly more interactions than expected, for a random set of proteins of similar size drawn from the human genome (PPI enrichment p-value: 2.42e−08). This enrichment indicates that the proteins are biologically connected. The five main functional enrichments in the PCOS-interacting protein network based on the highest false discovery rate are presented in Table 3. According to gene ontology (GO) analysis, these proteins participate in important biological processes and molecular functions related to response to chemical and organic stimuli, signaling pathways, and hormonal function, and regulate reproductive function metabolic disturbances pathways in PCOS.

STRING protein–protein interactions (PPI) network based on PCOS coding biomarkers reported. The line thickness indicates the strength of data support
Altered DNA methylation in genes related to androgen production, insulin resistance, and other metabolic alterations was reportedly observed in PCOS [106]. A genome-wide DNA methylation study of peripheral blood identified 52 differentially methylated CpG sites related to different functions such as significant functional pathways and important clinical characteristics of PCOS women, such as prolactin, estradiol, and progesterone levels, and menstrual cycle alterations [107]. Consecutive hypomethylated CpG sites have been identified in the promoter region of the EPHX1 gene in the peripheral blood of PCOS [88]. Hypomethylation of the repetitive element LINE-1 (Long interspersed nuclear elements 1) in blood leukocytes is strongly associated with the susceptibility and hormonal changes in PCOS [83].
The identification of epigenetic changes related to endocrine and metabolic disturbances in PCOS, such as IR, hyperinsulinemia, obesity, and other related conditions, contributes to a better understanding of the mechanisms involved in the pathogenesis of this disease. Insulin resistance (IR) is one of the main comorbidities in women with PCOS. In peripheral blood, Shen and colleagues (2013) identified 79 genes differentially methylated between PCOS with insulin resistance (PCOS/IR) and without IR, of which 40 genes were differentially methylated between PCOS and non-PCOS women [108]. Another study also showed that the LMNA gene, related to metabolic alterations such as hyperandrogenism and insulin resistance, was hypermethylated in PCOS women with IR [93]. Changes in DNA methylation of specific genes and promoter regions are closely related to PCOS development [87, 91, 109, 110]; however, global DNA methylation patterns in PCOS are poorly understood and controversial. No difference in global DNA methylation has been reported in PCOS-women when compared to non-PCOS controls [81]. In contrast, Sagvekar et al. (2017) demonstrated a reduction of ~25% in global DNA methylation in PCOS women [83].
Epigenetic modifications seem to be different in obese and nonobese PCOS. Obesity or overweight is a common characteristic of PCOS women, which also affects the clinical manifestations, such as IR, hyperinsulinemia, and hyperandrogenism [111]. Decreased DNA methylation and overexpression of the LHCGR gene were observed in subcutaneous adipocytes of non-obese PCOS, while the INSR gene showed decreased DNA methylation and underexpression in those with obesity [96]. DNA methylation profile may be useful in identifying the risk of developing PCOS metabolic disorders. Hypermethylation of the PPARGC1A promoter was observed in leukocytes from PCOS women, while mitochondrial DNA content decreased. The higher the metabolic risk, the greater the difference observed [95].
Besides altered DNA methylation, disruption in non-conding RNAs (ncRNAs) expression is related to PCOS and its related comorbidities. ncRNAs are epigenetic marks that control gene expression at the transcriptional level by binding to DNA or RNA sequences, and at posttranscriptional level by binding to the protein. The main classes of ncRNAs are microRNAs (~20 nt) and long ncRNAs (>200 nt), which are related to several biological processes [112, 113]. In this context, miR-93 was identified as overexpressed in the adipose tissues of women with PCOS, PCOS/IR, or non-PCOS/IR compared to that in the non-PCOS/non-IR controls, while its target, the MCM7 gene, was downregulated in PCOS and non-PCOS/IR, with the lowest expression levels being in the PCOS/IR [99]. A previous study reported the upregulation of miR-93, miR-133, and miR-223 in PCOS women; however, the predicted target for miR-93 was the GLUT4 gene, an insulin-sensitive glucose transporter that is downregulated by miR-93 overexpression and may be responsible for PCOS/IR [100]. In addition, the downregulation of lncRNA GAS5 and elevated expression of the IL-18 gene in the serum of PCOS patients might contribute to IR [114].
In addition to their influence on IR, the miRNAs are also related to obesity, which is often described as a PCOS comorbidity. The levels of miR-21, miR-27b, and miR-103, associated with metabolic disorders such as obesity and diabetes, are downregulated in the whole blood of obese women without PCOS; however, in those with PCOS, these miRNAs are overexpressed [104]. In women with PCOS, serum expression of miR-222, miR-146a, and miR-30c increases significantly and miR-222 levels are positively associated with insulin levels, while miR-146a levels are negatively associated with testosterone levels [103]. In addition, Qin and colleagues (2019) demonstrated that high expression levels of lncRNA H19 were associated with a high risk of PCOS, and its expression was positively correlated with fasting plasma glucose levels but not with total testosterone or insulin resistance [115].
Disruption of epigenetic control of gene expression through embryonic development, postnatal life, and adulthood is one of the main alterations behind the pathophysiology of complex diseases. Considering the multifaceted clinical manifestations of PCOS, an intricate network of interactions contributes to each different clinical manifestation that may have an epigenetic signature. However, each cell type presents its specific epigenetic programming, which is stably maintained during cell growth. Nevertheless, microenvironmental alterations in the ovary may change the epigenetic landscape of reproductive cells and may be related to chronic anovulation and infertility in PCOS.
Aberrant Epigenetic Reprogramming in Oocytes, Cumulus, and Granulosa Cells
Although the search for biomarkers in peripheral blood is important, the majority of studies have focused on female reproductive cells, such as oocytes, granulosa cells, and cumulus cells, since the main outcome of PCOS disorder is chronic ovulation and infertility. Increased DNA methylation level in the LINE-1 5'-UTR was observed in granulosa-cumulus cells, which was consistent with the results observed in blood cells [83]. Additionally, Pruksananonda et al. (2016) also reported increased DNA methylation levels in the LINE-1 in cumulus cells of mature oocytes from PCOS. LINE-1 hypomethylation downregulates the genes containing this element, while LINE-1 hypermethylation leads to an increase in gene expression profiles in cumulus cells [90]. Granulosa cells from PCOS women with hyperandrogenism showed hypermethylation in two CpG sites of PPARG1 and hypomethylation at five CpG sites in the NCOR1 promoter region. These results were consistent with the decrease in PPARG1 expression and increase in NCOR1 and HDAC3 mRNA levels [84].
Altered DNA methylation patterns in AKR1C3, GHRHR, MAMLD1, RETN, and TNF genes in granulosa cells indirectly contribute to excessive androgen levels in PCOS patients [12]. Furthermore, a genome-wide DNA methylation study comparing epigenetic changes in ovarian granulosa cells of women with polycystic ovaries versus normal ovarian morphology, identified 106 differentially methylated CpG sites associated with 88 genes, several of which are involved in endocrine, metabolic, and reproductive processes related to PCOS. In addition, 16 of the identified CpGs were mapped within six known PCOS susceptibility loci (YAP1, RAB5B/SUOX, HMGA2, KRR1, INSR, and SUMO1P1). Of these, five overlapped with known methylation quantitative trait loci (meQTLs) [116].
GWAS provide valuable data on the origins and progression of PCOS by identifying possible targets associated with the disease. Differential expression of 59 genes has been reported in cumulus cells from PCOS women; among them, important genes for embryogenesis, angiogenesis, and sexual development, such as LHCGR, ANGPTL1, and TNIK, showed increased expression, whereas genes related to adipogenesis, placental development, and adult uterine morphology and function, such as GRIN2A, SFRP4, and SOCS3, were downregulated [91]. Subsequently, loss of methylation in the promoter region of the LHCGR was identified in the peripheral blood and granulosa cells of PCOS women [82]. As the genes LHCGR, TNIK, and SOCS3 are related to the development of the embryo until the blastocyst stage, they were raised as possible biomarkers for PCOS embryonic viability [91].
Histone modifications are covalent posttranslational epigenetic modifications that alter the chromatin structure and consequently the gene expression. Histone proteins are responsible for packing the DNA into a small structure called nucleosome in the nucleus. Nucleosomes are primary chromatin subunits composed of a histone core, an octameric structure with two copies each of the histone proteins H2A, H2B, H3, and H4, wrapped with DNA and a linker histone (H1). Chemical modifications in histone tails, such as acetylation, methylation, phosphorylation, ubiquitylation, and sumoylation, modify chromatin accessibility to transcription factors, thereby affecting gene transcription and important cellular phenotypes [117]. Errors in histone posttranslational marks have been implicated in the pathogenesis of human disorders. However, this mechanism is poorly understood in PCOS. Hosseini et al. (2019) reported increased global MeCP2 (methyl-CpG binding protein 2) and a decrease in H3K9me2 (histone H3, lysine 9 di-methylation), which can both activate and repress transcription, and increased H3K9ac (histone H3, lysine 9 acetylation), which is associated with active transcription in cumulus cells of PCOS [92].
Epigenetic changes caused by excess androgen levels are also observed in mice with PCOS induced by dehydroepiandrosterone (DHEA), in which the oocytes show decreased polar corpuscle extrusion rates, increased oxidative stress, and abnormal morphology. Furthermore, reduction in global DNA methylation and dimethylation (me2) of histone H3 on lysine 9 (H3K9), besides an increase in H4K12 acetylation, is also observed in oocytes. Consistently, DNA methyltransferase-1 (DNMT1) and histone deacetylase-1 (HDAC1) gene expression decreased in these animals [118].
Disrupted ncRNA expression was also reported in cumulus cells from PCOS women. Fifty-nine microRNAs (miRNAs) were differentially expressed in PCOS women compared to those in non-PCOS controls, in which 21 miRNAs were overexpressed and 38 were downregulated. Many of these ncRNAs are involved in important pathways related to the clinical manifestations of PCOS, including hormonal regulation, energetic metabolism, and the Notch signaling pathway that regulates cell proliferation and differentiation and is involved in follicular growth and maturation [119]. In cumulus cells, 623 long ncRNAs (lncRNAs) were differentially expressed between PCOS and non-PCOS women (five were validated using qPCR: XLOC_011402, ENST00000454271, ENST00000433673, ENST00000450294, and ENST00000432431) [120].
The lncRNA PWRN2 plays an important role in oocyte nuclear maturation and is upregulated in PCOS cumulus cells; it may also be involved in the abnormal oocyte development commonly observed in PCOS [121]. Liu and colleagues (2017) showed 862 lncRNAs that were differentially expressed and the upregulation of the lncRNA HCG26 in granulosa cells from PCOS that was associated with antral follicle count [122]. Additionally, Zhao et al. (2019) found that the expression levels of lncRNA RP11-151A6.4 increased in granulosa cells from PCOS and suggested that this lncRNA might play an important role in IR, excess androgen, and adipose dysfunction [123]. These results were consistent with the increased expression of lncRNA LINC-01572:28 in PCOS granulosa cells, and this upregulation was associated with hyperandrogenism [124]. Moreover, lncRNA-ovarian cancer associated 1 (Lnc-OC1) was overexpressed in granulosa cells from PCOS. In a steroidogenic human granulosa-like tumor cell line (KGN), Lnc-OC1 knockdown inhibited cell viability, promoted apoptosis, and increased aromatase mRNA levels and estradiol biosynthesis. In PCOS mice, Lnc-OC1 promoted serum insulin release, production of angiogenesis-related factors, and IκBα phosphorylation, which was partially restored after knockdown with Lnc_OC1 shRNA [125].
Epigenetic Markers in Ovary and Follicular Fluid
Similar to oocytes and cumulus cells, the ovary also exhibits many epigenetic alterations that may compromise its morphology and viability. The analysis of more than 450,000 CpG sites and gene expression in the ovaries of PCOS women demonstrated 7929 CpG sites with differential methylation patterns and 650 differentially expressed transcripts, of which 54 genes showed altered expression related to DNA methylation changes [110]. High levels of DNA methylation in CpG island shores (regions flanking the CpG islands) and lower DNA methylation in the gene body were observed in PCOS women. In addition, the promoter regions of SLC2A8, NRIP1, IGF2BP2, CYP19A1, and AMHR2 showed increased DNA methylation, whereas the promoters of INSR and AMH were hypomethylated [87]. In accordance, CYP19A1 increased promoter methylation and reduced gene expression in the ovaries of PCOS women [88].
Sensitivity to epigenetic modifications may be so extreme that even a single differentially methylated CpG site may alter gene expression. PCOS women present hypermethylation at site cg10180092, a critical site in the regulation of TNIK transcription and a decreased H3K9me level, a repressive histone modification [110], leading to increased TNIK expression in PCOS ovarian tissue. This is a peculiar case in which a single point DNA hypermethylation affects histone methylation, thereby altering TNIK gene expression [89]. In contrast, epigenetic alterations may often not reflect differences in gene expression; for example, ovarian tissue from testosterone-induced PCOS mice showed hypomethylation at five sites in the AR gene promoter region and one site on the Cyp11a1 gene promoter. However, these differences were not sufficient to alter the expression of these genes [126].
MiRNAs also exert influence on the ovaries. Lin et al. (2015) identified 27 miRNAs differentially expressed in the ovaries of PCOS women compared to those in the controls. The downregulation of miR-92a and miR-92b in PCOS was confirmed using qPCR [97]. Increased expression of miR-93 and its target CDKN1A gene was downregulated in ovarian cortex immortalized granulosa cells, which might be due to the high concentration of insulin, a common condition in PCOS women [98].
The miRNAs in the follicular fluid (microvesicles and supernatant) and the overexpression of target genes in PCOS women are associated with reproduction, endocrine system, and metabolic processes [101, 102]. A positive relationship was observed among miR-132, miR-320, miR-520c-3p, miR-222, and estradiol concentrations, while a negative relationship was noted among miR-24, miR-193b, miR-483-5p, and progesterone concentrations [101]. In PCOS women, miR-132 and miR-320 are expressed at lower levels, whereas miR-32, miR-34c, miR-135a, miR-18b, and miR-9 show increased expression in the follicular fluid of PCOS [101, 102]. The lncRNA profile in follicular fluid from mature and immature ovarian follicles also differs between the PCOS and controls, and lncRNAs related to the metabolic process are highly enriched in the PCOS mature follicular fluid [127].
Mitochondrial Dysfunctions
Besides genetic, epigenetic, and environmental factors, mitochondrial dysfunction may play a role in the pathogenesis of PCOS. Ding et al. (2017) identified nine homoplasmic variants in mt-tRNAs that were associated with PCOS women with IR: mt-tRNALeu(UUR) A3302G and C3275A, mt-tRNAGln T4363C and T4395C, mt-tRNASer(UCN) C7492T, mt-tRNAAsp A7543G, mt-tRNALys A8343G, mt-tRNAArg T10454C, and mt-tRNAGlu A14693G. These mutations alter the secondary structure of mt-tRNAs, affecting mt-tRNA metabolism, thereby increasing the levels of 8-hydroxy-2'-deoxyguanosine (8-OHdG), malondialdehyde, and reactive oxygen species (ROS) in PCOS-IR women [128].
A previous study identified 16 variants in the noncoding region D-Loop, seven variations in the 12S rRNA gene, three variants in the 16S rRNA gene, and several variants of the oxidative phosphorylation (OXPHOS) complex in PCOS women [129]. The study also identified six variants in mitochondrial tRNA genes, including tRNAGln, tRNACys, tRNAAsp, tRNALys, tRNAArg, and tRNAGlu, localized in conserved regions important for tRNA stability and biochemical function [129]. Therefore, it may be possible that the cumulative effect of these mutations contributes to mitochondrial dysfunction, thereby contributing to PCOS pathogenesis.
Moreover, analysis of the mitochondrial displacement-loop (D-loop) of Indian PCOS women and the controls showed a significant association between D310 and A189G SNPs with PCOS, resulting in lower mtDNA copy number for patients carrying D310 and 189G alleles, and an elevated LH/FSH ratio for D310 carriers [130]. Mitochondrial D-loop is a hotspot region for mtDNA mutations; these mutations can interrupt the sequence in the promoter region, thereby altering the affinity for inducers/modifiers of mtDNA replication and/or transcription, which may further affect the electron transport chain machinery and consequently, cellular ROS generation [130].
Mutation analysis of the whole mitochondrial genes in a three-generation pedigree with maternally transmitted metabolic syndrome, combined with PCOS, identified multiple variants, especially in mt-tRNA genes (tRNALeu(UUR) C3275T, tRNAGln T4363C, and tRNALys A8343G). These mutations altered mt-tRNA metabolism, resulting in a deficiency in mitochondrial functions (lower levels of MMP, ATP production, and mtDNA copy number, and increase in ROS generation), which could be responsible for the clinical phenotypes [131].
Variants in nuclear-encoded genes regulating mitochondrial biogenesis, such as TFAM and PGC-1α, were also associated with PCOS, leading to reduced mtDNA copy number and higher levels of the luteinizing (LH) hormone [132]. Mitochondrial dysfunctions in the peripheral blood, leukocytes, follicular fluid, skeletal muscle cells, cumulus cells, and endometrial cells from PCOS women have also been described in several studies [133,134,135,136].
Conclusions and Perspectives
PCOS is an ancient, heterogeneous, multifactorial, and complex disorder; understanding the mechanisms underlying the pathophysiology of this syndrome and the consequences for female fertility, is challenging. The diversity of clinical manifestations observed in PCOS suggests that a combination of multiple factors is related to the pathophysiology of this syndrome (Fig. 2). Genetic predisposition (candidate genes) and a strong environmental contribution owing to lifestyle changes, obesity, and hormonal and metabolic disruptors lead to (epi)genetic susceptibility to PCOS development throughout life. Nevertheless, hyperandrogenism and metabolic disorders directly affect ovary morphology and folliculogenesis, which may alter the epigenetic landscape of reproductive cells, leading to chronic anovulation, impaired fertility, reduced embryo quality, and miscarriage. Environmentally induced epigenetic changes caused by an intrauterine adverse environment, as hyperandrogenism during early embryo development may trigger developmental PCOS or its associated clinical alterations after birth.

Lifestyles changes as unhealthy diet, sedentarism, and obesity, as well as hormonal and metabolic disturbances are environmental factors that combined with a genetic predisposition (candidate genes) changes epigenome landscape leading to (epi)genetic susceptibility to PCOS development throughout life. On the other hand, the hormonal and endocrine disturbances in PCOS directly affect folliculogenesis and gametogenesis, resulting in chronic anovulation, impaired fertilization, reduced implantation rates, and miscarriage. These clinical alterations affect embryo development since intrauterine environmental changes as hyperandrogenism, nutritional imbalance, and metabolic syndrome impact the epigenetic reprograming during fetal life that may trigger developmental PCOS or its associated clinical alterations after birth
However, the clinical alterations of PCOS and its associated health conditions have a great plasticity, changing or even disappearing throughout a lifetime. Lifestyle changes, such as physical activity and a healthy diet, directly affect metabolic disorders and improve fertility in PCOS through epigenetic changes in gene expression. The identification of possible molecular markers and epigenetic modifications related to the different phenotypes observed in PCOS, such as IR, hyperandrogenism, chronic anovulation, obesity, and other related alterations, could direct the treatment and improve the quality of life and fertility of women with PCOS. However, the incidence of PCOS is growing and many aspects of this syndrome and its effects on female fertility and quality of life remain unclear. Therefore, accurate investigation of the epigenetic mechanisms underlying this syndrome is of utmost importance for advancing the understanding and treatment of PCOS.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Azziz R, Dumesic DA, Goodarzi MO. Polycystic ovary syndrome: an ancient disorder? Fertil Steril. 2011;95:1544–8.
Vallisneri A. Storia della generazione dell’uomo e dell’animale; 1721. Cited in Cooke ID, Lunenfeld B. Res Clin Forums. 1989;11:109–13.
Szydlarska D, Machaj M, Jakimiuk A. History of discovery of polycystic ovary syndrome. Adv Clin Exp Med. 2017:555–8.
Stein IF, Leventhal ML. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol. 1935;29:181–91.
Zawadzki J, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif A, HR G, FP H, GR M, editors. Polycystic ovary syndr. Boston, MA: Blackwell Scientific; 1992. p. 377–384.
The Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81:19–25.
Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, et al. Position statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an androgen excess society guideline. J Clin Endocrinol Metab. 2006;91:4237–45.
Carmina E, Lobo RA. Use of fasting blood to assess the prevalence of insulin resistance in women with polycystic ovary syndrome. Fertil Steril. 2004;82:661–5.
Teede H, Deeks A, Moran L. Polycystic ovary syndrome: a complex condition with psychological, reproductive and metabolic manifestations that impacts on health across the lifespan. BMC Med. 2010:41.
Azziz R, Marin C, Hoq L, Badamgarav E, Song P. Health care-related economic burden of the polycystic ovary syndrome during the reproductive life Span. J Clin Endocrinol Metab. 2005:4650–8.
Azziz R, Carmina E, Chen Z, Dunaif A, Laven JSE, Legro RS, et al. Polycystic ovary syndrome. Nat. Rev. Dis. Prim. 2016:1–18.
Sagvekar P, Kumar P, Mangoli V, Desai S, Mukherjee S. DNA methylome profiling of granulosa cells reveals altered methylation in genes regulating vital ovarian functions in polycystic ovary syndrome. Clin Epigenetics. 2019;11:1–16.
Amato P, Simpson JL. The genetics of polycystic ovary syndrome. Best Pract Res Clin Obstet Gynaecol. 2004;18:707–18.
Kahsar-Miller MD, Nixon C, Boots LR, Go RC, Azziz R. Prevalence of polycystic ovary syndrome (PCOS) in first-degree relatives of patients with PCOS. Fertil Steril. 2001;75:53–8.
Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab. 2006;91:2100–4.
Hayes MG, Urbanek M, Ehrmann DA, Armstrong LL, Lee JY, Sisk R, et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun. 2015;6:1–12.
Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–45.
Hakonarson H, Qu HQ, Bradfield JP, Marchand L, Kim CE, Glessner JT, et al. A novel susceptibility locus for type 1 diabetes on chr12q13 identified by a genome-wide association study. Diabetes. 2008;57:1143–6.
Capalbo A, Sagnella F, Apa R, Fulghesu AM, Lanzone A, Morciano A, et al. The 312N variant of the luteinizing hormone/choriogonadotropin receptor gene (LHCGR) confers up to 2.7-fold increased risk of polycystic ovary syndrome in a Sardinian population. Clin Endocrinol (Oxf). 2012;77:113–9.
Almawi WY, Hubail B, Arekat DZ, Al-Farsi SM, Al-Kindi SK, Arekat MR, et al. Leutinizing hormone/choriogonadotropin receptor and follicle stimulating hormone receptor gene variants in polycystic ovary syndrome. J Assist Reprod Genet. 2015;32:607–14.
Shoham Z, Jacobs HS, Insler V. Luteinizing hormone: its role, mechanism of action, and detrimental effects when hypersecreted during the follicular phase. Fertil Steril. 1993;59:1153–61.
Mason H, Franks S. Local control of ovarian steroidogenesis. Baillieres Clin Obstet Gynaecol. 1997;11:261–79.
Shi Y, Zhao H, Shi Y, Cao Y, Yang D, Li Z, et al. Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat Genet. 2012;44:1020–5.
Day FR, Hinds DA, Tung JY, Stolk L, Styrkarsdottir U, Saxena R, et al. Causal mechanisms and balancing selection inferred from genetic associations with polycystic ovary syndrome. Nat Commun. 2015;6:1–7.
Goodarzi MO, Jones MR, Li X, Chua AK, Garcia OA, Chen YDI, et al. Replication of association of DENND1A and THADA variants with polycystic ovary syndrome in European cohorts. J Med Genet. 2012;49:90–5.
Jamnongjit M, Gill A, Hammes SR. Epidermal growth factor receptor signaling is required for normal ovarian steroidogenesis and oocyte maturation. PNAS. 2005.
LaVoie HA. The GATA-keepers of ovarian development and folliculogenesis. Biol. Reprod. 2014:38–9.
Qin K, Ehrmann DA, Cox N, Refetoff S, Rosenfield RL. Identification of a functional polymorphism of the human type 5 17β-hydroxysteroid dehydrogenase gene associated with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91:270–6.
Ju R, Wu W, Fei J, Qin Y, Tang Q, Wu D, et al. Association analysis between the polymorphisms of HSD17B5 and HSD17B6 and risk of polycystic ovary syndrome in Chinese population. Eur J Endocrinol. 2015;172:227–33.
Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–89.
Reddy KR, Deepika MLN, Supriya K, Latha KP, Rao SSL, Rani VU, et al. CYP11A1 microsatellite (tttta)n polymorphism in PCOS women from South India. J Assist Reprod Genet. 2014;31:857–63.
Tucci S, Futterweit W, Concepcion ES, Greenberg DA, Villanueva R, Davies TFTY. Evidence for association of polycystic ovary syndrome in caucasian women with a marker at the insulin receptor gene locus. J Clin Endocrinol Metab. 2001;86:446–9.
Zi-jiang Chen, Shi Y, Zhao Y, Li Y, Tang R, Zhao L, et al. Correlation between single nucleotide polymorphism of insulin receptor gene with polycystic ovary syndrome. Chin J Obs Gynecol. 2004;39:582–5.
Lee EJ, Oh B, Lee JY, Kimm K, Lee SH, Baek KH. A novel single nucleotide polymorphism of INSR gene for polycystic ovary syndrome. Fertil Steril. 2008;89:1213–20.
Goodarzi MO, Louwers YV, Taylor KD, Jones MR, Cui J, Kwon S, et al. Replication of association of a novel insulin receptor gene polymorphism with polycystic ovary syndrome. Fertil Steril. 2011;95:1736–41.
Ju R, Wu W, Tang Q, Wu D, Xia Y, Wu J, et al. Association analysis between the polymorphisms of HSD11B1 and H6PD and risk of polycystic ovary syndrome in Chinese population. PLoS One. 2015;10:1–10.
Sudo S, Kudo M, Wada SI, Sato O, Hsueh AJW, Fujimoto S. Genetic and functional analyses of polymorphisms in the human FSH receptor gene. Mol Hum Reprod. 2002;8:893–9.
Li T, Zhao H, Zhao X, Zhang B, Cui L, Shi Y, et al. Identification of YAP1 as a novel susceptibility gene for polycystic ovary syndrome. J Med Genet. 2012;49:254–7.
Jones MR, Wilson SG, Mullin BH, Mead R, Watts GF, Stuckey BGA. Polymorphism of the follistatin gene in polycystic ovary syndrome. Mol Hum Reprod. 2007;13:237–41.
Raja-Khan N, Kunselman AR, Demers LM, Ewens KG, Spielman RS, Legro RS. A variant in the fibrillin-3 gene is associated with TGF-β and inhibin B levels in women with polycystic ovary syndrome. Fertil Steril. 2010;94:2916–9.
Lin Y, Dion V, Wilson JH. Transcription and triplet repeat instability. Second Edi. Genet. Instab. Neurol. Dis. Second Ed. Elsevier Inc; 2006.
Lin LH, Baracat MCP, MacIel GAR, Soares JM, Baracat EC. Androgen receptor gene polymorphism and polycystic ovary syndrome. Int J Gynecol Obstet. 2013;120:115–8.
Li Q, Du J, Feng R, Xu Y, Wang H, Sang Q, et al. A possible new mechanism in the pathophysiology of polycystic ovary syndrome (PCOS): the discovery that leukocyte telomere length is strongly associated with PCOS. J Clin Endocrinol Metab. 2014;99:234–40.
Pedroso DCC, Miranda-Furtado CL, Kogure GS, Meola J, Okuka M, Silva C, et al. Inflammatory biomarkers and telomere length in women with polycystic ovary syndrome. Fertil Steril. 2015;103:542–547.e2.
Wei D, Xie J, Yin B, Hao H, Song X, Liu Q, et al. Significantly lengthened telomere in granulosa cells from women with polycystic ovarian syndrome (PCOS). J Assist Reprod Genet. 2017;34:861–6.
Calado RT, Yewdell WT, Wilkerson KL, Regal JA, Kajigaya S, Stratakis CA, et al. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood. 2009;114:2236–43.
Welt CK, Duran JM. The genetics of polycystic ovary syndrome. Semin Reprod Med. 2014;32:177–82.
Lee HJ, Hore TA, Reik W. Reprogramming the methylome: erasing memory and creating diversity. Cell Stem Cell. 2014:710–9.
Waddington CH. Canalization of development and the inheritance of acquired characters. Nat Publ Gr. 1942;150:563–5.
Monk D, Mackay DJG, Eggermann T, Maher ER, Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet Springer US. 2019;20:235–48.
Nageshwaran S, Festenstein R. Epigenetics and triplet-repeat neurological diseases. Front Neurol. 2015:1–9.
Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: moving forward. PLoS Genet. 2018;14:1–25.
Ling C, Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab. 2019;29:1028–44.
Filippou P, Homburg R. Is foetal hyperexposure to androgens a cause of PCOS? Hum Reprod Update. 2017;23:421–32.
Dumesic DA, Goodarzi MO, Chazenbalk GD, Abbott DH. Intrauterine environment and polycystic ovary syndrome. Semin Reprod Med. 2014;32:159–65.
Gur EB. Fetal programming of polycystic ovary syndrome. World J Diabetes. 2015;6:936.
Wu XY, Li ZL, Wu CY, Liu YM, Lin H, Wang SH, et al. Endocrine traits of polycystic ovary syndrome in prenatally androgenized female Sprague-Dawley rats. Endocr J. 2010;57:201–9.
Zhu JQ, Zhu L, Liang XW, Xing FQ, Schatten H, Sun QY. Demethylation of LHR in dehydroepiandrosterone-induced mouse model of polycystic ovary syndrome. Mol Hum Reprod. 2010;16:260–6.
Padmanabhan V, Veiga-Lopez A. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol. 2013:8–20.
Zhang D, Cong J, Shen H, Wu Q, Wu X. Genome-wide identification of aberrantly methylated promoters in ovarian tissue of prenatally androgenized rats. Fertil Steril. 2014;102:1458–67.
Xu N, Kwon S, Abbott DH, Geller DH, Dumesic DA, Azziz R, et al. Epigenetic mechanism underlying the development of polycystic ovary syndrome (PCOS)-like phenotypes in prenatally androgenized rhesus monkeys. PLoS One. 2011;6:1–9.
Das R, Hampton DD, Jirtle RL. Imprinting evolution and human health. Mamm. Genome. 2009:563–72.
Duncan CG, Grimm SA, Morgan DL, Bushel PR, Bennett BD, Roberts JD, et al. Dosage compensation and DNA methylation landscape of the X chromosome in mouse liver. Sci Rep. 2018;8:1–17.
Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–22.
Salehi Jahromi M, Hill JW, Ramezani Tehrani F, Zadeh-Vakili A. Hypomethylation of specific CpG sites in the promoter region of steroidogeneic genes (GATA6 and StAR) in prenatally androgenized rats. Life Sci Elsevier Inc. 2018;207:105–9.
Nilsson E, Klukovich R, Sadler-Riggleman I, Beck D, Xie Y, Yan W, et al. Environmental toxicant induced epigenetic transgenerational inheritance of ovarian pathology and granulosa cell epigenome and transcriptome alterations: ancestral origins of polycystic ovarian syndrome and primary ovarian insufiency. Epigenetics. 2018;13:875–95.
Albrecht ED, Pepe GJ. Estrogen regulation of placental angiogenesis and fetal ovarian development during primate pregnancy. Int J Dev Biol. 2010;54:397–407.
Fowler PA, Anderson RA, Saunders PT, Kinnell H, Mason JI, Evans DB, et al. Development of steroid signaling pathways during primordial follicle formation in the human fetal ovary. J Clin Endocrinol Metab. 2011;96:1754–62.
Fenichel P, Rougier C, Hieronimus S, Chevalier N. Le syndrome des ovaires polykystiques, est-il d’origine génétique, environnementale ou les deux ? Ann. Endocrinol. (Paris). Elsevier Masson SAS. 2017:176–85.
Steckler T, Wang J, Bartol FF, Roy SK, Padmanabhan V. Fetal programming: prenatal testosterone treatment causes intrauterine growth retardation, reduces ovarian reserve and increases ovarian follicular recruitment. Endocrinology. 2005;146:3185–93.
Xu N, Chua AK, Jiang H, Liu NA, Goodarzi MO. Early embryonic androgen exposure induces transgenerational epigenetic and metabolic changes. Mol Endocrinol Endocrine Soc. 2014;28:1329–36.
Echiburú B, Milagro F, Crisosto N, Pérez-Bravo F, Flores C, Arpón A, et al. DNA methylation in promoter regions of genes involved in the reproductive and metabolic function of children born to women with PCOS. Epigenetics Taylor Francis. 2020;00:1–17.
Melo AS, Vieira CS, Barbieri MA, Rosa-E-Silva ACJS, Silva AAM, Cardoso VC, et al. High prevalence of polycystic ovary syndrome in women born small for gestational age. Hum Reprod. 2010;25:2124–31.
Cherskov A, Pohl A, Allison C, Zhang H, Payne RA, Baron-Cohen S. Polycystic ovary syndrome and autism: a test of the prenatal sex steroid theory. Transl Psychiatry. 2018;8:1–10.
van den Berg IM, Laven JSE, Stevens M, Jonkers I, Galjaard RJ, Gribnau J, et al. X Chromosome inactivation is initiated in human preimplantation embryos. Am J Hum Genet. 2009;84:771–9.
Hickey T, Chandy A, Norman RJ. The androgen receptor CAG repeat polymorphism and X-chromosome inactivation in Australian Caucasian women with infertility related to polycystic ovary syndrome. J Clin Endocrinol Metab. 2002;87:161–5.
Hickey TE, Legro RS, Norman RJ. Brief Report: Epigenetic modification of the X chromosome influences susceptibility to polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91:2789–91.
Shah NA, Antoine HJ, Pall M, Taylor KD, Azziz R, Goodarzi MO. Association of androgen receptor CAG repeat polymorphism and polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93:1939–45.
Dasgupta S, Sirisha PVS, Neelaveni K, Anuradha K, Reddy AG, Thangaraj K, et al. Androgen receptor cag repeat polymorphism and epigenetic influence among the south indian women with polycystic ovary syndrome. PLoS One. 2010;5:1–8.
Laisk T, Haller-Kikkatalo K, Laanpere M, Jakovlev Ü, Peters M, Karro H, et al. Androgen receptor epigenetic variations influence early follicular phase gonadotropin levels. Acta Obstet Gynecol Scand. 2010;89:1557–63.
Xu N, Azziz R, Goodarzi MO. Epigenetics in polycystic ovary syndrome: a pilot study of global DNA methylation. Fertil Steril. 2010;94:781–3.
Wang P, Zhao H, Li T, Zhang W, Wu K, Li M, et al. Hypomethylation of the LH/choriogonadotropin receptor promoter region is a potential mechanism underlying susceptibility to polycystic ovary syndrome. Endocrinology. 2014;155:1445–52.
Sagvekar P, Mangoli V, Desai S, Patil A, Mukherjee S. LINE1 CpG-DNA hypomethylation in granulosa cells and blood leukocytes is associated with PCOS and related traits. J Clin Endocrinol Metab. 2017;102:1396–405.
Qu F, Wang FF, Yin R, Ding GL, El-prince M, Gao Q, et al. A molecular mechanism underlying ovarian dysfunction of polycystic ovary syndrome: hyperandrogenism induces epigenetic alterations in the granulosa cells. J Mol Med. 2012;90:911–23.
Pan JX, Tan YJ, Wang FF, Hou NN, Xiang YQ, Zhang JY, et al. Aberrant expression and DNA methylation of lipid metabolism genes in PCOS: a new insight into its pathogenesis. Clin Epigenetics. 2018;10:1–12.
Jiang L, Le XJK, Cui JQ, Wei D, Yin BL, Zhang YN, et al. Promoter methylation of yes-associated protein (YAP1) gene in polycystic ovary syndrome. Med (US). 2017;96:1–6.
Yu YY, Sun CX, Liu YK, Li Y, Wang L, Zhang W. Genome-wide screen of ovary-specific DNA methylation in polycystic ovary syndrome. Fertil Steril Elsevier Inc. 2015;104:145–53.
Yu YY, Sun CX, Liu YK, Li Y, Wang L, Zhang W. Promoter methylation of CYP19A1 gene in chinese polycystic ovary syndrome patients. Gynecol Obstet Invest. 2013;76:209–13.
Li D, Jiao J, Zhou YM, Wang XX. Epigenetic regulation of traf2- and nck-interacting kinase (Tnik) in polycystic ovary syndrome. Am J Transl Res. 2015;7:1152–60.
Pruksananonda K, Wasinarom A, Sereepapong W, Sirayapiwat P, Rattanatanyong P, Mutirangura A. Epigenetic modification of long interspersed elements-1 in cumulus cells of mature and immature oocytes from patients with polycystic ovary syndrome. Clin Exp Reprod Med. 2016;43:82–9.
Huang X, Hao C, Shen X, Liu X, Shan Y, Zhang Y, et al. Differences in the transcriptional profiles of human cumulus cells isolated from MI and MII oocytes of patients with polycystic ovary syndrome. Reproduction. 2013;145:597–608.
Hosseini E, Shahhoseini M, Afsharian P, Karimian L, Ashrafi M, Mehraein F, et al. Role of epigenetic modifications in the aberrant CYP19A1 gene expression in polycystic ovary syndrome. Arch Med Sci. 2019;15:887–95.
Ting W, Yanyan Q, Jian H, Keqin H, Duan M. The relationship between insulin resistance and CpG island methylation of LMNA gene in polycystic ovary syndrome. Cell Biochem Biophys. 2013;67:1041–7.
Sang Q, Li X, Wang H, Wang H, Zhang S, Feng R, et al. Quantitative methylation level of the EPHX1 promoter in peripheral blood DNA is associated with polycystic ovary syndrome. PLoS One. 2014;9:1–7.
Zhao H, Zhao Y, Ren Y, Li M, Li T, Li R, et al. Epigenetic regulation of an adverse metabolic phenotype in polycystic ovary syndrome: the impact of the leukocyte methylation of PPARGC1A promoter. Fertil Steril. 2017;107:467–474.e5.
Jones MR, Brower MA, Xu N, Cui J, Mengesha E, Chen YDI, et al. Systems genetics reveals the functional context of PCOS loci and identifies genetic and molecular mechanisms of disease heterogeneity. PLoS Genet. 2015;11:1–17.
Lin L, Du T, Huang J, Huang LL, Yang DZ. Identification of differentially expressed microRNAs in the ovary of polycystic ovary syndrome with hyperandrogenism and insulin resistance. Chin Med J (Engl). 2015;128:169–74.
Jiang L, Huang J, Li L, Chen Y, Chen X, Zhao X, et al. MicroRNA-93 promotes ovarian granulosa cells proliferation through targeting CDKN1A in polycystic ovarian syndrome. J Clin Endocrinol Metab Endocrine Soc. 2015;100:E729–38.
Wu HL, Heneidi S, Chuang TY, Diamond MP, Layman LC, Azziz R, et al. The expression of the miR-25/93/106b family of micro-RNAs in the adipose tissue of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2014;99:1–9.
Chen YH, Heneidi S, Lee JM, Layman LC, Stepp DW, Gamboa GM, et al. Mirna-93 inhibits glut4 and is overexpressed in adipose tissue of polycystic ovary syndrome patients and women with insulin resistance. Diabetes. 2013;62:2278–86.
Sang Q, Yao Z, Wang H, Feng R, Wang H, Zhao X, et al. Identification of MicroRNAs in human follicular fluid: characterization of microRNAs that govern steroidogenesis in vitro and are associated with polycystic ovary syndrome in vivo. J Clin Endocrinol Metab. 2013;98:3068–79.
Roth LW, McCallie B, Alvero R, Schoolcraft WB, Minjarez D, Katz-Jaffe MG. Altered microRNA and gene expression in the follicular fluid of women with polycystic ovary syndrome. J Assist Reprod Genet. 2014;31:355–62.
Long W, Zhao C, Ji C, Ding H, Cui Y, Guo X, et al. Characterization of serum microRNAs profile of PCOS and identification of novel non-invasive biomarkers. Cell Physiol Biochem. 2014;33:1304–15.
Murri M, Insenser M, Fernández-Durán E, San-Millán JL, Escobar-Morreale HF. Effects of polycystic ovary syndrome (PCOS), sex hormones, and obesity on circulating miRNA-21, miRNA-27b, miRNA-103, and miRNA-155 expression. J Clin Endocrinol Metab. 2013;98:1835–44.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–8.
Vázquez-martínez ER, Gómez-viais YI, García-gómez E, Reyes-mayoral C, Reyes-muñoz E, Camacho-arroyo I, et al. DNA methylation in the pathogenesis of polycystic ovary syndrome. Reproduction. 2019;158:R27–40.
Li S, Zhu D, Duan H, Ren A, Glintborg D, Andersen M, et al. Differential DNA methylation patterns of polycystic ovarian syndrome in whole blood of Chinese women. Oncotarget. 2017;8:20656–66.
Shen H ran, Qiu L hua, Zhang Z qing, Qin Y yuan, Cao C, Di W. Genome-wide methylated DNA immunoprecipitation analysis of patients with polycystic ovary syndrome. PLoS One. 2013;8:1–10.
Kokosar M, Benrick A, Perfilyev A, Fornes R, Nilsson E, Maliqueo M, et al. Epigenetic and transcriptional alterations in human adipose tissue of polycystic ovary syndrome. Sci Rep. 2016;6:1–17.
Wang XX, Wei JZ, Jiao J, Jiang SY, Yu DH, Li D. Genome-wide DNA methylation and gene expression patterns provide insight into polycystic ovary syndrome development. Oncotarget. 2014;5:6603–10.
Salehi M, Bravo-Vera R, Sheikh A, Gouller A, Poretsky L. Pathogenesis of polycystic ovary syndrome: what is the role of obesity? Metabolism. 2004;53:358–76.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. Nat Publ Group. 2016;17:47–62.
Lin H, Xing W, Li Y, Xie Y, Tang X, Zhang Q. Downregulation of serum long noncoding RNA GAS5 may contribute to insulin resistance in PCOS patients. Gynecol Endocrinol. 2018;34:784–8.
Qin L, Huang CC, Yan XM, Wang Y, Li ZY, Wei XC. Long non-coding RNA h19 is associated with polycystic ovary syndrome in Chinese women: a preliminary study. Endocr J. 2019;66:587–95.
Makrinou E, Drong AW, Christopoulos G, Lerner A, Chapa-Chorda I, Karaderi T, et al. Genome-wide methylation profiling in granulosa lutein cells of women with polycystic ovary syndrome (PCOS). Mol Cell Endocrinol Elsevier. 2020;500:1–11.
Barth TK, Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem Sci Elsevier Ltd. 2010;35:618–26.
Eini F, Novin MG, Joharchi K, Hosseini A, Nazarian H, Piryaei A, et al. Intracytoplasmic oxidative stress reverses epigenetic modifications in polycystic ovary syndrome. Reprod Fertil Dev. 2017;29:2313–23.
Xu B, Zhang YW, Tong XH, Liu YS. Characterization of microRNA profile in human cumulus granulosa cells: identification of microRNAs that regulate Notch signaling and are associated with PCOS. Mol Cell Endocrinol Elsevier Ireland Ltd. 2015;404:26–36.
Huang X, Hao C, Bao H, Wang M, Dai H. Aberrant expression of long noncoding RNAs in cumulus cells isolated from PCOS patients. J Assist Reprod Genet. 2016;33:111–21.
Huang X, Pan J, Wu B, Teng X. Construction and analysis of a lncRNA (PWRN2)-mediated ceRNA network reveal its potential roles in oocyte nuclear maturation of patients with PCOS. Reprod Biol Endocrinol. 2018;16:1–13.
Liu YD, Li Y, Feng SX, Ye DS, Chen X, Zhou XY, et al. Long noncoding RNAs: potential regulators involved in the pathogenesis of polycystic ovary syndrome. Endocrinology. 2017;158:3890–9.
Zhao J, Huang J, Geng X, Chu W, Li S, Chen ZJ, et al. Polycystic ovary syndrome: Novel and hub lncRNAs in the insulin resistance-associated lncRNA–mRNA network. Front Genet. 2019;10:1–12.
Zhao J, Xu J, Wang W, Zhao H, Liu H, Liu X, et al. Long non-coding RNA LINC-01572:28 inhibits granulosa cell growth via a decrease in p27 (Kip1) degradation in patients with polycystic ovary syndrome. EBioMedicine Authors. 2018;36:526–38.
Wu G, Yang Z, Chen Y, Li X, Yang J, Yin T. Downregulation of Lnc-OC1 attenuates the pathogenesis of polycystic ovary syndrome. Mol Cell Endocrinol, Elsevier. 2020;506:1–7.
Xia Y, Shen S, Zhang X, Deng Z, Xiang Z, Wang H, et al. Epigenetic pattern changes in prenatal female Sprague-Dawley rats following exposure to androgen. Reprod Fertil Dev. 2015;28:1414–23.
Jiao J, Shi B, Wang T, Fang Y, Cao T, Zhou Y, et al. Characterization of long non-coding RNA and messenger RNA profiles in follicular fluid from mature and immature ovarian follicles of healthy women and women with polycystic ovary syndrome. Hum Reprod. 2018;33:1735–48.
Ding Y, Xia BH, Zhang CJ, Zhuo GC. Mutations in mitochondrial tRNA genes may be related to insulin resistance in women with polycystic ovary syndrome. Am J Transl Res. 2017;9:2984–96.
Zhuo G, Ding Y, Feng G, Yu L, Jiang Y. Analysis of mitochondrial DNA sequence variants in patients with polycystic ovary syndrome. Arch Gynecol Obstet. 2012;286:653–9.
Reddy TV, Govatati S, Deenadayal M, Sisinthy S, Bhanoori M. Impact of mitochondrial DNA copy number and displacement loop alterations on polycystic ovary syndrome risk in south Indian women. Mitochondrion Elsevier. 2019;44:35–40.
Ding Y, Xia BH, Zhang CJ, Zhuo GC. Mitochondrial tRNALeu(UUR) C3275T, tRNAGln T4363C and tRNALys A8343G mutations may be associated with PCOS and metabolic syndrome. Gene. 2018;642:299–306.
Reddy TV, Govatati S, Deenadayal M, Shivaji S, Bhanoori M. Polymorphisms in the TFAM and PGC1-α genes and their association with polycystic ovary syndrome among South Indian women. Gene, Elsevier. 2018;641:129–36.
Zhao H, Zhao Y, Li T, Li M, Li J, Li R, et al. Metabolism alteration in follicular niche: the nexus among intermediary metabolism, mitochondrial function, and classic polycystic ovary syndrome. Free Radic Biol Med, Elsevier. 2015;86:295–307.
Cree-Green M, Rahat H, Newcomer BR, Bergman BC, Brown MS, Coe GV, et al. Insulin resistance, hyperinsulinemia, and mitochondria dysfunction in nonobese girls with polycystic ovarian syndrome. J Endocr Soc. 2017;1:931–44.
Wang T, Zhang J, Hu M, Zhang Y, Cui P, Li X, et al. Differential expression patterns of glycolytic enzymes and mitochondria-dependent apoptosis in PCOS patients with endometrial hyperplasia, an early hallmark of endometrial cancer, in vivo and the impact of metformin in vitro. Int J Biol Sci. 2019;15:714–25.
Azhary JMK, Harada M, Takahashi N, Nose E, Kunitomi C, Koike H, et al. Endoplasmic reticulum stress activated by androgen enhances apoptosis of granulosa cells via induction of death receptor 5 in PCOS. Endocrinology. 2019;160:119–32.
Acknowledgements
We would like to thank the members of the Human Reproduction Division at Department of Gynecology and Obstetrics of the Ribeirao Preto Medical School, University of Sao Paulo (FMRP-USP), and the members of the Laboratory of Experimental Oncology (LOE) at Drug Research and Development Center, Federal University of Ceara (NPDM-UFC). Fig. 2 was made in part using BioRender. The English was revised by Editage.
Funding
This study was supported by the Sao Paulo Research Foundation (FAPESP, Fundação de Amparo à Pesquisa do Estado de São Paulo) with the grant 2015/14031-0 (RMR) and fellowship 2012/11069-9 (CLMF), National Council for Scientific and Technological Development (CNPq, Conselho Nacional de Desenvolvimento Científico e Tecnológico), Coordination for the Improvement of Higher Education Personnel (CAPES, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior; Academic Excellence Program - PROEX and Graduate Support Program - PROAP) and by the National Institutes of Science and Technology (INCT, Institutos Nacionais de Ciência e Tecnologia).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics Statement
Since this is a review article any human or animal samples were used in this manuscript, so no ethical consents are required.
Consent to Participate
Not applicable.
Consent for Publication
All authors approved the final version and consent to the publication of this study.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 14 kb)
Rights and permissions
About this article

Cite this article
Eiras, M.C., Pinheiro, D.P., Romcy, K.A.M. et al. Polycystic Ovary Syndrome: the Epigenetics Behind the Disease. Reprod. Sci. 29, 680–694 (2022). https://doi.org/10.1007/s43032-021-00516-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43032-021-00516-3