
Abstract
Autophagy represents an intracellular defense mechanism equipped within each eukaryotic cells to enable them to cope with variety of physical, chemical, and biological stresses. This mechanism helps to restore the homeostasis and preserve the cellular integrity and function of the cells. In these conditions, such as hypoxia, nutrient deprivation, inhibition of protein synthesis or microbial attack, the process of autophagy is upregulated to maintain cellular homeostasis. The role of autophagy in cancer is an intriguing topic which needs further exploration. This process of autophagy has been many times referred as a double-edged sword in the process of tumorigenesis. In the initial stages, it may act as a tumor suppressor and enable to quench the damaged organelles and harmful molecules generated. In more advanced stages, autophagy has been shown to act as a tumor-promoting system as it may help the cancer cells to cope better with stressful microenvironments. Besides this, autophagy has been associated with development of resistance to anticancer drugs as well as promoting the immune evasion in cancer cells, representing a serious obstacle in cancer treatment and its outcome. Also, autophagy is associated with hallmarks of cancer that may lead to activation of invasion and metastasis. The information on this twin role needs further exploration and deeper understanding of the pathways involved. In this review, we discuss the various aspects of autophagy during tumor development, from early to late stages of tumor growth. Both the protective role of autophagy in preventing tumor growth and the underlying mechanisms adopted with evidence from past studies have been detailed. Further, the role of autophagy in conferring resistance to distinct lung cancer treatment and immune shielding properties has also been discussed. This is essential for further improving on treatment outcome and success rates.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lung cancer represents one of the leading causes of deaths, making up almost 25% of all cancer-related deaths (WHO 2018). Worldwide, lung cancer is the second most commonly diagnosed cancer (Sung et al. 2021). It is broadly divided into two categories: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), both of which receive different therapeutic interventions. About 13% of all lung cancers are SCLC, and 84% are NSCLC. In 2020, an estimated 1,796,144 people died worldwide from the disease (Ye et al. 2022).
Lung cancer as discussed can be broken down into two main types: SCLC and NSCLC.SCLC is typically caused by tobacco intake and smoking. It starts in the bronchi or the airways and branches off to smaller structures. It quickly spreads to other parts of the body, and thus, aggressive treatment is required to block its metastasis formation. It can further be divided as small cell carcinoma and the affected cells look flat under the microscope (Dela Cruz et al. 2011; Zheng 2016). The second type of SCLC is the combined small cell carcinoma that refers to tumor growth containing flat small cell carcinoma cells along with some cells of small cell lung cancer cells. Talking of NSCLC, this category represents the most common type of lung cancers and has a higher survival rate as well. Also, it typically grows at a slower rate than SCLC (Chen et al. 2014; Oser et al. 2015). There are three main types of NSCLC: adenocarcinoma of the lung, squamous cell, and large-cell undifferentiated carcinoma (LCUC). Adenocarcinoma of the lung is the most common type (40–70%) and mainly affects the outer regions of lungs and in mucus-secreting glands (Myers and Wallen 2022). The second type, i.e., squamous cell cancer accounts for 30% of the reported cases of non-small lung cancers and this type is mostly associated with smoking starting from the center of the lung (Sabbula et al. 2022). The third category, i.e., LCUC (10–15% of cases) can develop in any part of the lung and is considered to spread and progress very fast. NSCLC is less aggressive, but it is mostly identified at later stages which again makes the treatment a challenge (Chen et al. 2014; Liu et al. 2017a, b, c).
The challenges associated with lung cancer are manyfold and arise from the late occurrence of symptoms by the patients leading to delayed diagnosis and poor prognosis. Further, resistance to both radiation and chemotherapy adds to the existing complication. In this review, we focus on the essential role of autophagy in lung cancer and its underlying mechanisms which changes as per the stage of cancer. Determining these molecular mechanisms of NSCLC will enable to discover new targets and biomarkers aiding in the development of better, more specific therapies.
Autophagy: overview
Before diving deep into the connection between autophagy and cancer cell metabolism, let’s have a brief overview of the physiological role played by autophagy. Autophagy represents a cellular mechanism for the removal of damaged proteins/organelles allowing the turnover and recycling, through a lysosome-dependent degradation pathway in the cell (Parzych and Klionsky 2014). In this process, autophagy first sequesters cargo from the cytosol into double membrane structures, i.e., autophagosomes. These then fuse to lysosomes, wherein the contents get degraded and recycled into metabolic precursors given back to enter the metabolic cycles. This way, autophagy plays a crucial role in clearance of subcellular materials and its recycling enabling cellular detoxification (Glick et al. 2010; Das et al. 2012). Dysfunction in autophagic mechanism eventually leads to pathogenic alterations thus linking to onset of many diseases. Autophagy impacts cell metabolism, cell growth, cell death, organelle numbers and their quality, various proteins, essential pathways, etc., and helps to maintain cell homeostasis in diverse ways (Das et al. 2012; Chun and Kim 2018).
Detailing out the mechanism and regulatory pathways of autophagy, its proceeds through a series of sequential steps. This involves the rearrangements of membrane components derived from the Golgi bodies, endoplasmic reticulum (ER), plasma membrane and mitochondrial membranes. The nucleation of the autophagic membrane of ER and Golgi occurs which converges the degraded components, misfolded or damaged proteins, carcinogenic molecules, etc. This elongates forming the isolation membrane of phagophore. The phagophore further expands and matures forming autophagosome. It then fuses with the lysosomes to form a single membrane structure called autolysosomes. This protein involves participation of proteins such as class C subset of vacuolar protein sorting (Vps) proteins, small GTPases, lysosome-associated membrane proteins (e.g., LAMP2), etc. The cargo within the autolysosomes is then acted upon by lysosomal enzymes for degradation under acidic conditions. This is done through proton pump that reside in the lysosomal membranes that pump H + ions from the cytosol into the lysosome (Mizushima 2007; Kinchen and Ravichandran 2008). Finally, the degraded products consisting of simple sugars, free amino acids, nucleotides, free fatty acids, etc., get released for recycling and reuse by lysosomal permeases back into the cell cytoplasm (Hurley and Young 2017; Chun and Kim 2018; Dikic and Elazar 2018).
Regulatory mechanism involved
Autophagy is a highly conserved pathway through evolution and plays an important role in maintaining cellular homeostasis. It has been best characterized in yeast and drosophila. Autophagy is upregulated in response to extra- or intracellular stimuli and signals such as hypoxia, glucose starvation or amino acid starvation, growth factor deprivation, ER stress, and pathogen infection (Mizushima and Komatsu 2011). The regulation of autophagy involves the participation of many autophagy-related proteins (Atg) at various steps. The Atg proteins function during initiation, cargo recognition, fusion and vesicle formation, final breakdown, and final release, etc. (Feng et al. 2014). Autophagy under normal conditions occurs at low levels owing to the central inhibition of autophagy by the serine/threonine protein kinase TOR (target of rapamycin). The induction of autophagy requires efficient mechanism to inhibit the TOR pathway (He and Klionsky 2009). In yeast, TOR inhibition occurs by starvation or by rapamycin, this leads to activation of another serine /threonine protein kinase, i.e., Atg1 which binds to Atg13 and Atg17. This complex formed in yeast promotes the formation of PAS, which is the site of autophagosome formation and is also termed the phagophore assembly site (Cheong et al. 2005; Kabeya et al. 2005).
The mammalian homologs of Atg1 are the Unc-51-like kinase 1 (ULK1) and -2 (ULK2). The homolog of yeast Atg17 in mammals is the FIP200 (the focal adhesion kinase family interacting protein of 200 kD). This forms a complex with ULK-1 and -2 and also with mammalian Atg13 (Jung et al. 2009). This kinase complex is essential for the formation of phagophore. Upon normal condition (nutrient-rich conditions), mTOR phosphorylates ULKs and Atg13 leading to their inhibition. But under stress, such as starvation, intracellular levels of AMP increase that leads to activation of the kinase of AMP (i.e., AMPK). AMPK then causes the activation of ULK 1/2 which further lead to activation and complex formation with Atg13 and FIP200. The ULK1-complex includes ULK1, ULK2, Atg 101, FIP200, and Atg 13. This leads to phagophore nucleation (Hosokawa et al. 2009). Next, this activated complex so formed localizes to the phagophore and activates another complex, i.e., Beclin-1-class III phosphatidylinositol 3-kinase (PI3K) complex which includes PI, VPS34, Atg14L, VPS15, and beclin 1(BECN1). This activated complex promotes the conversion of phosphatidylinositol to phosphatidylinositol 3-phosphate (PI3P) producing a pool of PI3P. PI3P recruits additional accessory protein factors such as the double FYVE-containing protein-1 (DFCP1) and the WD-repeat protein in association with phosphoinositides (WIPI) proteins, i.e., WIP2I. WIP2I simultaneously recruits the Atg 12–Atg16L conjugation complex thus promoting expansion of the phagophore (Wirth et al. 2013; Dooley et al. 2014).
The next step is the elongation of isolation membrane and this process involves participation of two ubiquitin-like conjugation systems: Atg5-Atg12 and type II light chain 3 (LC3) pathways. In the first pathway, Atg12 activation occurs by Atg7 which leads to formation of Atg5-Atg12 complex. This complex conjugates to Atg16 to form a complex called Atg5-Atg12-Atg16 which has E3 ligase activity (Romanov et al. 2012). In the LC3 pathway, firstly, the C-terminus cleavage of LC3 occurs by Atg4B to generate soluble LC3-I. LC3-I is then conjugated to phosphatidylethanolamine (PE) by Atg7, Atg3 and the Atg5-Atg12-Atg16 complex, producing the LC3-II conjugated form. LC3 II is located in the inner and outer membrane of the autophagosome to bind substrates selected for degradation (Kroemer et al. 2010). In phagosome fusion and closure, members of the endosomal sorting complex required for transport (ESCRT), mainly CHMP2A and the vacuolar protein sorting-associated-4 (VPS4) actively participate. The translocation of CHMP2A to the edge of phagophore promotes closure of the membranes. Also, VPS4 locates on the outer leaf of nascent autophagosomal membranes to promote disassembly of ESCRT molecule. Microtubule motors of the dynein families also participate in the mobilization of autophagosomes (Rai et al. 2016). Experiments show that inhibition of CHMP2A or VPS4 is associated with impaired phagophore closure and late or no fusion with lysosomes (Takahashi et al. 2018). Finally, in the maturation step of autophagosomes, LC3-II gets delipidated and autophagolysosomal structures are formed due to fusion, leading to degradation of autophagosome content by the various hydrolytic enzymes. Rab GTPases promote the process of docking and fusion of lysosomes and recruit soluble NSF attachment protein receptors, i.e., SNARE family (Nair et al. 2011; Takáts et al. 2013). LC3B-II located in the outer membrane gets recycled back by Atg4B and the LC3B-II associated within the inner membrane of the autophagosome is degraded by lysosomal enzymes (Yang et al. 2010). A schematic diagram of the various steps involved in autophagy and important proteins involved is depicted in Fig. 1.

Schematic overview of autophagy and steps involved: initiation, nucleation, elongation, maturation (PAS), autophagosome completion, fusion with lysosome and final degradation of cargo and recycling. (ULK: Unc-51 Like Autophagy Activating Kinase; ATG: autophagy-related proteins, FIP: the focal adhesion kinase family interacting protein, LC3: type II light chain 3, ER: endoplasmic reticulum, Vps: vacuolar protein sorting proteins)
Autophagy and apoptosis: crosstalk
Both apoptosis and autophagy are important self-regulatory systems through which our body cells respond to stress conditions and maintain homeostatic balance. They both act antagonistically under specific conditions. Normally, autophagy precedes the event of apoptosis but there is debate and some declare autophagy as a mechanism of apoptosis itself. Talking of apoptosis, it is a subtype of programmed cell death mechanisms involved in removing damaged cells from the healthy tissue and limiting their proliferation. The intrinsic pathways of apoptosis may be triggered by DNA damage, replication errors, accumulation of reactive oxygen radicals, mitotic defects, etc., while the extrinsic pathways get activated by external stress factors inducing the expression of specific death signaling receptors on damaged cell surfaces such as tumor necrosis factor receptor, i.e., TNFR1A and FAS (Elmore et al. 2007). This leads to death-inducing signaling complex formation and release of apoptogenic molecules causing final cell death. The crosstalk between autophagy and apoptosis is still unclear. However, we discuss the key regulatory molecules shared by the two processes in this crosstalk platform. Under nutrient starvation conditions, C-Jun N-terminal protein kinase-1 (JNK-1) is activated which causes phosphorylation of Bcl-2 loop thus preventing the interaction between Beclin-1 and Bcl-2. The isolated Beclin-1 promotes autophagy by activation of Beclin-1/VPS34/Vps15 core complex (Morris et al. 2015). In addition to this, cardiac glycoside ouabain which is a Na/K-ATPase inhibitor also disrupts Bcl-2/Beclin-1 interaction by causing reduction of Bcl-2 thus resulting in autophagic cell death in non-small-cell lung cancer cells. Thus, Beclin-1 plays an important role in early stages of autophagy and mitigating DNA damage and inducing cell repair pathways essential for protecting the cell from external insult (Kang et al. 2011). Here, Beclin-1 is an important component of crosstalk between autophagy and apoptosis. Beclin-1 ± mice showed a higher incidence of developing spontaneous carcinomas of lung and liver (Yue et al. 2003). Also, studies have shown that in 50–70% of human breast, prostrate, liver and lung cancers, beclin-1 gene is epigenetically silenced (Li et al. 2010; Marinkovic et al. 2018). This shows that Beclin-1 is important for tumor suppression. Loss of Beclin-1 also blocks the activation of autophagy. This impairment of beclin-1 expression seen in cancer cells may hinder with their timely degradation and clearance enhancing the chances of tumor progression. Beclin-1 is also involved in apoptotic cell death. Its overexpression seen in glioblastoma cell lines causes release of pro-apoptotic molecules such as Bcl-2-associated X protein (BAX) and Bcl-2 homologous antagonist/killer (BAK) from Bcl-2 leading to apoptosis (Huang et al. 2014; Wang et al. 2017). Also, caspase activation causes cleavage of Beclin-1 into distinct fragments and the c-terminal fragment enters the mitochondria and induces the release of pro-apoptotic molecules. Another protein FLIP (FADD-like IL-1_-converting enzyme-inhibitory protein) is an anti-apoptotic protein associated with extrinsic pathway (Safa 2012). In addition, FLIP competes with Atg3 thus inhibiting the lipidation of LC3 blocking autophagy and under induction of autophagy involves suppression of interaction between Atg3 and FLIP (Lee et al. 2009).
Next, the Atg members are also involved in this crosstalk. Atg complex is part of the ubiquitin-like conjugation system participating in the elongation phase of autophagy. Both Atg12 and Atg5 are essential for autophagy induction and also in induction of apoptosis. Under apoptotic stimuli, Atg5 undergoes cleavage by calpains and the amino terminal portion enters the mitochondria associating with Bcl-X1 and release of cytochrome c to the cytoplasm leading to apoptosis (Yousefi et al. 2006). The non-conjugated ATG12 gets involved via its BH-3 domain and binds to anti-apoptotic Bcl-2 molecules thus inhibiting their action and this causes release of pro-apoptotic molecules leading to intrinsic apoptosis (Rubinstein et al. 2011).
In addition to these key molecules, caspases also are involved in both the processes. Caspases, a family of cysteinyl aspartate-requiring proteases, have a central role in the transduction of apoptotic signals (Lavrik et al. 2005). The caspases may function either as the initiator caspases or the effector caspases. Studies have reported that activated caspases are involved in degradation of main autophagy-related proteins (i.e., Beclin-1, Atg5, and Atg7) that may lead to shut down of the autophagic process (Liu et al. 2017a, b, c). Caspase-8 acts as a well-known initiator caspase that undergoes dimerization and autoproteolysis finally leading to events causing extrinsic apoptosis. In addition to this, caspase-8 regulates autophagy by cleavage of Atg3, leading to the pro-autophagic inhibition (Oral et al. 2012). Results of in vitro studies demonstrate the protective role of caspase-8 in preventing and rescuing T cells from hyperactive autophagy. The inhibition of this caspase-8 cleavage action was associated with hyperactive autophagy seen in T cells (Yu et al. 2004; Oral et al. 2012). Another initiator caspase, i.e., caspase-9 is associated with apoptosome formation and intrinsic apoptosis pathway. Upon activation, caspase-9 acts on its downstream effector caspases (caspase-3, caspase-6, and caspase-7) leading to key events bringing about apoptotic cell death. This caspase-9 also helps and induces autophagy by facilitating autophagosome formation and promotes LC3 lipidation process by interaction with Atg7 (Han et al. 2014; Li et al. 2017). Jeong and team demonstrated the potential role of caspase-9 in autophagy-mediated cell survival of breast cancer MCF-7 cells (Jeong et al. 2011). Han and co-workers stated that caspase-9 and its interaction with Atg7 may play an important cross-regulatory role determining the participation of caspase-9 either in autophagy or apoptosis (Han et al. 2014). In addition, caspase-10 downregulates autophagy by cleaving ATG5, and Beclin-1. Also, it has been shown to inhibit autophagy by cleaving the BCL2-interacting protein BCLAF1. This action impairs Bcl-2-Beclin-1 dissociation upon autophagy induction (Guo et al. 2016; Tsapras and Nezis 2017).
Next, is the most conserved initiator caspase, i.e., Caspase-2. In addition to its role in apoptosis, evidence suggests that caspase-2 also affects the process of autophagy. Loss of caspase-2 was associated with enhanced levels of autophagic proteins and autophagy seen in embryonic fibroblast cells of mice (Tiwari et al. 2014). Among the effector caspases, Caspase-3 is an important player in apoptotic cell death but besides this, it also has been shown to be involved in autophagic regulation. Caspase-3 cleaves Beclin-1 yielding fragmented portions thus abrogating the pro-autophagic effect of Beclin-1 in HeLa cells (Zhu et al. 2010).Further, cleavage of Beclin-1 exposes its BH-3 domain to other anti-apoptotic Bcl-2 family members thus initiating intrinsic apoptotic cascades (Oberstein et al. 2007; Erlich et al. 2007).Therefore, activated caspases are not only important for apoptotic process but also regulate the process of autophagy at various levels hence playing as an important crosstalk switch between the two regulatory processes.
Another key modulator of autophagy and apoptosis is the tumor suppressor protein TP53. Under normal conditions, this protein also referred as p53 gets degraded in the cytoplasm under the action of E3-ubiquitin system. However, under stressed conditions which can be due to genotoxic agents or activation of oncogenes, p53 gets localized in the nucleus and upregulates the transcription of various other genes involved in repair, cell cycle control, induction of autophagy and apoptotic events (Kumari et al. 2014). Rosenfeldt and team (Rosenfeldt et al. 2013) developed a genetically modified mouse model of pancreatic ductal adenocarcinoma (PDAC) and showed that p53 plays a tumor suppressive role. Studies were done in mice with pancreas containing an activated oncogenic allele of Kras (also called Ki-Ras)—the most common mutational event in PDAC that led to the development of small number of pre-cancerous lesions. Further, the findings indicated that mice lacking the essential autophagy genes Atg5 or Atg7 developed smaller and low-grade, pre-malignant pancreatic lesions that did not progress to high-grade aggressive phenotype of PDAC. However, with the concomitant loss of p53, mice developed more aggressive pancreatic tumor. Thus, with loss of autophagy and loss of p53, tumor progression was accelerated suggesting the protective role of p53 as tumor suppressor. Other aspect is that p53 present in nucleus also regulates transcription and expression of the damage-regulated autophagy modulator (DRAM), a TP53 target gene that modulates both autophagy and apoptosis. Zhang and team (Zhang et al. 2013) elucidated on the role of DRAM 1 in regulating autophagy flux in A549 lung cancer cell lines. DRAM 1 gets localized in lysosomes following exposure to mitochondrial inhibitor or any genotoxic insult. Specifically, DRAM is involved in LC3-I to LC3-II conversion, participation of acidification of lysosomes, fusion of lysosomes with autophagosomes and clearance of autophagosomes. But, under sustained stress, DRAM also participates in apoptosis as well. Guan et al. (2015) showed that DRAM1 regulates apoptosis involving BAX and lysosomes after investigating in cancer cell lines. BAX is a pro-apoptotic protein and is degraded by autophagy under basal condition. However, upon treatment of A549 or HeLa cells with carcinogenic agents or mitochondrial complex II inhibitor, DRAM1 interacted with BAX and this interaction was able to enhance prolong the half-life of BAX. Furthermore, DRAM1 upregulation led to recruitment of BAX to the lysosomes and the simultaneous releases of cathepsin B from lysosomes with cleavage of BID (BH3-interacting domain death agonist). The cleavage of BID into t-BID provokes the concomitant release of apoptogenic molecules from mitochondria. Thus, it seems that during early stages if tumor development, both autophagy and apoptosis work in sync and cooperate to minimize and prevent damage at cellular level eliminating the damaged cell. But this role may not be steady with tumor progression. As the tumor evolves conditions surrounding it change, the role of autophagy and apoptosis is bound to change and work differently. This has been discussed in the following section with special reference to lung cancer.
Autophagy: anti-tumor role
Autophagy represents an essential process to maintain optimal balance within the cell even under stress or nutrient deprivation. Autophagy constantly works to mitigate the oxidative stress and avoiding any malignant transformation by preventing the accumulation of damaged organelles or proteins within the cell which act as source of ROS (Guo et al. 2013). Autophagy genes (Atgs) deficiency is directly linked to accumulation of DNA damage, higher levels of ROS and accumulation of damaged mitochondria and other proteins all leading to genomic instability thus increasing the chances of oncogenic transformations (Mathew et al. 2009). Autophagy deficiency is associated with increased oxidative stress and activates the master regulator of antioxidant defense, nuclear factor, erythroid-2-like 2 (NRF2), that stimulates tumor growth (White 2015).
Autophagy acts as a source of supplying nucleotides for DNA replication thus preventing genome damage (Rabinowitz and White 2010). Robert and team (2011) showed that autophagy plays an important role in degradation of Sae2 (target of cyclin-dependent kinase 1 and is involved in meiotic recombination) in response to histone deacetylase inhibition (HDAC), which is key protein associated chromosome dynamics and homologous recombination-mediated DNA repair. Qu and co-workers (2003) demonstrated using targeted mutant mouse model (with monoallelic deletion of beclin-1 autophagy gene) that in such mice, the frequency of developing spontaneous malignant lesions significantly increased. Furthermore, beclin-1 heterozygous disruption results in increased cellular proliferation and reduced autophagy in vivo. In a similar study (Yue et al. 2003), it was observed that beclin 1-/- mutant mice die early in embryogenesis and beclin 1 ± mutant mice suffered from a high incidence of spontaneous tumors establishing a role for autophagy in tumor suppression. Ding and team (2008) found decreased expression of beclin1 in 44 tissue samples taken from patients suffering from hepatocellular carcinoma (HCC) as compared with adjacent nontumor tissues. Also, aggressive malignant HCC cell lines and HCC tissues with recurrent disease displayed much lower autophagic levels. Conversely, in genetically engineered mouse models (GEMMs) for hereditary breast cancer, allelic loss of beclin1 promoted p53 activation and reduced tumorigenesis, showing the opposite role of beclin1 here (Huo et al. 2013).
The p62, also called sequestosome 1 (SQSTM1) in humans, represents a selective substrate of autophagy. During intact autophagy, the p62/SQSTM1 has a LC3 interaction region (LIR) that directly interacts with LC3 which leads to degradation of p62. However, in defective autophagy, there occurs p62 upregulation which is commonly seen in many human tumor. Hence, autophagy deficiency or inhibition leads to accumulation of autophagy cargo receptors including p62 and this has been linked to promotion of lung and mammary tumorigenesis (Komatsu et al. 2007; Pankiv et al. 2007; Moscat and Diaz-Meco 2012). Also, p62 activates nuclear factor erythroid 2-related factor 2 (NRF2) and also activates mTOR thus activating the oncogenic signaling pathways in autophagy-deficient tissues (Komatsu et al. 2010). Aberrant accumulation of p62 correlates with cancer development and high levels of p62 have been found in HCC (Saito et al. 2016), breast cancer (Thompson et al. 2003), lung cancer (Inoue et al. 2012) suggesting that autophagy plays anti-tumorigenic role by preventing/limiting p62 accumulation (Mathew et al. 2009).
ATG gene products are also the main players in the autophagy process. Kang and team (Kang et al. 2009) analyzed the mononucleotide repeats in ATG2B, ATG5, ATG9B and ATG12 in gastric carcinomas and colorectal carcinomas using single-strand conformation polymorphism (SSCP) method. Results showed that frameshift mutations in ATG genes with mononucleotide repeats were common in both types of carcinomas high microsatellite instability suggesting that mutated ATG genes may contribute to dysregulation in the autophagy process linking to cancer development. In a animal study by Takamura and team (Takamura et al. 2011), a novel mice model was developed in which there was systemic mosaic deletion of Atg5 and liver-specific Atg7-/-. Results showed that such mice developed multiple benign liver adenomas. In addition, there was concomitant accumulation of p62, abnormally swollen mitochondria seen by electron microscopy in hepatocytes, induction of oxidative stress and genomic damage responses. Similar observation was made by Mathew and team (Mathew et al. 2009) highlighting that autophagy defective tumor cells showed p62 accumulation sufficient to cause oxidative stress and increased ROS levels with damaged mitochondria and genome damage contributes directly to tumorigenesis. Thus, autophagy loss creates conditions conducive toward promoting oncogenic activation and promoting tumorigenesis. This includes all those factors that are well known to create a cancer-prone environment such as high levels of ROS, oxidative stress markers, chronic tissue damage, increased genomic instability, and inflammation. (White 2012).
Rao and co-workers (Rao et al. 2014) reported dual role of autophagy in a mouse model of KRasG12D− driven lung cancer. The team found that the inactivation autophagy gene ATG5 at early stage showed an increase in number of adenomas in the mouse model of KRAS-driven NSCLC. But, at later stages, autophagy was involved in progression of adenomas to adenocarcinomas. Mice with tissue-specific inactivation of ATG5, impaired the progression of KRasG12D− driven lung cancer, giving advantage of tumor-bearing mice. These findings indicate that Atg5-dependent autophagy plays a dual role in lung cancer first as an inhibitor of initial oncogenesis and then as a facilitator of tumor progression.
Autophagy in cancer progression & metastasis: dual role
Cancer cell metastasis represents the process of invasion and colonization to secondary sites via the vascular and the lymphatic systems. This aggressive spread of the tumor to new sites is a major treatment challenge. The process of metastasis is activated due to the changes in the tumor microenvironment and certain metabolic alterations (Zhuyan et al. 2020). There are studies which depict the role of autophagy in inhibiting EMT; while several studies implicate autophagy in the promotion of EMT Autophagy plays an important role in this metastasis cascade (Mowers et al. 2017). It is involved in both pro-metastatic and anti-metastatic effects as discussed.
Autophagy may play anti-metastatic role as evidenced by past studies. In a study on primary melanoma patients (158 patients) showed that patients with low levels of Atg5 in their tumor were associated with reduction in progression-free survival (Liu et al. 2013). The team also employed an in vitro model of melanoma tumorigenesis (wherein normal melanocytes were transduced with BRAF oncogene) and reported that lowering of Atg5 expression levels promoted proliferation by preventing oncogene-induced senescence. This indicates that during early stages of cancer, autophagy contributes toward establishment of senescence and upon Atg5 down-regulation, cells tend to progress toward aggressive proliferation via precluding the Atg5-induced senescence. The PI3K/AKT/mTOR pathway is responsible for lung cancer aggressiveness and metastatic spread.Based on this pathway, many drug candidates are undergoing clinical or preclinical trials such as buparlisib (PI3K inhibitor), MK2206 (AKT inhibitor), sirolimus (mTOR inhibitor), and perifosine (dual PI3K/AKT inhibitor (Iksen et al. 2021). In a previous study by Hashimoto and team (2008), it was shown that blocking mTOR signaling induces autophagic cell death and inhibits metastasis spread and tumor progression in gastric cancer cells. This study also highlighted that chemokine, CXCL12 strongly phosphorylated Akt and also induced the activation of p70S6K (S6K) and eukaryotic initiation factor 4E binding protein 1 (4E-BP1), located downstream of Akt, resulting in cancer metastasis and migration seen in gastric cancer, NUGC4 cells. Further, by blocking this mTOR signaling pathway (by rapamycin), autophagic death was induced and cell migration, MMP production was significantly inhibited.
Majority of natural products show antimetastatic potential by targeting the PI3K/AKT/mTOR pathway and their downstream signaling intermediates, such as p70S6K, 4EBP1, and HIF1α causing autophagy induction, suppression of EMT, inhibition of migration and invasion. Curcumin, a natural polyphenol present in Curcuma longa L. (Zingiberaceae), suppresses cell proliferation in NSCLC cell lines through up-regulation of miR-206 and suppression of PI3K/AKT/mTOR signaling pathway. miR-206 is a microRNA and in humans is a member of the myo-miR family of miRNAs. miR-206 has been shown to suppress NSCLC growth and further invasion. Wang and team (2020a, b) showed that curcumin treatment was accompanied with inhibition of migration and invasion in A549 cells and elevated expression of miR-206 levels. This heightened expression of miR-206 significantly decreased the phosphorylation levels of mTOR and AKT inhibiting mTOR pathways and inducing autophagic cell death thus decreasing migration and further invasion. Also, the inhibition of miR-206 was associated with increased cell migration and increased phosphorylation levels of mTOR and AKT. Another Chinese medicine, i.e., Sotetsuflavone shows potent anti-cancer effects, especially in lung cancer. This drug was shown to exert its anti-metastatic effect in A549 cancer cells by suppression of hypoxia-inducible factor-1α (HIF-1α) transcription factor, an essential downstream effect intermediate of PI3K/AKT/mTOR pathway which is involved cell motility and invasion in NSCLC. This, by downregulating the PI3K/AKT/mTOR pathway, the drug induces autophagy in NSCLC cells and inhibits metastasis (Wang et al. 2018).
Now, coming to the second role played by autophagy, i.e., acting as pro-metastatic. The metastatic process includes the ability of cancer cells to detach from the extracellular matrix (ECM) and disseminate to the secondary sites. However, ECM detachment of primary cancer cells induces metabolic stress and activates another process called anoikis, i.e., programmed cell death. Evidence suggests that autophagy here plays a protective role toward these cancer cells by avoiding anoikis (Fung et al. 2008; Guadamillas et al. 2011). In a study by Avivar-Valdera et al. (2011), the team studied the underlying mechanism and showed the important role of protein kinase RNA-like endoplasmic reticulum kinase (PERK). This kinase PERK is related to promoting autophagy in ECM-detached cells via activation of AMPK levels that result in downstream inhibition of mTORC1-p70(S6K) signaling pathway. This process leads to rapid autophagy induction and protect cells from anoikis during loss of ECM adhesion. Peng and co-workers (2013) also showed that autophagy plays a role in hepatocellular carcinoma (HCC) metastasis due to its pro-survival effect. The team demonstrated that with stable silencing of expression of two of the main autophagic proteins, i.e., Beclin-1 and Atg5 in HCC cells, there was suppression in the incidence of pulmonary metastasis as seen in orthotopic mouse model via facilitating anoikis resistance and lung colonization of HCC cells. A study (using an in vivo metastasis assay) revealed that transfection with autophagy-related gene ATG16L1-300 T (vs. 300A) significantly increased brain metastasis in NSCLC patients. Also, transfection with ATG16L1-300 T (vs. 300A) stimulated the migration and invasion of A549 cells. Reduced Beclin1 expression was seen in NSCLC tissues and further low expression of Beclin1 has been seen in NSCLC patients with advanced stages of cancer. This is associated with low survival rates seen in NSCLC, thus suggesting that autophagy may be suppressed in these patients (Jiang et al. 2012; Du et al. 2020). In another study on NSCLC, it was found that autophagy induction inhibits the process of metastasis in NSCLC cells involving the role of a transcription factor 21, i.e., TCF21. Promoter methylation of TCF21 has been observed in the early stage of NSCLC (Richards et al. 2011) and has been found to be associated with tumor stage, metastasis and invasion of NSCLC cells. Therefore, TCF21 expression may inhibit autophagy and by suppressing both Atg9 and Beclin-1, and this factor is clinically related to the further progress of lung cancer. TCF21 knockdown cells exhibited significantly upregulated Atg9 and Beclin-1 expression with enhanced autophagy. Also, inhibition of autophagy by 3-methyladenine (3-MA) is associated with elevated TCF21 expression and increased cell apoptosis and invasion. This indicates the role of autophagy in regulation of TCF21 expression and suppressing tumor invasion and progression.
Autophagy is involved in cancer cell metastasis by regulating EMT. Epithelial-to-mesenchymal transition (EMT) which represents an important biological process that occurs during normal embryonic development, tissue regeneration, wound healing process, etc. (Kalluri et al. 2009). During EMT, the epithelial cells covert into a mesenchymal phenotype. This process is also involved in metastasis of cancer, movement of cancer cells from primary site to adjacent or distant sites, cell–cell adhesion, increased cell motility, polarity and also leading to resistance to cancer treatment (Thiery et al. 2009; Roche 2018).
There are studies which depict the role of autophagy in inhibiting EMT; while several studies implicate autophagy in the promotion of EMT. Studies depict that EMT-activated cells show high level of autophagy that may aid cancer cells to survive under the stressful condition faced during spread and invasion to new environment (Xiao and He 2010; Mikhaylova et al. 2012; Avivar-Valderas et al. 2011). DRAM1 is a highly conserved transmembrane protein that localizes itself to lysosomes. It is also a potential target of TP53-mediated autophagy and programmed cell death. Chen and team (2018) used small interfering (si)RNA or short hairpin RNA technique for knockdown of DRAM1 in hepatoblastoma cells. The migration and invasion potencies were studied including the levels of EMT markers. The results showed that DRAM1 knockdown inhibited cell autophagy, as well as inhibited the migration and invasion of HepG2 cells in transwell assays. Further, under EMT-mediated changes, cells show a decreased expression E-cadherins and increased expression of vimentin, fibronectin, etc. But, in DRAM1 knockdown HepG2 cells, there was upregulation of E-cadherin levels and decreased expression of EMT markers, i.e., Vimentin. Thus, autophagy here works in sync with EMT via regulatory role of DRAM1 to promote invasion of HepG2 cells. EMT promotes tumor metastasis and autophagy supports cancer cell viability during this metastatic spread along with helping the cancer cells in body’s immune surveillance escape. Akalay and colleagues (2013) reported that acquisition of EMT phenotype by breast cancer cells was associated with an inhibition of cytotoxic t-cell (CTL)-mediated tumor cell lysis. EMT cells showed enhanced autophagy and attenuation in CTL-mediated immunologic synapse. Also, with the silencing of Beclin-1, autophagy was inhibited and the cells showed susceptibility to T-cell cytotoxicity. Thus, suggesting that autophagy worked toward the helping EMT-mediated cancer spread.
Autophagy has been shown to negatively regulate and even inhibit EMT in many studies as well. Catalano and team (Catalano et al. 2015) studied the effect of autophagy on EMT in glioblastoma cells. It was observed that under nutrient deprivation condition or mTOR inhibition process, autophagy was induced and there was reduced cell migration and invasion seen. Also, there was downregulated of two master transcription factors of the EMT process, i.e., SNAIL and SLUG and an up-regulation of N- and R-cadherins. But, in BECLIN 1-silenced blastoma cells, up-regulation of SNAIL and SLUG and decline in N- and R-cadherin mRNAs expression were observed by the team. CDCA4, also known as HEPP/SEI-3/TRIP-Br3, is a member of cell division cycle associated (CDCA) gene family that have been shown to play different roles in various types of cancers. For example, in breast cancer and hepatocellular carcinoma, it promotes cancer cell progression (Xu et al. 2018; Gu et al. 2020) while in cervical cancer, knockdown of CDCA4 promoted cell proliferation showing its tumor suppressor role (Hayashi et al. 2006).In study (Xu et al. 2021), the team investigated the role of CDCA4 in NSCLC using cell lines with gene knockdown for CDCA4 and cell lines overexpressing CDCA4.The team found that in cell line with CDCA4 deletion, there was associated with induced EMT, migration and invasion of NSCLC cells and inhibition of autophagy of NSCLC cells. While, in overexpressing cell lines, reverse was observed with increased autophagy and minimal invasion of NSCLC cells. Also, CDCA4-inhibited EMT, migration and invasion were partially aggravated in the presence of an autophagy activator, i.e., rapamycin, and reversed on treatment with an autophagy inhibitor. This study highlights not only on the fact that CDCA4 could inhibit EMT, migration and invasion of NSCLC cells but also focusses on the essential role for autophagy in inhibiting EMT-mediated NSCLC migration and progression.
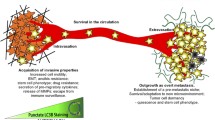
Thus, the twin role of autophagy in cancer metastasis is dependent on the stage of tumor growth. In early stages, autophagy acts in an anti-metastatic fashion creating conditions against tumor spread. However, when tumor cells enter hematogenous circulation, autophagy protects the circulating tumor cells from anoikis and this augments the tumor spread (Chavez-Dominguez et al. 2020). Autophagy switches to promote the proliferation of macro-metastasis by helping tumor cells to adapt to the stressful microenvironment and thus promotes cancer-cell survival and its successful colonization to the new site.
Autophagy: chemoresistance of nscl cancer
Cancer chemotherapy remains a mainstay and central to the treatment of lung cancer. But, the success rate of chemotherapy is often hindered due to the development of chemoresistance by cancer cells and the same is true in case of treating lung cancer. Many studies have shown that anticancer drugs cause upregulation of autophagy and this autophagy plays a protective role in making cancer cells resist many of these drugs and the same has been seen in lung cancer cells. Thus, the induction of protective autophagy that acts as a shield for the cancer cells represents a major challenge in cancer chemotherapy. For example, 5-Fluorouracil (5FU), a common anticancer drug used in treatment of many solid cancers, works by inhibiting thymidylate synthetase and DNA synthesis (Zhang et al. 2008). However, 5FU is also associated with induction of protective autophagy which is induced by upregulation of beclin-1 expression, followed by conversion of LC3I to LC3II cells. Similarly, epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs), i.e., gefitinib and erlotinib have been widely used in patients with NSCLC. Han and team (Han et al. 2011) reported that both gefitinib and erlotinib can induce a high level of autophagy, which was accompanied by the inhibition of the PI3K/Akt/mTOR signaling pathway. Further, the team showed that EGFR-TKIs can still induce autophagy even after EGFR expression was reduced by EGFR-specific siRNAs. This indicates the underlying role of autophagy in acquired drug resistance and the fact that autophagy inhibition may improve the efficacy of cancer drugs representing a potential target for further exploration and clinical testing.
Hypoxia-induced autophagy may play an important role in induced drug resistance. Hypoxic conditions prevail in growing tumors as the normal process of angiogenesis cannot supply enough oxygen to the rapidly growing tumor masses (Cosse et al. 2007; Semenza 2012). Hypoxia induces many changes in various cancer-related signaling pathways and leads to autophagy activation. Hypoxia causes stabilization of hypoxia-inducible factor 1-alpha (HIF-1α) transcription factor and inhibition of the mTOR kinase signaling pathway with induced autophagy. This HIF-1α enables to promote tumor cell growth and survival by controlling both glycolysis and the process of angiogenesis under harsh hypoxic conditions. Lee and team (Lee et al. 2015) stressed on the activation of hypoxia-induced autophagic pathways being involved in the developed resistance seen in NSCLC cells. It was seen that upon hypoxic exposure with 1% O2 given to A549 cell, autophagic induction was observed as shown by increase of LC3BI to LC3BII conversion and significant decline of p62/sequestosome1 in western blot assay with increased appearance of double-membrane autophagic vacuoles as seen in electron micrographs of treated A549 cells. Further, the team reported that treatment with LC3B siRNA was able to restore back the sensitivity of cancer cells to cisplatin hinting on the role of hypoxia-related pathway involved in autophagy induction in lung cancer cells. This also advocates on the use of hypoxia modification therapy (as an adjunct therapy) to maintain oxygen levels in solid tumor masses so as to suppress autophagic resistance for improved efficacy of cancer drugs (Horsman et al. 2021).
Cisplatin resistance also represents one of the main causes of treatment failure in NSCLC and sadly, autophagy is one the important players associated with development of cisplatin resistance in lung cancer patients. This platinum-based drug, i.e., cisplatin is a standard drug treatment given to lung cancer patients. It works and elaborates its anti-cancer effects by the generating DNA damage and mitochondrial apoptosis in cancer cells leading to their death (Guclu et al. 2018). Pan and his team (2019) studied that tripartite motif (TRIM)-containing proteins (proteins involved in development, immunity, carcinogenesis, autophagy) are involved in the regulation of autophagy and chemoresistance to cisplatin in NSCLC cells. The team showed that there were increased levels of TRIM65 in cisplatin-resistant NSCLC cell line (A549/DDP) as compared to the parental cell line (A549). Further, upon knockdown of TRIM6, autophagy was inhibited in A549/DDP cells, as shown by Annexin V/PI staining, caspase3 activity and immunofluorescence staining of LC3-II. Also, with knockdown of TRIM65, there was a significantly decreased levels of another important autophagy mediator, i.e., Atg7. This Atg7 is a target of miR-138-5p. This microRNA target 3' UTR of the Atg7 gene. Also, with a miR-138-5p inhibitor, the effects of TRIM65 knockdown on autophagy and cisplatin-induced apoptosis were significantly abolished. Furthermore, NSCLC tissues which were cisplatin-resistant showed higher expression of TRIM65 mRNA and lower expression of miR-138-5p as compared to cisplatin non-resistant NSCLC tissue. This indicates that TRIM65 knockdown attenuated autophagy-mediated cisplatin resistance in A549/DDP cells via regulating miR-138-5p. Goldberg and co-workers (Goldberg et al. 2012) also demonstrated that hydroxychloroquine (HCQ), an autophagy inhibitor when used was associated with enhanced sensitivity to chemotherapy drug, i.e., erlotinib as seen in twenty-seven patients with advanced non-small-cell lung cancer. Similarly, in another finding, it was reported that HCQ was able to prevent the development of paclitaxel resistance in A549 cells over time while potentiating the effect of paclitaxel by increased ROS generation and elevated accumulation of superoxide-producing mitochondria. Also, alleviation of the metastatic potential of NSCLC cells was observed (Datta et al. 2019) Hence, autophagy inhibition by HCQ represents fruitful strategy for overcoming the paclitaxel resistance development as well as metastasis in lung cancer. Thus, these findings highlight the protective role of autophagy in cancer survival and escape from drugs. Further, detailed investigation into the autophagy pathways involved is essential for overcoming the issue of chemoresistance and use of novel drugs targeting pathways of protective autophagy to enhance the anticancer drug efficacy. Table 1 lists some of the major studies done lately focussing on using autophagy inhibitors in combination with antitumor drugs representing a beneficial approach against NSCLC progression.
Autophagy: role in cancer cell immune evasion
Apart from participating in development of chemoresistant to anticancer drugs, evidence suggests the potential involvement of autophagy in protecting the cancer cells against immune attack and clearance.
Tumor microenvironment, i.e., nutrient deprivation and most importantly hypoxia has been found to be related to autophagy-mediated immune escape by cancer cells. In this direction, Noman and his team (Noman et al. 2011) elucidated that hypoxia-induced autophagy in tumor cells could, in theory, help in escaping CTL-mediated elimination and thus hypoxic conditions impaired elimination of NSCLC cells by T-cells. The team found that tumor cells exposed to hypoxia (1% PO2) caused hypoxia-inducible factor-1α (HIF-1α) induction HIF-1α and increased phosphorylation of signal transducer and activator of transcription 3 (pSTAT3). This was functionally linked to alteration of NSCLC in their susceptibility to CTL-mediated killing. Further, gene silencing of STAT3 by small interfering RNA as well as knockdown of HIF-1α, both restored the target cell lysis under hypoxic condition. This indicates toward targeting HIF-1α and STAT3 as a novel approach for further exploration while emphasizing on the role of hypoxic environmental as a crucial player in developing immune resistance.
SKIL (also known as SnoN) is a mediator of the transforming growth factor-β (TGF-β) signaling pathway and exerts pro-oncogenic activity. Ma and team (Ma et al. 2020) found that SKIL facilitates tumorigenesis and immune escape of NSCLC. The team observed that SKIL expression was higher in NSCLC tissue compared to adjacent normal tissue. Upon SKIL silencing, formation of malignant phenotypes of NSCLC cells was inhibited and T cell infiltration was promoted. SKIL-knockdown inhibited autophagy and activated the stimulator of interferon (IFN) gene STING pathway (plays and important role in anticancer immunity) in NSCLC cells through down-regulation of TAZ (an important transcriptional regulator which works toward cancer cell growth and initiation).The study thus concluded on the finding that SKIL promoted tumorigenesis and immune escape of NSCLC cells via upregulating the TAZ/autophagy axis and by inhibiting the STING pathway resulting in decreased T cell infiltration and release of chemokines such as CXCL10, CCL5 and IFN-β.
The interaction of tumor antigens and Class-I MHC molecules on the surface of TCR is the prime step in the CTL-mediated killing of tumor cells. An interesting finding (Yamamoto et al. 2020) showed that in pancreatic ductal adenocarcinoma (PDAC), lysosomal degradation involving the autophagy cargo receptor NBR1 was responsible for selective targeting of MHC-I molecules. Further, with the inhibition of autophagy, the surface levels of MHC-I were increased leading to improved antigen presentation, enhanced anti-tumor T cell responses and reduced tumor growth in syngeneic host mice. Also, with systemic autophagy inhibition by chloroquine as well as tumor-specific autophagy inhibition, infiltration of CTL’s and an increase of MHC-I molecules on the surface of cancer cell occurred making them sensitive toward immune checkpoint blockade (ICB) (Yamamoto et al. 2020; Duan et al. 2021).
Besides degrading MHC molecules, autophagy also been linked with degradation of cytotoxic granules released from CD8 + T and natural killer cells under hypoxic conditions (Baginska et al. 2013). The team showed that breast cancer cells evade effective NK-mediated killing under hypoxia by activating autophagy causing the degradation of NK-derived granzyme B. Further, blocking autophagy restored NK-mediated lysis in vitro and facilitated breast tumor elimination by NK cells in mice. Liang and team (Liang et al. 2012) showed that autophagy inhibitor such as chloroquine when given along with IL-2 therapy showed enhanced tumor regression with decreased toxicity and increase in immune infiltration in liver and spleen as seen in mice model of metastatic liver tumor. In addition to this, autophagy also makes the tumor microenvironment free from accumulated reactive oxygen species (ROS) thereby promoting tumor survival. Under hypoxic conditions, cancer cells produce ROS and the transfer of ROS to cancer-associated fibroblasts promotes autophagy, thereby providing nutrition for the growth of cancer cells.
Concluding remarks
Autophagy is a complex intracellular process and plays an important role in cancer metabolism. The regulation of autophagy is dependent on multiple factors including nutrient deprivation, the presence of damaged organelles, cancer cell microenvironment, stage of cancer, anticancer therapy, immune cell participation, etc. The role is dual with one side showing that autophagy participates in tumor suppression and prevents its further invasion to new sites. However, the second aspect is that under altered conditions, the same phenomenon of autophagy starts protecting cancer cells from stressful conditions, helps in immune evasion, makes the cancer cells drug resistant and thus aids in cancer cell metastasis and survival. Findings from various studies demonstrate that in the early stages of tumorigenesis, autophagy acts as a control mechanism and suppresses cancer initiation and progression working toward maintaining homeostasis. But, once tumor growth enters late stage, autophagy acts as a recycling platform providing conditions promoting survival and spread of the tumor by facilitating metastasis. Autophagy represents a potential therapeutic target to address the issue of chemoresistance and treatment failures in NSCLC patients as well as in suppressing metastasis and recurrence. But, needless to say, great importance needs to be given to understand the role of autophagy based on the right staging and grading of tumor from the perspective of clinical application and for this a comprehensive understanding of tumor dependence on the autophagy pathway in relation to the tumor microenvironment is essential. Also, more animal-based studies and clinical trials involving the use of autophagy modulators along with standard anticancer drug therapy are required to gather more data on the therapeutic effect in early stage and during late-stage lung cancer patients for achieving higher clinical success.
Data availability
All the data pertaining the above study are available within the manuscript.
References
Akalay I, Janji B, Hasmim M, Noman MZ, Thiery JP, Mami-Chouaib F, Chouaib S (2013) EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy 9(7):1104–1106. https://doi.org/10.4161/auto.24728
Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, Aguirre-Ghiso JA (2011) PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol 31(17):3616–3629. https://doi.org/10.1128/MCB.05164-11
Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, Medves S, Zimmer J, Oudin A, Niclou SP, Bleackley RC, Goping IS, Chouaib S, Janji B (2013) Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A 110(43):17450–17455. https://doi.org/10.1073/pnas.1304790110
Catalano M, D’Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, Faienza F, Esposito V, Limatola C, Cecconi F, Di Bartolomeo S (2015) Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol 9(8):1612–1625. https://doi.org/10.1016/j.molonc.2015.04.016
Chavez-Dominguez R, Perez-Medina M, Lopez-Gonzalez JS, Galicia-Velasco M, Aguilar-Cazares D (2020) The double-edge sword of autophagy in cancer: from tumor suppression to pro-tumor activity. Front Oncol 10:578418. https://doi.org/10.3389/fonc.2020.578418
Chen C, Liang QY, Chen HK, Wu PF, Feng ZY, Ma XM, Wu HR, Zhou GQ (2018) DRAM1 regulates the migration and invasion of hepatoblastoma cells via autophagy-EMT pathway. Oncol Lett 16(2):2427–2433. https://doi.org/10.3892/ol.2018.8937
Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK (2014a) Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer 14(8):535–546. https://doi.org/10.1038/nrc3775.Erratum.In:NatRevCancer.2015Apr;15(4):247
Cheong H, Yorimitsu T, Reggiori F, Legakis JE, Wang CW, Klionsky DJ (2005) Atg17 regulates the magnitude of the autophagic response. Mol Biol Cell 16(7):3438–3453. https://doi.org/10.1091/mbc.e04-10-0894
Chun Y, Kim J (2018a) Autophagy: an essential degradation program for cellular homeostasis and life. Cells 7(12):278. https://doi.org/10.3390/cells7120278
Cosse JP, Sermeus A, Vannuvel K, Ninane N, Raes M, Michiels C (2007) Differential effects of hypoxia on etoposide-induced apoptosis according to the cancer cell lines. Mol Cancer 6:61. https://doi.org/10.1186/1476-4598-6-61
Das G, Shravage BV, Baehrecke EH (2012) Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol 4(6):a008813. https://doi.org/10.1101/cshperspect.a008813
Datta S, Choudhury D, Das A, Mukherjee DD, Dasgupta M, Bandopadhyay S, Chakrabarti G (2019) Autophagy inhibition with chloroquine reverts paclitaxel resistance and attenuates metastatic potential in human non-small lung adenocarcinoma A549 cells via ROS mediated modulation of β-catenin pathway. Apoptosis 24(5–6):414–433. https://doi.org/10.1007/s10495-019-01526-y
Dela Cruz CS, Tanoue LT, Matthay RA (2011) Lung cancer: epidemiology, etiology, and prevention. Clin Chest Med. 32(4):605–644. https://doi.org/10.1016/j.ccm.2011.09.001
Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19(6):349–364. https://doi.org/10.1038/s41580-018-0003-4
Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, Shi GM, Wang XY, Ke AW, Wu B, Fan J (2008) Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res 68(22):9167–9175. https://doi.org/10.1158/0008-5472.CAN-08-1573
Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA (2014) WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell 55(2):238–252. https://doi.org/10.1016/j.molcel.2014.05.021
Du H, Chen L, Luo F, Chen X, Li Y, Cheng Q (2020) Beclin-1 expression is associated with prognosis in a Bcl-2-dependent manner in non-small cell lung cancer. Oncol Lett 20(4):9. https://doi.org/10.3892/ol.2020.11870
Duan Y, Tian X, Liu Q, Jin J, Shi J, Hou Y (2021) Role of autophagy on cancer immune escape. Cell Commun Signal 19(1):91. https://doi.org/10.1186/s12964-021-00769-0
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516. https://doi.org/10.1080/01926230701320337
Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, Hirsch JA, Stein R, Pinkas-Kramarski R (2007) Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 3(6):561–568. https://doi.org/10.4161/auto.4713
Feng Y, He D, Yao Z, Klionsky DJ (2014) The machinery of macroautophagy. Cell Res 24(1):24–41. https://doi.org/10.1038/cr.2013.168
Fung C, Lock R, Gao S, Salas E, Debnath J (2008) Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 19(3):797–806. https://doi.org/10.1091/mbc.e07-10-1092
Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221(1):3–12. https://doi.org/10.1002/path.2697
Goldberg SB, Supko JG, Neal JW, Muzikansky A, Digumarthy S, Fidias P, Temel JS, Heist RS, Shaw AT, McCarthy PO, Lynch TJ, Sharma S, Settleman JE, Sequist LV (2012) A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J Thorac Oncol 7(10):1602–1608. https://doi.org/10.1097/JTO.0b013e318262de4a
Gu Y, Li J, Guo D, Chen B, Liu P, Xiao Y, Yang K, Liu Z, Liu Q (2020) Identification of 13 key genes correlated with progression and prognosis in hepatocellular carcinoma by weighted gene co-expression network analysis. Front Genet 11:153. https://doi.org/10.3389/fgene.2020.00153
Guadamillas MC, Cerezo A, Del Pozo MA (2011) Overcoming anoikis–pathways to anchorage-independent growth in cancer. J Cell Sci 124(Pt 19):3189–3197. https://doi.org/10.1242/jcs.072165
Guan JJ, Zhang XD, Sun W, Qi L, Wu JC, Qin ZH (2015) DRAM1 regulates apoptosis through increasing protein levels and lysosomal localization of BAX. Cell Death Dis 6(1):e1624. https://doi.org/10.1038/cddis.2014.546
Guçlu H, Doganlar ZB, Gürlü VP, Özal A, Dogan A, Turhan MA, Doganlar O (2018) Effects of cisplatin-5-fluorouracil combination therapy on oxidative stress, DNA damage, mitochondrial apoptosis, and death receptor signalling in retinal pigment epithelium cells. Cutan Ocul Toxicol 37(3):291–304. https://doi.org/10.1080/15569527.2018.1456548
Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, Snyder E, Santanam U, Dipaola RS, Jacks T, Rabinowitz JD, White E (2013) Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 27(13):1447–1461. https://doi.org/10.1101/gad.219642.113
Guo W, Dong A, Pan X, Lin X, Lin Y, He M, Zhu B, Jin L, Yao R (2016) Role of caspase-10 in the death of acute leukemia cells. Oncol Lett 12(2):1623–1629. https://doi.org/10.3892/ol.2016.4785
Han J, Hou W, Goldstein LA, Stolz DB, Watkins SC, Rabinowich H (2014) A complex between Atg7 and caspase-9: a novel mechanism of cross-regulation between autophagy and apoptosis. J Biol Chem 289(10):6485–6497. https://doi.org/10.1074/jbc.M113.536854
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, Ge W, Feng L, Lin X, Wang X, Wang X, Jin H (2011) EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One 6(6):e18691. https://doi.org/10.1371/journal.pone.0018691
Hashimoto I, Koizumi K, Tatematsu M, Minami T, Cho S, Takeno N, Nakashima A, Sakurai H, Saito S, Tsukada K, Saiki I (2008) Blocking on the CXCR4/mTOR signalling pathway induces the anti-metastatic properties and autophagic cell death in peritoneal disseminated gastric cancer cells. Eur J Cancer 44(7):1022–1029. https://doi.org/10.1016/j.ejca.2008.02.043
Hayashi R, Goto Y, Ikeda R, Yokoyama KK, Yoshida K (2006) CDCA4 is an E2F transcription factor family-induced nuclear factor that regulates E2F-dependent transcriptional activation and cell proliferation. J Biol Chem 281(47):35633–35648. https://doi.org/10.1074/jbc.M603800200
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 43:67–93. https://doi.org/10.1146/annurev-genet-102808-114910
Horsman MR, Sørensen BS, Busk M, Siemann DW (2021) Therapeutic modification of hypoxia. Clin Oncol (r Coll Radiol) 33(11):e492–e509. https://doi.org/10.1016/j.clon.2021.08.014
Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N (2009) Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5(7):973–979. https://doi.org/10.4161/auto.5.7.9296
Hsieh MJ, Lin CW, Yang SF, Sheu GT, Yu YY, Chen MK, Chiou HL (2017) A combination of pterostilbene with autophagy inhibitors exerts efficient apoptotic characteristics in both chemosensitive and chemoresistant lung cancer cells. Toxicol Sci 156(2):549. https://doi.org/10.1093/toxsci/kfx050
Huang X, Qi Q, Hua X, Li X, Zhang W, Sun H, Li S, Wang X, Li B (2014) Beclin 1, an autophagy-related gene, augments apoptosis in U87 glioblastoma cells. Oncol Rep 31(4):1761–1767. https://doi.org/10.3892/or.2014.3015
Huo Y, Cai H, Teplova I, Bowman-Colin C, Chen G, Price S, Barnard N, Ganesan S, Karantza V, White E, Xia B (2013) Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov 3(8):894–907. https://doi.org/10.1158/2159-8290.CD-13-0011
Hurley JH, Young LN (2017) Mechanisms of autophagy initiation. Annu Rev Biochem 86:225–244. https://doi.org/10.1146/annurev-biochem-061516-044820
Iksen, Pothongsrisit S, Pongrakhananon V (2021) Targeting the PI3K/AKT/mTOR Signaling pathway in lung cancer: an update regarding potential drugs and natural products. Molecules, 26(13): 4100. https://doi.org/10.3390/molecules26134100
Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S et al (2012) Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci 103(4):760–766. https://doi.org/10.1111/j.1349-7006.2012.02216.x
Jeong HS, Choi HY, Lee ER, Kim JH, Jeon K, Lee HJ, Cho SG (2011) Involvement of caspase-9 in autophagy-mediated cell survival pathway. Biochim Biophys Acta 1813(1):80–90. https://doi.org/10.1016/j.bbamcr.2010.09.016
Jiang ZF, Shao LJ, Wang WM, Yan XB, Liu RY (2012) Decreased expression of Beclin-1 and LC3 in human lung cancer. Mol Biol Rep 39(1):259–267. https://doi.org/10.1007/s11033-011-0734-1
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signalling to the autophagy machinery. Mol Biol Cell. 20(7):1992–2003. https://doi.org/10.1091/mbc.e08-12-1249
Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y (2005) Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell 16(5):2544–2553. https://doi.org/10.1091/mbc.e04-08-0669
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119(6):1420–1428. https://doi.org/10.1172/JCI39104.Erratum.In:JClinInvest.2010;120(5):1786
Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS, Ahn CH, Yoo NJ, Lee SH (2009) Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol 217(5):702–706. https://doi.org/10.1002/path.2509
Kang R, Zeh HJ, Lotze MT, Tang D (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18(4):571–580. https://doi.org/10.1038/cdd.2010.191
Kinchen JM, Ravichandran KS (2008) Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol 9(10):781–795
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y et al (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12(3):213–223. https://doi.org/10.1038/ncb2021
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T et al (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131(6):1149–1163. https://doi.org/10.1016/j.cell.2007.10.035
Kroemer G, Mariño G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40(2):280–293. https://doi.org/10.1016/j.molcel.2010.09.023
Kumari R, Kohli S, Das S (2014) p53 regulation upon genotoxic stress: intricacies and complexities. Mol Cell Oncol. 1(3):e969653. https://doi.org/10.4161/23723548.2014.969653
Lavrik IN, Golks A, Krammer PH (2005) Caspases: pharmacological manipulation of cell death. J Clin Invest 115(10):2665–2672. https://doi.org/10.1172/JCI26252
Lee JG, Shin JH, Shim HS, Lee CY, Kim DJ, Kim YS, Chung KY (2015) Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir Res 16:138. https://doi.org/10.1186/s12931-015-0285-4
Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU (2009) FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol 11(11):1355–1362. https://doi.org/10.1038/ncb1980
Li Z, Chen B, Wu Y, Jin F, Xia Y, Liu X (2010) Genetic and epigenetic silencing of the beclin 1 gene in sporadic breast tumors. BMC Cancer 10:98. https://doi.org/10.1186/1471-2407-10-98
Li P, Zhou L, Zhao T, Liu X, Zhang P, Liu Y, Zheng X, Li Q (2017) Caspase-9: structure, mechanisms and clinical application. Oncotarget 8(14):23996–24008. https://doi.org/10.18632/oncotarget.15098
Liang X, De Vera ME, Buchser WJ, de Vivar Romo, Chavez A, Loughran P, Beer Stolz D, Basse P, Wang T, Van Houten B, Zeh HJ, Lotze MT (2012) Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res 72(11):2791–801. https://doi.org/10.1158/0008-5472.CAN-12-0320
Liu B, Oltvai ZN, Bayır H, Silverman GA, Pak SC, Perlmutter DH, Bahar I (2017a) Quantitative assessment of cell fate decision between autophagy and apoptosis. Sci Rep 7(1):17605. https://doi.org/10.1038/s41598-017-18001-w
Liu G, Pei F, Yang F, Li L, Amin AD, Liu S, Buchan JR, Cho WC (2017b) Role of autophagy and apoptosis in non-small-cell lung cancer. Int J Mol Sci 18(2):367. https://doi.org/10.3390/ijms18020367
Liu H, He Z, von Rütte T, Yousefi S, Hunger RE, Simon HU (2013) Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med 5(202):202ra123. https://doi.org/10.1126/scitranslmed.3005864
Liu JT, Li WC, Gao S, Wang F, Li XQ, Yu HQ, Fan LL, Wei W, Wang H, Sun GP (2015) Autophagy inhibition overcomes the antagonistic effect between gefitinib and cisplatin in epidermal growth factor receptor mutant non–small-cell lung cancer cells. Clin Lung Cancer 16(5):e55-66. https://doi.org/10.1016/j.cllc.2015.03.006
Liu X, Wang P, Zhang C, Ma Z (2017c) Epidermal growth factor receptor (EGFR): a rising star in the era of precision medicine of lung cancer. Oncotarget 8(30):50209–50220. https://doi.org/10.18632/oncotarget.16854
Lv X, Liu F, Shang Y, Chen SZ (2015) Honokiol exhibits enhanced antitumor effects with chloroquine by inducing cell death and inhibiting autophagy in human non-small cell lung cancer cells. Oncol Rep 34(3):1289–1300. https://doi.org/10.3892/or.2015.4091
Ma F, Ding MG, Lei YY, Luo LH, Jiang S, Feng YH, Liu XL (2020) SKIL facilitates tumorigenesis and immune escape of NSCLC via upregulating TAZ/autophagy axis. Cell Death Dis 11(12):1028. https://doi.org/10.1038/s41419-020-03200-7
Marinković M, Šprung M, Buljubašić M, Novak I (2018) Autophagy modulation in cancer: current knowledge on action and therapy. Oxid Med Cell Longev 2018:8023821. https://doi.org/10.1155/2018/8023821
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E (2009) Autophagy suppresses tumorigenesis through elimination of p62. Cell 137(6):1062–1075. https://doi.org/10.1016/j.cell.2009.03.048.Erratum.In:Cell.2011;145(2):322
Meng J, Chang C, Chen Y, Bi F, Ji C, Liu W (2019) EGCG overcomes gefitinib resistance by inhibiting autophagy and augmenting cell death through targeting ERK phosphorylation in NSCLC. Onco Targets Ther 12:6033–6043. https://doi.org/10.2147/OTT.S209441
Mikhaylova O, Stratton Y, Hall D, Kellner E, Ehmer B, Drew AF, Gallo CA, Plas DR, Biesiada J, Meller J, Czyzyk-Krzeska MF (2012) VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 21(4):532–546. https://doi.org/10.1016/j.ccr.2012.02.019
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147(4):728–741. https://doi.org/10.1016/j.cell.2011.10.026
Mizushima N (2007) Autophagy: process and function. Genes Dev 21(22):2861–2873. https://doi.org/10.1101/gad.1599207
Morris DH, Yip CK, Shi Y, Chait BT, Wang QJ (2015) Beclin 1-Vps34 complex architecture: understanding the nuts and bolts of therapeutic targets. Front Biol (Beijing) 10(5):398–426. https://doi.org/10.1007/s11515-015-1374-y
Moscat J, Diaz-Meco MT (2012) p62: a versatile multitasker takes on cancer. Trends Biochem Sci 37(6):230–236. https://doi.org/10.1016/j.tibs.2012.02.008
Mowers EE, Sharifi MN, Macleod KF (2017) Autophagy in cancer metastasis. Oncogene 36(12):1619–1630. https://doi.org/10.1038/onc.2016.333
Myers DJ, Wallen JM. Lung Adenocarcinoma. (2022) In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022. PMID: 30137862
Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, Baba M, McNew JA, Jiang X, Reggiori F, Melia TJ, Klionsky DJ (2011) SNARE proteins are required for macroautophagy. Cell 146(2):290–302. https://doi.org/10.1016/j.cell.2011.06.022
Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, Buart S, Berchem G, Romero P, Mami-Chouaib F, Chouaib S (2011) Blocking hypoxia-induced autophagy in tumor restores cytotoxic T-cell activity and promotes regression. Cancer Res 71(18):5976–5986. https://doi.org/10.1158/0008-5472.CAN-11-1094
Oberstein A, Jeffrey PD, Shi Y (2007) Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem 282(17):13123–13132. https://doi.org/10.1074/jbc.M700492200
Oral O, Oz-Arslan D, Itah Z, Naghavi A, Deveci R, Karacali S, Gozuacik D (2012) Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis 17(8):810–820. https://doi.org/10.1007/s10495-012-0735-0
Oser MG, Niederst MJ, Sequist LV, Engelman JA (2015) Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol 16(4):e165–e172. https://doi.org/10.1016/S1470-2045(14)71180-5
Pan X, Chen Y, Shen Y, Tantai J (2019) Knockdown of TRIM65 inhibits autophagy and cisplatin resistance in A549/DDP cells by regulating miR-138-5p/ATG7. Cell Death Dis 10(6):429. https://doi.org/10.1038/s41419-019-1660-8
Pan X, Zhang X, Sun H, Zhang J, Yan M, Zhang H (2013) Autophagy inhibition promotes 5-fluorouraci-induced apoptosis by stimulating ROS formation in human non-small cell lung cancer A549 cells. PLoS One 8(2):e56679. https://doi.org/10.1371/journal.pone.0056679
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282(33):24131–24145. https://doi.org/10.1074/jbc.M702824200
Parzych KR, Klionsky DJ (2014) An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20(3):460–473. https://doi.org/10.1089/ars.2013.5371
Peng YF, Shi YH, Ding ZB, Ke AW, Gu CY, Hui B, Zhou J, Qiu SJ, Dai Z, Fan J (2013) Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 9(12):2056–2068. https://doi.org/10.4161/auto.26398
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112(12):1809–1820. https://doi.org/10.1172/JCI20039
Rabinowitz JD, White E (2010) Autophagy and metabolism. Science 330(6009):1344–1348. https://doi.org/10.1126/science.1193497
Rai A, Pathak D, Thakur S, Singh S, Dubey AK, Mallik R (2016) Dynein clusters into lipid microdomains on phagosomes to drive rapid transport toward lysosomes. Cell 164(4):722–734. https://doi.org/10.1016/j.cell.2015.12.054
Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R et al (2014) A dual role for autophagy in a murine model of lung cancer. Nat Commun 5:3056. https://doi.org/10.1038/ncomms4056
Richards KL, Zhang B, Sun M, Dong W, Churchill J, Bachinski LL, Wilson CD, Baggerly KA, Yin G, Hayes DN, Wistuba II, Krahe R (2011) Methylation of the candidate biomarker TCF21 is very frequent across a spectrum of early-stage non-small cell lung cancers. Cancer 117(3):606–617. https://doi.org/10.1002/cncr.25472
Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, Foiani M (2011) HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 471(7336):74–79. https://doi.org/10.1038/nature09803
Roche J (2018) The epithelial-to-mesenchymal transition in cancer. Cancers (Basel) 10(2):52. https://doi.org/10.3390/cancers10020052
Romanov J, Walczak M, Ibiricu I, Schüchner S, Ogris E, Kraft C, Martens S (2012) Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J 31(22):4304–4317. https://doi.org/10.1038/emboj.2012.278
Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, Adams PD, Anderson KI, Gottlieb E, Sansom OJ, Ryan KM (2013) p53 status determines the role of autophagy in pancreatic tumour development. Nature 504(7479):296–300. https://doi.org/10.1038/nature12865
Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A (2011) The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell 44(5):698–709. https://doi.org/10.1016/j.molcel.2011.10.014
Sabbula BR, Gasalberti DP, Anjum F. Squamous Cell Lung Cancer (2022) In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022. PMID: 33232091
Safa AR (2012) c-FLIP, a master anti-apoptotic regulator. Exp Oncol 34(3):176–184
Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K et al (2016) p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun 7:12030. https://doi.org/10.1038/ncomms12030
Semenza GL (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148(3):399–408. https://doi.org/10.1016/j.cell.2012.01.021
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–249. https://doi.org/10.3322/caac.21660
Takahashi Y, He H, Tang Z, Hattori T, Liu Y, Young MM, Serfass JM, Chen L, Gebru M, Chen C, Wills CA, Atkinson JM, Chen H, Abraham T, Wang HG (2018) An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat Commun 9(1):2855. https://doi.org/10.1038/s41467-018-05254-w
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N (2011) Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25(8):795–800. https://doi.org/10.1101/gad.2016211
Takats S, Nagy P, Varga Á, Pircs K, Kárpáti M, Varga K, Kovács AL, Hegedűs K, Juhász G (2013) Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol 201(4):531–539. https://doi.org/10.1083/jcb.201211160
Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139(5):871–890. https://doi.org/10.1016/j.cell.2009.11.007
Thompson HG, Harris JW, Wold BJ, Lin F, Brody JP (2003) p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene 22(15):2322–2333. https://doi.org/10.1038/sj.onc.1206325
Tiwari M, Sharma LK, Vanegas D, Callaway DA, Bai Y, Lechleiter JD, Herman B (2014) A nonapoptotic role for CASP2/caspase 2: modulation of autophagy. Autophagy 10(6):1054–1070. https://doi.org/10.4161/auto.28528.Erratum.In:Autophagy.2017;13(3):637
Tsapras P, Nezis IP (2017) Caspase involvement in autophagy. Cell Death Differ 24(8):1369–1379. https://doi.org/10.1038/cdd.2017.43
Wang M, Yu H, Wu R, Chen ZY, Hu Q, Zhang YF, Gao SH, Zhou GB (2020a) Autophagy inhibition enhances the inhibitory effects of ursolic acid on lung cancer cells. Int J Mol Med 46(5):1816–1826. https://doi.org/10.3892/ijmm.2020.4714
Wang N, Feng T, Liu X, Liu Q (2020b) Curcumin inhibits migration and invasion of non-small cell lung cancer cells through up-regulation of miR-206 and suppression of PI3K/AKT/mTOR signalling pathway. Acta Pharm 70(3):399–409. https://doi.org/10.2478/acph-2020-0029
Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L et al (2017) Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32(1):42-56.e6. https://doi.org/10.1016/j.ccell.2017.06.003.Erratum.In:CancerCell.2018;33(1):152
Wang S, Hu Y, Yan Y, Cheng Z, Liu T (2018) Sotetsuflavone inhibits proliferation and induces apoptosis of A549 cells through ROS-mediated mitochondrial-dependent pathway. BMC Complement Altern Med 18(1):235. https://doi.org/10.1186/s12906-018-2300-z
White E (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12(6):401–410. https://doi.org/10.1038/nrc3262
White E (2015) The role for autophagy in cancer. J Clin Invest 125(1):42–46. https://doi.org/10.1172/JCI73941
Wirth M, Joachim J, Tooze SA (2013) Autophagosome formation-The role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol 23:301–309
World Health Organization. Cancer Fact Sheet, 2018
Xiao D, He J (2010) Epithelial mesenchymal transition and lung cancer. J Thorac Dis 2(3):154–159. https://doi.org/10.3978/j.issn.2072-1439.2010.02.03.7.PMID:22263037;PMCID:PMC3256459
Xu C, Cao H, Sui Y, Zhang H, Shi C, Wu J, Ma R, Feng J (2021) CDCA4 suppresses epithelial-mesenchymal transition (EMT) and metastasis in Non-small cell lung cancer through modulating autophagy. Cancer Cell Int 21(1):48. https://doi.org/10.1186/s12935-021-01754-w
Xu Y, Wu X, Li F, Huang D, Zhu W (2018) CDCA4, a downstream gene of the Nrf2 signaling pathway, regulates cell proliferation and apoptosis in the MCF-7/ADM human breast cancer cell line. Mol Med Rep 17(1):1507–1512. https://doi.org/10.3892/mmr.2017.8095
Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, Sohn ASW, Mukhopadhyay S, Lin EY, Parker SJ, Banh RS, Paulo JA, Wen KW, Debnath J, Kim GE, Mancias JD, Fearon DT, Perera RM, Kimmelman AC (2020) Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581(7806):100–105. https://doi.org/10.1038/s41586-020-2229-5
Yang Z, Klionsky DJ (2010) Eaten alive: a history of macroautophagy. Nat Cell Biol 12(9):814–822. https://doi.org/10.1038/ncb0910-814
Ye W, Lu W, Li X, Chen Y, Wang L, Zeng G, Xu C, Ji C, Cai Y, Yang L, Luo Z (2022) Long-term changes in the premature death rate in lung cancer in a developed region of china: population-based study. JMIR Public Health Surveill 8(4):e3363
Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU (2006) Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 8(10):1124–1132. https://doi.org/10.1038/ncb1482
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304(5676):1500–1502. https://doi.org/10.1126/science.1096645
Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 100(25):15077–15082. https://doi.org/10.1073/pnas.2436255100
Zhang L, Qiang P, Yu J, Miao Y, Chen Z, Qu J, Zhao Q, Chen Z, Liu Y, Yao X, Liu B, Cui L, Jing H, Sun G (2019) Identification of compound CA-5f as a novel late-stage autophagy inhibitor with potent anti-tumor effect against non-small cell lung cancer. Autophagy 15(3):391–406. https://doi.org/10.1080/15548627.2018.1511503
Zhang N, Yin Y, Xu SJ, Chen WS (2008) 5-Fluorouracil: mechanisms of resistance and reversal strategies. Molecules 13(8):1551–1569. https://doi.org/10.3390/molecules13081551
Zhang XD, Qi L, Wu JC, Qin ZH (2013) DRAM1 regulates autophagy flux through lysosomes. PLoS One 8(5):e63245. https://doi.org/10.1371/journal.pone.0063245
Zheng M (2016) Classification and pathology of lung cancer. Surg Oncol Clin N Am 25(3):447–468. https://doi.org/10.1016/j.soc.2016.02.003
Zhu Y, Zhao L, Liu L, Gao P, Tian W, Wang X, Jin H, Xu H, Chen Q (2010) Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 1(5):468–477. https://doi.org/10.1007/s13238-010-0048-4
Zhuyan J, Chen M, Zhu T, Bao X, Zhen T, Xing K, Wang Q, Zhu S (2020) Critical steps to tumor metastasis: alterations of tumor microenvironment and extracellular matrix in the formation of pre-metastatic and metastatic niche. Cell Biosci 10:89
Zou Y, Ling YH, Sironi J, Schwartz EL, Perez-Soler R, Piperdi B (2013) The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J Thorac Oncol 8(6):693–702. https://doi.org/10.1097/JTO.0b013e31828c7210
Acknowledgements
Not applicable.
Funding
This work received no specific grant from any funding agency.
Author information
Authors and Affiliations
Contributions
SZ, YQ, LY were involved in literature search, data collection and extraction, draft writing, editing and approval of the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that there is no conflict of interest regarding the publication of this article.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, S., Qian, Y. & Ye, L. Delineating the twin role of autophagy in lung cancer. BIOLOGIA FUTURA 74, 119–135 (2023). https://doi.org/10.1007/s42977-023-00165-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42977-023-00165-4