
Abstract
Umbilical cord mesenchymal stem cells (UCMSCs) have been proposed as an ideal source for cell-based therapy to promote endometrial repair and regeneration. Furthermore, increasing evidence has indicated that UCMSC-derived exosomes (UCMSC-exos) act as important paracrine mediators to recapitulate the features of MSCs and may play a vital role in this process. UCMSCs and human endometrial stromal cells (ESCs) were isolated and characterized. ESCs were cocultured with UCMSCs and further assessed by flow cytometry and EdU incorporation assays. UCMSC-exos were extracted by differential ultracentrifugation and identified by western blots, transmission electron microscopy, and nanoparticle tracking analysis. The internalization of UCMSC-exos by ESCs was observed under a confocal microscope. ESCs were treated with UCMSC-exos at different concentrations and for different durations, with cell viability evaluated by CCK-8 assays. The cell cycle analysis showed that the percentage of ESCs in S phase significantly increased after coculture with UCMSCs, whereas it significantly decreased after inhibition of UCMSC-exo secretions. EdU incorporation assays also showed a similar trend. The isolated UCMSC-exos had a typical cup-shaped morphology with a monolayer membrane, expressed the specific exosomal markers Alix, CD63, and TSG101 and were approximately 60 to 200 nm in diameter. The PKH26-labeled UCMSC-exos were incorporated into ESCs. Moreover, UCMSC-exos enhanced the cell growth and viability of ESCs in a dose-dependent manner, and the effects occurred in a short period of time. UCMSC-exos promote the proliferation of ESCs in a dose-dependent manner; thus, they could be used as a potential treatment to promote endometrial repair.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Intrauterine adhesions (IUAs), namely, Asherman syndrome, are typically secondary to iatrogenic trauma of the basal layer of the endometrium, resulting in the absence of a functional endometrium, the formation of fibrotic tissues, and the obliteration of the uterine cavity. These pathological changes trigger various symptoms, such as oligomenorrhea, amenorrhea, recurrent miscarriage, pelvic pain, and abnormal placentation, leading to subfertility and adverse outcomes of pregnancy [1]. Hysteroscopic adhesiolysis is commonly recommended under these clinical conditions to restore the uterine cavity and facilitate menstrual recovery [2, 3]. However, the recurrence rate of adhesions after surgical intervention can reach up to 62.5%, especially among moderate to severe IUA patients [4]. Multiple ancillary treatments, such as oral conjugated estrogen, amnion graft, intrauterine balloon, and hyaluronic acid gel, are separately or synergistically applied postoperatively to improve the prognosis of patients [5,6,7,8]. Unfortunately, none of these treatments can completely prevent de novo adhesions and effectively increase the regeneration of the endometrium. Several recent studies have suggested that endometrial stromal cells (ESCs) differentiate and incorporate into the endometrial epithelium and contribute to the regeneration of both the stromal and epithelial cell compartments of the uterus, indicating the important role of ESCs and mesenchymal–epithelial transition (MET) in endometrial regeneration [9,10,11,12,13]. Thus, exploring efficient methods to enhance the proliferation of ESCs may be the key to addressing this difficult issue.
Circulating stem cells have been shown to home and infiltrate into the endometrium in response to injury, and endometrium stroma may be the primary target during this process; thus, stem cell therapy, particularly therapy based on mesenchymal stem cells (MSCs), has been considered a potential treatment strategy for the reconstruction of the endometrium following tissue damage and IUA formation [14,15,16]. Although several sources of MSCs are available and have proven to be efficacious, umbilical cord MSCs (UCMSCs), a type of neonatal MSCs, are regarded as an ideal source of stem cells for endometrial repair both in vitro and in vivo [17,18,19]. Compared with other types of MSCs, UCMSCs are noninvasively isolated from medical waste and exhibit various superior biological characteristics, including a greater capacity for self-renewal and differentiation, lower immunogenicity, and better separation efficiency [20]. Our team observed that UCMSC transplantation reduced fibrosis and improved endometrial regeneration in IUA rats, but the associated mechanism remained unclear [18].
Notably, emerging studies have confirmed the crucial effect of MSC-derived exosomes (MSC-exos) as a principal form of cellular paracrine communication in the regeneration of multiple tissues, including the lungs, kidney, liver, spinal cord, cartilage, bone, skin, and heart [21,22,23,24,25,26,27,28]. MSCs-exos are nano-sized, lipid bilayer membrane-enclosed vesicles secreted by MSCs that contain lipids, proteins, and various nucleic acids; these components not only mediate intercellular communication but also mimic the immunomodulatory function and the regenerative capacity of MSCs [29, 30]. However, whether MSC-exos promote the proliferation of endometrial cells remains unclear.
Therefore, in this study, we established a coculture system between primary ESCs and UCMSCs and performed flow cytometry and EdU assays to investigate whether UCMSCs stimulated the DNA synthesis and promoted the cell cycle in allogeneic ESCs via secreted exosomes. In addition, through exosomes uptake assays and CCK-8 assays, we investigated whether ESCs could directly take up USMSCs-exos and whether ESCs treated with USMSC-exos at different concentrations and for different durations exhibited differences in cell viability. We thus demonstrated a novel therapeutic approach for IUAs.
Materials and Methods
Endometrial Tissue Collection and Primary Culture of ESCs
Proliferative phase endometrial tissues (n = 3) were obtained from premenopausal patients who underwent laparoscopic or laparotomic hysterectomy for uterine myomas or early-stage cervical cancer and had no endometrial pathological changes, which was verified by postoperative pathological diagnosis in the Beijing Obstetrics and Gynecology Hospital. Patients with a history of endometriosis, reproductive endocrine disorders, and administration of any hormones or gonadotropin-releasing hormone (GnRH) agonist therapy within 3 months of surgery were excluded. This study complied with the terms listed in the Declaration of Helsinki, and all participants provided informed consent in accordance with a protocol approved by the ethics committee of our hospital before specimen collection (No. IEC-B-03-V01-FJ1). All specimens were separated under sterile conditions and immediately transferred to the laboratory in normal saline.
After being rinsed with PBS several times to remove red blood cells, the tissue was minced into pieces less than 1 mm3 and digested with 0.2% type I collagenase containing 0.005% deoxyribonuclease in an incubator with 5% CO2 at 37 °C for 1 h with continuous shaking. Subsequently, DMEM/F12 containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin was added to an equal volume of cell suspension to stop the digestion. After successive filtration through 100 and 40 μM cell strainers, ESCs were separated from the resulting cell suspension and seeded in culture dishes [31]. After reaching 90% confluence, the primary ESCs were trypsinized, reseeded, and passaged every 2 to 3 days. ESCs within five passages were used for the subsequent experiments.
Identification of ESCs
To identify the specific marker of ESCs and verify their purity, we performed immunofluorescence of cells at the second passage. Briefly, ESCs (5 × 105 cells/well) were grown on cover slides in a 6-well plate. Then, the cells were fixed with 4% paraformaldehyde (PFA) for 30 min and permeabilized with 0.2% Triton X-100 for 10 min at room temperature. Subsequently, the cells were blocked with 0.8% bovine serum albumin at 37 °C for 30 min and incubated with a mixture of primary antibodies against vimentin (mouse,1:100, Proteintech, Wuhan, China) and pan cytokeratin (rabbit, 1:100, Santa Cruz Biotechnology, USA) overnight at 4 °C. Thereafter, the cells were incubated with the secondary antibodies, donkey anti-mouse IgG, Alexa Fluor 488 (1:1000, Invitrogen, USA) and donkey anti-rabbit IgG, Alexa Fluor 594 (1:1000, Invitrogen, USA), for 1 h at room temperature and further immersed in FluoroShield with DAPI (Sigma-Aldrich, USA) to visualize the nuclei. Images were scanned and captured with a laser scanning confocal microscope (TCS SP5, Leica, Germany).
Isolation and Identification of UCMSCs
The primary UCMSCs were generously provided by Lei Wang, Ph.D. (Beijing Cord Blood Bank) and isolated by explant culture as previously described [32]. Briefly, the human umbilical cords were collected from full-term healthy cesarean deliveries and were immediately transferred to the laboratory in sterile normal saline. After being washed with PBS several times to remove blood and mucus, the surface membranes and vessels were isolated and removed. Then, the remaining tissues were dissected into 2 mm × 2 mm pieces and tiled in culture dishes with DMEM/F12 containing 10% FBS and 1% penicillin-streptomycin with 5% CO2 at 37 °C for 7–10 days. Once reached 90% confluence, the supernatant and remnant tissues were removed, and cells adhering to the dishes were digested and passaged.
After the second passage, the differentiation abilities and immunophenotype of the UCMSCs were assessed as follows. For analysis of the adipogenic and osteogenic abilities of cells, UCMSCs were cultured in adipogenic medium for 14 days and osteogenic medium for 21 days following the manufacturer’s instructions (Biological Industries, Israel). Briefly, 6 × 104 UCMSCs at passage 3 were seeded in a 24-well plate and then incubated with induction medium. Subsequently, the induced UCMSCs were fixed with 4% PFA for 30 min at room temperature and then stained with Oil Red O or Alizarin Red solution (Sigma-Aldrich, USA). The cells were observed under a phase-contrast microscope (Nikon, Japan).
For determination of the phenotypic markers of MSCs, the UCMSCs were harvested at passage 3, and surface antigen expression was examined by flow cytometry as previously described [33]. Briefly, the UCMSCs were incubated with mouse antihuman monoclonal antibodies against CD73, CD90, CD105, CD19, CD34, CD45, CD11b, and HLA-DR (Bio Legend, USA) at room temperature for 30 min before flow cytometric analysis (Beckman Coulter, USA).
Coculture of ESCs and UCMSCs
ESCs were cocultured with UCMSCs, UCMSCs+GW4869 (Sigma-Aldrich, USA), or DMEM/F12 medium using a transwell system with a 0.4 μM pore polyester membrane (Costar, Corning, NY, USA) (Fig. 2a). Briefly, UCMSCs were seeded in 6-well plates (5 × 105 per well) or 24-well plates (1 × 105 per well) and incubated for cell adherence. Then, the cell culture medium was changed to exosome-depleted medium containing 20 μM GW4869 or an equal volume of DMSO as a control. Meanwhile, matched transwell chambers with ESCs were inserted into the 6-well plates (2.5 × 105 per chamber) or 24-well plates (1.8 × 104 per chamber) and cocultured with the UCMSCs for 24 h. Subsequently, the supernatant in the upper chambers of the transwell system was removed, and the ESCs were harvested for the following tests.
Cell Proliferation and Cell Cycle Assays
After the ESCs were cocultured with UCMSCs, the cell proliferation was evaluated by 5-ethynyl-2′-deoxyuridine (EdU) incorporation assays and cell cycle analysis.
For the EdU incorporation assays, an EdU cell proliferation kit with Alexa Fluor 488 (Beyotime, Shanghai, China) was used to examine the DNA synthesis of the ESCs in the upper inserts of 24-well plates according to the manufacturer’s instructions. Briefly, the ESCs were incubated with 10 μM EdU for 2 h before the click reaction. Additionally, cell nuclei were stained with 1 × Hoechst 33342 for 10 min before observation under a fluorescence microscope (Nikon, Japan). The cells were counted in three random fields per chamber, and the percentage of EdU-positive cells was calculated using ImageJ software.
For cell cycle analysis, the ESCs were fixed and permeabilized with 70% cold ethanol at −20 °C overnight after they were harvested from the upper inserts of 6-well plates. Subsequently, a cell cycle kit (KeyGEN, Nanjing, China) was used for the following procedures according to the manufacturer’s instructions. Briefly, fixed cells resuspended in 500 μl of propidium iodide/ ribonuclease A (PI/RNase; 9:1) buffer were incubated in the dark at room temperature for 40 min. The samples were analyzed by flow cytometry (EPICS@XL, Beckman Coulter, USA), and debris and doublets were gated out. The DNA contents in the G0/G1, S, and G2/M phases of the cell cycles were estimated using ModFit LT software, and the percentage of cells in the S phase was calculated using the eq. S% = S/(G0/1 + S + G2M).
Isolation and Characterization of UCMSC-Exos
UCMSCs at passage 4 to10 were used to produce exosomes and were cultured in the standard medium until they reached 70–80% confluence. 36 to 48 h before supernatant collection, the culture medium was changed to medium containing exosome-depleted FBS, which was prepared by overnight ultracentrifugation at 100,000×g and 4 °C. UCMSCs-exos were isolated and purified from the culture supernatant by differential ultracentrifugation according to a previously published protocol [34]. Briefly, the culture supernatant was successively centrifuged at 300×g for 10 min, 2000×g for 10 min, and 10,000×g for 30 min at 4 °C to remove dead cells and cell debris. Then, the exosome pellets were collected by ultracentrifugation at 100,000×g for 70 min at 4 °C and washed with sterile PBS followed by another ultracentrifugation step (SW32Ti rotor, Beckman Coulter). Finally, the pellet was resuspended in 100–200 μl PBS and aliquoted for storage at − 80 °C.
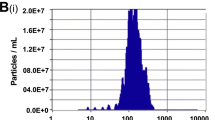
The quantity of exosomes was measured by total protein concentration using the BCA Protein Assay Kit (Pierce, Thermo Scientific, USA) following the manufacturer’s instructions. The exosomes size was determined by nanoparticle tracking analysis (NTA) with ZetaView (Particle Metrix, German). The exosomal specific markers TSG101 (Abcam, USA), Alix (Abcam, USA), and CD63 (Abcam, USA) were examined by western blotting. Furthermore, the exosome morphology was observed via transmission electron microscopy (TEM) (HT7700, Hitachi, Japan).
UCMSC-Exos Labeling and Internalization by ESCs
UCMSC-exos were labeled with the red fluorescent dye PKH26 (Sigma-Aldrich, USA) according to the manufacturer’s instructions. Briefly, PKH26 was diluted in Diluent C before being mixed with exosomes resuspended in sterile PBS. After 10 min of incubation at 37 °C in the dark, exosome-free medium was added to quench the dyeing reaction. Then, the labeled exosomes were enriched by ultracentrifugation at 100,000 × g for 70 min at 4 °C and then washed with sterile PBS followed by another ultracentrifugation step (MLA-150 rotor, Beckman Coulter). Subsequently, the labeled exosomes were added to the subconfluent ESCs seeded in a confocal dish and incubated for 24 h at 37 °C. Finally, the ESCs were fixed with 4% PFA for 10 min, and the cytoskeleton and nuclei of ESCs were stained with Actin-Tracker Green (Beyotime, Shanghai, China) and DAPI, respectively. Images were scanned and captured with a laser scanning confocal microscope (TCS SP5, Leica, Germany).
Cell Viability
ESCs (4 × 103 per well) were seeded in a 96-well plate and incubated for cell adherence. Then, the cells were treated with UCMSC-exos at different concentrations and for different durations, after which the cell viability was measured using a Cell Counting Kit-8 (CCK-8; Dojindo, Japan). Briefly, 10 μl of CCK-8 reagent was added to each test well followed by an incubation at 37 °C for 2 h. Finally, the absorbance was measured at 450 nm with a microplate reader (ELx800, BioTek Instruments Inc., Highland Park, FL, USA).
Statistical Analysis
All experimental data are presented as the means ± standard deviation (SD). Statistical significance was evaluated by Student’s (two-tailed) t test or one-way ANOVA followed by Tukey’s multiple comparison test using SPSS 23.0 and GraphPad Prism 5 software. P < 0.05 was considered to indicate a significant difference.
Results
Characterization of ESCs and UCMSCs
The primary ESCs adhered to the plastic culture dishes after 24 h; short, spindle-shaped and fibroblast-like cells, which reached 90% confluence 1 to 2 days later, were observed (Fig. 1a). The phenotype and purity of the ESCs was detected by immunofluorescence double staining of the intermediate filaments. The primary ESCs expressed the stromal cell marker vimentin and did not express the epithelial cell marker pan-cytokeratin (Fig. 1b).

Characterization of ESCs and UCMSCs. a Morphological characteristics of ESCs. Scale bar = 200 μm. b The isolated ESCs were stained with anti-vimentin antibody, anti-cytokeratin antibody, and DAPI to detect the origin of the cells. Merged images showing that the cells displayed ESC features. Scale bar = 75 μm. c Morphological characteristics of UCMSCs. Scale bar = 200 μm. d Multidifferentiation potential of UCMSCs toward osteogenic and adipogenic lineages. Adipogenesis was determined by the formation of lipid droplets and staining with Oil Red-O. Osteogenesis was determined by the formation of calcium nodules and staining with Alizarin Red. Scale bar = 100 μm. e Flow cytometry analysis of UCMSC surface markers. The cells were positive for CD73, CD90, and CD105 (> 95%) and negative for CD19, CD34, CD45, CD11b, and HLA-DR (< 5%)
Phase-contrast microscopy showed that the primary UCMSCs exhibited a fibroblast-like morphology, a long spindle shape, a whirlpool arrangement, and adherent growth (Fig. 1c). To evaluate the multipotency of these cells, we incubated UCMSCs in differentiation induction medium. The cells incubated in osteogenic medium developed calcified nodules and showed Alizarin Red staining, whereas the cells incubated in adipogenic medium accumulated lipid droplets in the cytoplasm and showed Oil Red O staining (Fig. 1d). Furthermore, flow cytometric analysis showed that the cells had high expression of CD73, CD90, and CD105 and low expression of CD19, CD34, CD45, CD11b, and HLA-DR (Fig. 1e). All of these characteristics were consistent with those of MSCs [35].
UCMSCs Regulate the Proliferation of ESCs Via Exosomes
To explore the ability of UCMSCs-derived exosomes to enhance ESC proliferation, we cocultured ESCs with UCMSCs, UCMSCs with 20 μM GW4869, or medium in a transwell system that only allowed the passage of molecules with a diameter < 0.4 μm, such as exosomes (Fig. 2a). Subsequently, cell cycle analysis and EdU incorporation assays were conducted on ESCs harvested from transwell inserts after 24 h of coculture. The cell cycle analysis results showed that the proportion of ESCs in S phase after coculture with UCMSCs significantly increased from 2.75 ± 0.38% to 19.26 ± 0.85% compared to that of cells cultured without UCMSCs (P < 0.01, Fig. 2b, c), whereas this value significantly decreased from 19.26 ± 0.85% to 16.91 ± 1.26% after coculture with UCMSCs and GW4869 (P < 0.05, Fig. 2b, c). Moreover, the EdU incorporation assays showed a similar trend; the EdU-labeled cells in the UCMSCs group increased from 9.31 ± 0.09% to 23.35 ± 0.97% compared with those of the nil control group (P < 0.01, Fig. 2d, e), while this percentage decreased from 23.35 ± 0.97% to 19.86 ± 1.58% after addition of GW4869 (P < 0.05, Fig. 2d, e). In general, these results indicated that UCMSCs promote the proliferation of ESCs at least partly through exosomes.

UCMSCs regulate the proliferation of ESCs via exosomes. a Diagrams of the coculture system of ESCs with UCMSCs, UCMSCs+GW4869, or nil control medium. b Flow cytometry for cell cycle analysis of ESCs from the upper inserts of the coculture system. c Percentages of the cell population in the G0/G1, S, and G2/M phases of the cell cycle. d EdU incorporation assays to assess the cell proliferation of ESCs from the upper inserts of the coculture system. e Percentages of EdU positive cells. Bars = the means ± SD, n = 3. *, P < 0.05, **, P < 0.01
Characterization and Internalization of UCMSC-Exos
To investigate the direct communication between UCMSC-exos and ESCs, we extracted exosomes from the culture supernatant of UCMSCs through differential ultracentrifugation and identified them by western blot analysis, TEM, and NTA. As expected, the western blot results demonstrated that the specific exosomal marker proteins Alix, CD63, and TSG101 were present in these exosomes (Fig. 3a). The TEM revealed that the purified UCMSC-exos had a typical cup-shaped morphology with a monolayer membrane and a diameter of approximately 100 nm (Fig. 3b). Additionally, the NTA showed that the particle size mainly ranged from 60 to 200 nm in diameter, representing 99.3% of isolated exosomes in the tested sample, and the main peak of particle size was in the typical size range of exosomes (Fig. 3c). Finally, the scanning confocal microscopy analysis showed that UCMSC-exos labeled with PKH26 were incorporated into ESCs after incubation for 24 h (Fig. 3d). In summary, these results confirmed that the particle preparations derived from UCMSCs were exosomes, and could be internalized by ESCs.

Characterization and internalization of UCMSC-derived exosomes. a Detection of specific exosomal marker proteins, including Alix, CD63, and TSG101, via western blot analyses of UCMSC-exosomes; whole cell lysate was used as a control. b Representative morphology of UCMSC exosomes by transmission electron microscopy. Scale bar = 100 nm. c UCMSC-exosomes concentration and particle size distribution were determined by nanoparticle tracking analysis. d UCMSC-exosomes were labeled with PKH26 (red), and the cytoskeleton and nuclei of ESCs were stained with Actin-Tracker Green and DAPI, respectively. Merged images showing that the UCMSC-exosomes were internalized by ESCs after 24 h incubation. Bar = 50 μm
UCMSC-Exos Promote the Proliferation of ESCs in a Dose-Dependent Manner
To elucidate the mechanism of UCMSC-exos-stimulated ESC growth, we performed CCK-8 assays after the ESCs were directly treated with UCMSC-exos. First, the ESCs were treated with UCMSC-exos at different concentrations (0, 2.5, 5, 10, 20, and 40 μg/ml) for 24 h. The absorbance values at 450 nm were 0.94 ± 0.02, 0.95 ± 0.07, 0.98 ± 0.04, 1.00 ± 0.06, 1.04 ± 0.03, and 1.09 ± 0.03, showing that the viability of ESCs in the group treated with high doses (20 or 40 μg/ml) of UCMSC-exos significantly increased compared with that of the negative control and low-dose groups (0, 2.5 or 5 μg/ml) (Fig. 4a). Next, 20 μg/ml was chosen as the concentration for the UCMSC-exos treatment group and the viability of the ESCs was determined at six different time points (0, 12, 24, 36, 48, and 60 h). The absorbance values at 450 nm in the UCMSC-exos group were 0.44 ± 0.02, 0.79 ± 0.02, 1.08 ± 0.13, 1.31 ± 0.10, 1.66 ± 0.02, and 1.70 ± 0.06, while the control group exhibited values of 0.44 ± 0.03, 0.68 ± 0.05, 0.84 ± 0.10, 1.12 ± 0.03, 1.42 ± 0.13, and 1.34 ± 0.10. Thus, the effect of UCMSC-exos was detected at 12 h and continued to 60 h, resulting in a significant improvement in the proliferation potential of ESCs compared with that of the control group (Fig. 4b). These findings revealed that UCMSC-exos rapidly promoted the proliferation of ESCs after they were added to the culture system and that the effect lasted for 60 h. Additionally, UCMSCs might play a key role in activating ESC growth through their secreted exosomes in a dose-dependent manner.

UCMSC-derived exosomes promote proliferation of ESCs in a dose-dependent manner. a The viability of the ESCs was measured by CCK-8 assays after treatment with UCMSC-exosomes (UCMSCs-exos) at different concentrations for 24 h. The absorbance values at 450 nm in the control group and group treated with the 2.5, 5, 10, 20, and 40 μg/ml UCMSCs-exos were 0.94 ± 0.02, 0.95 ± 0.07, 0.98 ± 0.04, 1.00 ± 0.06, 1.04 ± 0.03, and 1.09 ± 0.03, respectively. Bars = the means ± SD, n = 4. *, P < 0.05, **, P < 0.01. b The viability of ESCs was measured by a CCK-8 assay at different time points after treatment with 20 μg/ml UCMSC-exos. The absorbance values at 450 nm in UCMSC-exos group after 0, 12, 24, 36, 48, 60 h were 0.44 ± 0.02, 0.79 ± 0.02, 1.08 ± 0.13, 1.31 ± 0.10, 1.66 ± 0.02, and 1.70 ± 0.06. The absorbance values at 450 nm in control group after 0, 12, 24, 36, 48, 60 h were 0.44 ± 0.03, 0.68 ± 0.05, 0.84 ± 0.10, 1.12 ± 0.03, 1.42 ± 0.13, 1.34 ± 0.10, respectively. Bars = the means ± SD, n = 4. *, P < 0.05, **, P < 0.01
Discussion
In this study, for the first time, we demonstrated that UCMSCs could accelerate the proliferation of ECSs via secreted exosomes by enhancing DNA synthesis and promoting the cell cycle. Moreover, we observed that UCMSC-exos could be internalized by ESCs directly and stimulate ESC growth in a dose-dependent manner, with the effects occurring in a short period of time.
Over the past decade, MSCs have been proposed to be an ideal source for cell-based therapy to promote the scar-free healing of injured endometrium during the acute stage and the chronic phase of IUAs [18, 36]. A pilot clinical trial using MSCs was recently conducted, but the underlying mechanism associated with their efficacy is still unclear [19]. At present, the paracrine effects, differentiation capacity, and immunomodulation potential of MSCs are believed to be the main therapeutic mechanisms through which they support the regenerative processes in injured tissues [37]. Alawadhi et al. reported that bone marrow MSCs were recruited to endometrium in response to injury and improved fertility outcomes of IUA mice for the first time, but only a small number of exogenous MSCs were detected in the endometrium of the transplanted mice [38]; in addition, several studies have indicated that stem cell engraftment of non-hematopoietic lineages did not contribute to endometrial cell lineages in rodent models [15, 39]. Hence, the previous studies excluded the differentiation and clonal expansion of MSCs in the injured endometrium and highlighted MSCs as site-specific producers of trophic factors in endometrial regeneration. Although Shi et al. reported that MSCs could differentiate into endometrial-like cells inducing them in conditioned medium containing 17β-estradiol and 8-Br-cAMP, this study did not confirm that allogeneic MSCs could spontaneously differentiate into functional endometrium [40], and the paracrine effects of MSCs that might impact the proliferation of endometrial cells should be investigated in further detailed analyses. Thus, we used a coculture system that only allowed for the passage of small molecules and confirmed that UCMSCs could promote ESC growth mainly via paracrine factors, not by direct replacement of ESCs.
Recently, mounting evidence has shown that exosomes secreted from MSCs, acting as paracrine mediators, could recapitulate some of the features of MSCs [29, 30]. Thus, to explore the function of UCMSC-exos, we used GW4869, a known inhibitor of neutral sphingomyelinase, which is required for exosome biogenesis, to disturb the exosomes secretion of UCMSCs [41]. The results revealed that the DNA synthesis and cell cycle progression of ESCs was inhibited in the coculture system, whereas the proliferation of ESCs in the exosome-depletion group was significantly greater than that in the control group. These results indicated that UCMSC-exos secretion might not be completely blocked by an inhibitor of neutral sphingomyelinase, and other methods, such as Rab GTPase knockout, should be applied to inhibit exosome biogenesis if necessary [42]; in addition, other soluble trophic factors (growth factors, cytokines, and chemokines, etc.) secreted by UCMSCs might simultaneously affect ESC growth simultaneously during this process [37].
Next, we successfully isolated and purified exosomes from UCMSC culture supernatants, and UCMSCs-exos showed several potential advantages compared to MSC-based cell therapy [30, 43]. First, NTA showed that their diameters were much smaller than those of UCMSCs, which allowed them to circulate easily and avoided capillary blockage by MSCs. Second, TEM revealed that UCMSC-exos were encapsulated in a monolayer membrane, which could protect their integrity and contents and confer pharmacokinetic advantages versus purified cytokine administration. Third, UCMSC-exos could be quantified by BCA assays after extraction and purification, allowing for an accurate and convenient measurement of the exosome dosage and demonstrating their potential as off-the-shelf products in the future. Furthermore, we observed that the UCMSC-exos stained with PKH26, a lipophilic membrane dye, were taken up by ESCs after incubation, supporting the direct effects of UCMSC-exos on proliferation of ECSs. In summary, all the above features would be conducive to UCMSC-exos participating in endometrial healing and reconstruction.
Finally, we showed that UCMSCs-exos enhanced ESC viability and growth in a dose-dependent manner, with the effects occurring rapidly and lasting at least 60 h. This mechanism of UCMSC-exos was consistent with the findings of several previous studies that examined exosome efficacy [44,45,46,47]. Notably, multiple studies have reported that the stromal region might be the major target when MSCs migrate to the endometrium in response to injury, and ESCs support the regeneration of both the stromal and epithelial cell compartments of the uterus [9,10,11,12,13, 15]. These results suggested that the proliferation of ESCs and MET might be responsible for the mechanisms underlying the MSCs involvement in endometrial regeneration. Furthermore, recent studies have demonstrated that MSC-derived exosomes could reverse epithelial–mesenchymal transition (EMT) and ameliorate fibrosis in IUA rodent models [48, 49]. Therefore, the ability of UCMSC-exos to promote ESC proliferation in vitro will help elucidate the novel mechanisms associated with MSC-exo therapy in damaged endometrium repair and provide a new alternative for IUA therapy.
Although this is the first study showing that allogeneic UCMSC-exos promote the proliferation of ESCs in vitro, this study had several limitations. Initially, the primary ESCs expressed vimentin, indicating that these cells are originated from mesenchyme of endometrium, but it was insufficient to distinguish them from endometrial mesenchymal stem cells (eMSCs). The eMSCs are a specific subpopulation of ESCs, which function as pericytes and might be the principal component of ESCs that participate in MET and promote endometrial repair and regeneration [9, 50]. Thus, immunofluorescence and flow cytometry sorting using specific markers of eMSCs, such as CD146, PDGFR-β, and SUSD2, should be carried out to isolate them from the primary ESCs for further study on this topic in the future. What is more, because our study only focused on in vitro experiments, further in vivo experiments should be conducted to explore whether UCMSC-exos stimulate the proliferation of ESCs in situ and whether MET occurs after UCMSC-exos treatment in future studies. Finally, UCMSC-exos predominantly function via horizontal transfer of mRNAs, miRNAs, and proteins that subsequently modulate the expression of various genes in target cells and result in multiple phenotypes [43]. Consequently, sequencing and analysis of the transcriptome in both UCMSCs and ESCs is necessary to identify the specific signaling pathways and molecular mechanisms involved in this process.
Taken together, our findings suggested that UCMSCs could promote the proliferation of ESCs via secreted exosomes and that UCMSCs-exos could be internalized by ESCs and enhance their viability and growth in a dose-dependent manner with a rapid onset. These results might represent a promising strategy for endometrial repair and regeneration.
References
Khan Z, Goldberg JM. Hysteroscopic Management of Asherman's syndrome. J Minim Invasive Gynecol. 2018;25(2):218–28.
AAGL Elevating Gynecologic Surgery. AAGL practice report: practice guidelines on intrauterine adhesions developed in collaboration with the European Society of Gynaecological Endoscopy (ESGE). J Minim Invasive Gynecol. 2017;24(5):695–705.
Duan H. Chinese Society of Obstetrics and Gyneocology, Chinese Medical Association. Expert consensus on the diagnosis and management of intrauterine adhesions in China. Zhonghua Fu Chan Ke Za Zhi. 2015;50:881–7 [in Chinese].
Yu D, Wong YM, Cheong Y, Xia E, Li TC. Asherman syndrome-one century later. Fertil Steril. 2008;89:759–79.
Healy MW, Schexnayder B, Connell MT, Terry N, DeCherney A, Csokmay JM, et al. Intrauterine adhesion prevention after hysteroscopy: a systematic review and meta-analysis. Am J Obstet Gynecol. 2016;215(3):267–75.
Gan L, Duan H, Sun FQ, Xu Q, Tang YQ, Wang S. Efficacy of freeze-dried amnion graft following hysteroscopic adhesiolysis of severe intrauterine adhesions. Int J Gynaecol Obstet. 2017;137(2):116–22.
Zhu R, Duan H, Gan L, Wang S. Comparison of intrauterine suitable balloon and Foley balloon in the prevention of adhesion after Hysteroscopic Adhesiolysis. Biomed Res Int. 2018;2018:9494101.
Hooker AB, De LR, Pm VDV, et al. Prevalence of intrauterine adhesions after the application of hyaluronic acid gel after dilatation and curettage in women with at least one previous curettage: short-term outcomes of a multicenter, prospective randomized controlled trial. Fertil Steril. 2017;107(5):1223–31.
Yin M, Zhou HJ, Lin C, et al. CD34(+)KLF4(+) stromal stem cells contribute to endometrial regeneration and repair. Cell Rep. 2019;27(9):2709–24.
Owusu-Akyaw A, Krishnamoorthy K, Goldsmith LT, Morelli SS. The role of mesenchymal-epithelial transition in endometrial function. Hum Reprod Update. 2019;25(1):114–33.
Huang CC, Orvis GD, Wang Y, Behringer RR. Stromal-to-epithelial transition during postpartum endometrial regeneration. PLoS One. 2012;7:e44285.
Patterson AL, Zhang L, Arango NA, Teixeira J, Pru JK. Mesenchymal-to-epithelial transition contributes to endometrial regeneration following natural and artificial decidualization. Stem Cells Dev. 2013;22:964–74.
Cousins FL, Murray A, Esnal A, Gibson DA, Critchley HOD, Saunders PTK. Evidence from a mouse model that epithelial cell migration and mesenchymal–epithelial transition contribute to rapid restoration of uterine tissue integrity during menstruation. PLoS One. 2014;9:e86378.
Taylor HS. Endometrial cells derived from donor stem cells in bone marrow transplant recipients. JAMA. 2004;292(1):81–5.
Du H, Naqvi H, Taylor HS. Ischemia/reperfusion injury promotes and granulocyte-colony stimulating factor inhibits migration of bone marrow-derived stem cells to endometrium. Stem Cells Dev. 2012;21(18):3324–31.
Santamaria X, Mas A, Cervelló I, Taylor H, Simon C. Uterine stem cells: from basic research to advanced cell therapies. Hum Reprod Update. 2018;24(6):673–93.
Yang X, Zhang M, Zhang Y, Li W, Yang B. Mesenchymal stem cells derived from Wharton jelly of the human umbilical cord ameliorate damage to human endometrial stromal cells. Fertil Steril. 2011;96(4):1029–36.
Tang YQ, Gan L, Xu Q, Wang S, Li JJ, Duan H. Effects of human umbilical cord mesenchymal stem cells on intrauterine adhesions in a rat model. Int J Clin Exp Pathol. 2016;9(11):12119–29.
Cao Y, Sun H, Zhu H, Zhu X, Tang X, Yan G, et al. Allogeneic cell therapy using umbilical cord MSCs on collagen scaffolds for patients with recurrent uterine adhesion: a phase I clinical trial. Stem Cell Res Ther. 2018;9(1):192.
Araújo AB, Salton GD, Furlan JM, Schneider N, Angeli MH, Laureano ÁM, et al. Comparison of human mesenchymal stromal cells from four neonatal tissues: amniotic membrane, chorionic membrane, placental decidua and umbilical cord. Cytotherapy. 2017;19(5):577–85.
Khatri M, Richardson LA, Meulia T. Mesenchymal stem cell-derived extracellular vesicles attenuate influenza virus-induced acute lung injury in a pig model. Stem Cell Res Ther. 2018;9(1):17.
Eirin A, Zhu XY, Puranik AS, Tang H, McGurren K, van Wijnen A, et al. Mesenchymal stem cell-derived extracellular vesicles attenuate kidney inflammation. Kidney Int. 2017;92(1):114–24.
Haga H, Yan IK, Takahashi K, Matsuda A, Patel T. Extracellular vesicles from bone marrow-derived Mesenchymal stem cells improve survival from lethal hepatic failure in mice. Stem Cells Transl Med. 2017;6(4):1262–72.
Ruppert KA, Nguyen TT, Prabhakara KS, Toledano Furman NE, Srivastava AK, Harting MT, et al. Human Mesenchymal stromal cell-derived extracellular vesicles modify microglial response and improve clinical outcomes in experimental spinal cord injury. Sci Rep. 2018;8(1):480.
Zhang S, Chu WC, Lai RC, Lim SK, Hui JH, Toh WS. Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration. Osteoarthr Cartil. 2016;24(12):2135–40.
Cosenza S, Toupet K, Maumus M, et al. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics. 2018;8(5):1399–410.
Zhang J, Guan J, Niu X, et al. Exosomes released from human induced pluripotent stem cells-derived MSCs facilitate cutaneous wound healing by promoting collagen synthesis and angiogenesis. J Transl Med. 2015;13:49.
Chen CW, Wang LL, Zaman S, Gordon J, Arisi MF, Venkataraman CM, et al. Sustained release of endothelial progenitor cell-derived extracellular vesicles from shear-thinning hydrogels improves angiogenesis and promotes function after myocardial infarction. Cardiovasc Res. 2018;114(7):1029–40.
Witwer KW, Van Balkom BWM, Bruno S, et al. Defining mesenchymal stromal cell (MSC)-derived small extracellular vesicles for therapeutic applications. J Extracell Vesicles. 2019;8(1):1609206.
Wiklander OPB, Brennan MÁ, Lötvall J, Breakefield XO, El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci Transl Med. 2019;11(492).
Li HWR, Li YX, Li TT, Fan H, Ng EH, Yeung WS, et al. Effect of ulipristal acetate and mifepristone at emergency contraception dose on the embryo-endometrial attachment using an in vitro human trophoblastic spheroid and endometrial cell co-culture model. Hum Reprod. 2017;32(12):2414–22.
Mushahary D, Spittler A, Kasper C, Weber V, Charwat V. Isolation, cultivation, and characterization of human mesenchymal stem cells. Cytometry A. 2018;93(1):19–31.
Thaweesapphithak S, Tantrawatpan C, Kheolamai P, Tantikanlayaporn D, Roytrakul S, Manochantr S. Human serum enhances the proliferative capacity and immunomodulatory property of MSCs derived from human placenta and umbilical cord. Stem Cell Res Ther. 2019;10(1):79.
Théry C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006; Chapter 3:Unit 3.22.
Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–7.
Zhang L, Li Y, Guan CY, et al. Therapeutic effect of human umbilical cord-derived mesenchymal stem cells on injured rat endometrium during its chronic phase. Stem Cell Res Ther. 2018;9(1):36.
Andrzejewska A, Lukomska B, Janowski M. Concise review: Mesenchymal stem cells: from roots to boost. Stem Cells. 2019;37(7):855–64.
Alawadhi F, Du H, Cakmak H, Taylor HS. Bone marrow-derived stem cell (BMDSC) transplantation improves fertility in a murine model of Asherman's syndrome. PLoS One. 2014;9(5):e96662.
Ong YR, Cousins FL, Yang X, Mushafi AAAA, Breault DT, Gargett CE, et al. Bone marrow stem cells do not contribute to endometrial cell lineages in chimeric mouse models. Stem Cells. 2018;36(1):91–102.
Shi Q, Gao J, Jiang Y, et al. Differentiation of human umbilical cord Wharton's jelly-derived mesenchymal stem cells into endometrial cells. Stem Cell Res Ther. 2017;8(1):246.
Xiao C, Wang K, Xu Y, Hu H, Zhang N, Wang Y, et al. Transplanted Mesenchymal stem cells reduce Autophagic flux in infarcted hearts via the Exosomal transfer of miR-125b. Circ Res. 2018;123(5):564–78.
Song L, Tang S, Han X, et al. KIBRA controls exosome secretion via inhibiting the proteasomal degradation of Rab27a. Nat Commun. 2019;10(1):1639.
Phinney DG, Pittenger MF. Concise review: MSC-derived Exosomes for cell-free therapy. Stem Cells. 2017;35(4):851–8.
Zhu Z, Zhang Y, Zhang Y, et al. Exosomes derived from human umbilical cord mesenchymal stem cells accelerate growth of VK2 vaginal epithelial cells through MicroRNAs in vitro. Hum Reprod. 2019;34(2):248–60.
Bari E, Ferrarotti I, Di Silvestre D, et al. Adipose Mesenchymal Extracellular Vesicles as Alpha-1-Antitrypsin Physiological Delivery Systems for Lung Regeneration. Cells. 2019;8(9):E965.
Stronati E, Conti R, Cacci E, Cardarelli S, Biagioni S, Poiana G. Extracellular Vesicle-Induced Differentiation of Neural Stem Progenitor Cells. Int J Mol Sci. 2019;20(15):3691.
Sjöqvist S, Ishikawa T, Shimura D, et al. Exosomes derived from clinical-grade oral mucosal epithelial cell sheets promote wound healing. J Extracell Vesicles. 2019;8(1):1565264.
Ebrahim N, Mostafa O, El Dosoky RE, et al. Human mesenchymal stem cell-derived extracellular vesicles/estrogen combined therapy safely ameliorates experimentally induced intrauterine adhesions in a female rat model. Stem Cell Res Ther. 2018;9(1):175.
Yao Y, Chen R, Wang G, Zhang Y, Liu F. Exosomes derived from mesenchymal stem cells reverse EMT via TGF-β1/Smad pathway and promote repair of damaged endometrium. Stem Cell Res Ther. 2019;10(1):225.
Gargett CE, Schwab KE, Deane JA. Endometrial stem/progenitor cells: the first 10 years. Hum Reprod Update. 2016;22(2):137–63.
Funding
This study was funded by the National Key Research and Development Program of China (2018YFC1004803) and the Natural Science Foundation of China (Grant No. 81801403), and was supported by Capital Medical University (1192070309) and Beijing Obstetrics and Gynecology Hospital, Capital Medical University (FCYY201823).
Author information
Authors and Affiliations
Contributions
Cheng-Xiao Lv conceived, drafted, and revised the article, carried out the experiments, and performed the statistical analysis; Hua Duan participated in the study design and revision; Sha Wang, Lu Gan and Qian Xu participated in patient recruitment and sample collection. All authors approved the final version of the article.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lv, CX., Duan, H., Wang, S. et al. Exosomes Derived from Human Umbilical Cord Mesenchymal Stem Cells Promote Proliferation of Allogeneic Endometrial Stromal Cells. Reprod. Sci. 27, 1372–1381 (2020). https://doi.org/10.1007/s43032-020-00165-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43032-020-00165-y