
Abstract
Bovine coronavirus (BCoV) has dual tropisms that can trigger enteric and respiratory diseases in cattle. Despite its global distribution, BCoV field strains from Brazil remain underexplored in studies investigating the virus's worldwide circulation. Another research gap involves the comparative analysis of S protein sequences in BCoV isolates from passages in cell lines versus direct sequencing from clinical samples. Therefore, one of the objectives of our study was to conduct a comprehensive phylogenetic analysis of BCoV strains identified from Brazil, including a respiratory strain obtained during this study, comparing them with global and ancestral BCoV strains. Additionally, we performed a comparative analysis between wild-type BCoV directly sequenced from the clinical sample (nasal secretion) and the cell culture-adapted strain, utilizing the Sanger method. The field strain and multiple cell passage in cell culture (HRT-18) adapted BCoV strain (BOV19 NS) detected in this study were characterized through molecular and phylogenetic analyses based on partial fragments of 1,448 nt covering the hypervariable region of the S gene. The analyses have demonstrated that different BCoV strains circulating in Brazil, and possibly Brazilian variants, constitute a new genotype (putative G15 genotype). Compared with the ancestral prototype (Mebus strain) of BCoV, 33 nt substitutions were identified of which 15 resulted in non-synonymous mutations (nine transitions and six transversions). Now, compared with the wild-type strain was identified only one nt substitution in nt 2,428 from the seventh passage onwards, which resulted in transversion, neutral-neutral charge, and one substitution of asparagine for tyrosine at aa residue 810 (N810Y).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bovine coronavirus (BCoV) is a positive-sense single-stranded RNA virus classified in the Coronaviridae family, genus Betacoronavirus, and species Betacoronavirus 1 [1]. BCoV infections are associated with enteric and respiratory diseases in cattle, such as winter dysentery (WD) in adult animals, neonatal diarrhea in calves, and respiratory diseases in all ages. BCoV isolates from respiratory and enteric tracts are commonly referred to as bovine respiratory coronavirus (BRCoV) and bovine enteric coronavirus (BECoV), respectively [2, 3].
The BCoV genome encodes five major structural proteins, nucleocapsid (N), transmembrane (M), hemagglutinin esterase (HE), spike (S), small membrane, or envelope (E) proteins, and several non-structural proteins (nsp). The N protein is highly conserved among BCoV isolates and is one of the main targets for molecular diagnosis. The S protein is a type 1 viral fusion protein involved in viral attachment and entry into target cells and contains epitopes that are targets for neutralizing antibodies [4]. The S protein is cleaved into the S1 and S2 subunits by host cell proteases during processing in the Golgi Complex. The S1 subunit of coronaviruses contains a receptor-binding domain (RBD) that recognizes host cell receptors [5, 6]. Mutations in the RBD of the spike protein of coronaviruses may alter viral host tropism and even cause cross-species spillover. Bovine coronaviruses can infect a broad host range, including domestic and wild ruminants and humans [7,8,9].
The hypervariable region (HVR), also located within the S1 subunit and close to the RBD, has a greater tendency to accumulate mutations that may result in immune escape or altered viral fitness [10,11,12]. Phylogenetic analysis of the complete S gene of BCoV strains from America, Europe, and Asia has revealed that BCoVs are primarily distinguished into two major types: European and American types [13, 14].
Due to the absence of a correction mechanism in its RNA polymerase, BCoV, like to most RNA viruses, can frequently mutate. Therefore, it is crucial to consider these intrinsic factors when adapting and isolating clinical specimens prior to sequencing [15]. The extensively studied coronavirus pandemic, severe acute respiratory syndrome coronavirus (SARS-CoV-2), has led to the identification of eight single nucleotides (nt) polymorphisms after the first passage, which persist even after 12 cell culture passages, resulting in four non-synonymous amino acids (aa) substitutions in the S protein [16]. These findings have substantial implications for interpreting molecular characterization data because virus isolation in cell culture can lead to adaptive mutations that may not reflect naturally circulating viruses [17]. However, comparative studies of S protein mutations in BCoV isolates from cell lines are limited, and direct sequencing of clinical samples is not commonly performed [18]. In this study, we report the detection and isolation of a BCoV associated with respiratory disease and establish phylogenetic relationships based on partial sequences of the S gene. Furthermore, we compared wild-type BCoV sequenced directly from a clinical sample (nasal secretion) with a cell culture-adapted strain.
Methods
Animals and samples
The study was conducted in a herd of dairy cattle from Paraná State, southern Brazil, consisting of Holstein and Jersey breeds and their crossbreeds. The cattle herd had a documented history of enteric and respiratory diseases and mortality among adult and young animals. During the sample collection period in August 2016, animals up to five months old, exhibited dyspnea, serous nasal secretion, and no clinical signs of diarrhea.
This study was approved by the Ethics Committee for the Use of Animals of the Federal University of Paraná, under protocol n°. 34/2016.
Nasal secretions from 11 heifer calves with respiratory clinical signs were collected by inserting sterile cotton swabs deep into the nostrils and subsequently placing them in tubes containing 2 mL phosphate-buffered saline (PBS; 137 mM NaCl, 3 mM KCl, 8 mM Na2HPO4, and 15 mM KH2PO4; pH 7.2).
BCoV molecular diagnosis
Nuclei acid was extracted from 400 μL of sample suspensions using a combination of the phenol/chloroform/isoamyl alcohol and silica/guanidine isothiocyanate methods [19].
Nucleic acid extracts were subjected to a semi-nested RT-PCR (snRT-PCR) for the BCoV N gene partial amplification. The primers used and the conditions of the snRT-PCR were according to the method described by [20]. Samples that tested positive for the N gene were subjected to an RT-PCR for partial amplification of the BCoV-S gene partial amplification and viral isolation in HRT-18 cells.
To validate the extraction and genomic amplification, diethylpyrocarbonate (DEPC) water aliquots were used as a negative control, and the BCoV-Kakegawa prototype strain propagated in HRT-18 cells was used as a positive control.
Virus isolation
HRT-18 cells were cultured in Dulbecco's modified Eagle’s medium (Gibco BRL®, USA), supplemented with 10% fetal bovine serum (Gibco BRL®), 55 μg/mL gentamicin (Sigma Co., USA), and 2.5 μg/mL amphotericin B (Bristol-Meyers Squibb Co., Brazil). The confluent monolayers were washed with calcium- and magnesium-free PBS. Prior to inoculation, suspensions of the biological samples selected for viral isolation were treated with gentamicin (100 mg/mL) and amphotericin B (10 mg/mL). Inoculated cells were maintained at 37 °C for observation of the cytopathic effect, along with uninoculated (negative control) HRT-18 cells, and cells inoculated with the BCoV prototype Kakegawa strain as positive controls. To evaluate the possible mutations induced by in vitro replication of BCoV, the procedure was repeated for nine cell passages. Aliquots of 50 μL of the inoculated cell supernatant from each passage were separated, and 400 μL of PBS was added for viral nucleic acid extraction and subsequent snRT-PCR (N gene) and RT-PCR (S gene) analyses.
RT-PCR for partial amplification of the BCoV S gene
Samples previously identified as BCoV-positive for the N gene were subjected to RT-PCR to partially amplify the S glycoprotein gene. Three pairs of primers were used, which flanked a 1,448 nt region, including the HVR region and the proteolytic cleavage site of the S gene. The RT-PCR primers are described by Takiuchi et al. [18] and are provided in Online Resource 1.
Purification and sequencing
The amplified products were purified using the PureLink® Quick Gel Extraction Kit (Invitrogen, Carlsbad, CA, USA), quantified with the Qubit ™ fluorometer (Invitrogen Life Technologies, USA), and sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems®, Carlsbad, CA, USA) on the ABI 3500 genetic analyzer (Applied Biosystems®), using the forward and reverse primers for their respective targets of interest (N or S gene).
Phylogenetic analysis
Sequence analysis and consensus sequences were obtained using Phred and CAP3 software (http://asparagin.cenargen.embrapa.br/phph/). GenBank homology searches were performed using the Basic Local Alignment Search Tool (BLAST; http://blast.ncbi.nlm.nih.gov/Blast.cgi). The nt and aa sequences obtained were aligned and compared with the sequences of prototype BCoV strains and other BECoV, BRCoV, and wild-type field strains from the European, Asian, and American continents, totaling 80 sequences. This comparison was conducted using BioEdit software version 7.2.6.1. Phylogenetic trees were constructed with the Maximum Likelihood method (Tamura-Nei model) using MEGAX software. A bootstrap value of 1,000 replicates was used to determine significant differences between strains. Potential N-glycosylation sites in the partial S protein sequence were identified using the CBS NetNGlyc 1.0 server (http://www.cbs.dtu.dk/services/NetNGlyc/).
Results
BCoV was detected in the nasal secretions of all (n = 11) sampled animals using snRT-PCR targeting the N gene. Eight of these samples showed positive results in the first round of amplification, producing a product of 454 bp. The remaining three nasal secretion samples tested positive only after the second round of genomic amplification, resulting in amplicons of the expected size of 251 bp.
Direct Sanger sequencing confirmed the identity of all amplified products as BCoV concerning the N gene. Sequences obtained from 10 nasal secretions were of good quality for molecular characterization, showing that they were identical.
Of the ten nasal secretion samples that tested positive for the N gene, three (BOV13-NS, BOV19-NS, and BOV21-NS) were selected for S gene amplification. Among them, successful amplification of the three contiguous fragments of the S gene was achieved only with the BOV19-NS, which was selected for viral isolation in cell culture. Sequencing of BOV19-NS resulted in a consensus sequence of 1,448 nt (nt 1,258 to 2,705 in the Mebus strain), corresponding to the 3’-half of the S1 gene. Samples BOV13 and BOV21 amplified the region flanked by primers SPK6-FOR and SPK7-REV (998 nt) and SPK7-FOR and SPK7-REV (636 nt), respectively. Alignment of the consensus sequences obtained from the three nasal secretion samples demonstrated 100% identity. Therefore, all molecular characterization and phylogenetic assays were performed based on BOV19-NS.
Sequence and phylogenetic analysis
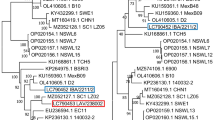
Analysis of the BCoV S gene involved two steps. In the first step, it was analyzed a 1,448 nt fragment spanning the cleavage site region of the S protein (nt 1,258 to 2,705 in the Mebus strain) from 55 BCoV sequences, including classical and vaccine strains (Kakegawa/Japan, Quebec/Canada, Mebus/USA, Ly-138/USA, F15/France, Norden-vaccine/USA, BC94-Vaccine/South Korea), and isolates from America (USA and Brazil), Europe (Sweden, Denmark, Italy, and France), and Asia (Japan and South Korea). This analysis included at least one sequence from each of the 14 BCoV genotypes, as described by Suzuki et al. [13]. According to the identity matrix, the BOV19-NS strain shared the highest nt (98.8%) and aa (98.5%) identity with Brazilian BECoV (DQ479421.1, DQ479422.1, and DQ479423.1) isolates collected in 2004 [18]. In contrast, the lowest nt (97.1%) and aa (95.8%) sequence identities were observed in the F15 (D00731.1) strain, a classical BECoV strain from France. The nt percentage identity among the Brazilian strains was in the range of 98.3–98.8% (Online Resource 2). In phylogenetic analysis were formed two large clades, separating the European and American types into 11 (G1–G3 and G5–G12) and three (G4, G13, and G14) genotypes, respectively, as proposed by Suzuki et al. [13]. The BOV19-NS strain was classified within the American-type clade; however, it was grouped into a separate subclade composed exclusively of Brazilian isolates. Based on this analysis, this subclade was identified as the putative G15 genotype (Fig. 1). Based on this grouping, the nt sequence identities among the Brazilian strains (putative G15 genotype) and Asian-American-type and European-type clades were in the range of 96.6–98.8% and 96.5–98.6%, respectively.

Phylogenetic analysis based on the partial sequence of 1,448 nt (nt 1,258 to 2,705 of Mebus strain) of the S gene of BCoV strains. The phylogenetic tree was constructed using the maximum likelihood method, Tamura-Nei evolutionary model, with Gamma 5 distribution, invariant sites (ML + TN93 + G + I), and 1,000 bootstrap replicates. Bootstrap values > 70 are indicated next to nodes. The isolates included in the analysis are identified using the GenBank accession number / strain name / country of origin / year of isolation. Respiratory isolates are indicated with (R), and the enteric isolates with (E). BCoV strains, as classified by Suzuki et al. [13], are indicated by an asterisk (*) followed by the corresponding genotype number. The sequence obtained in this study is indicated by the symbol ■
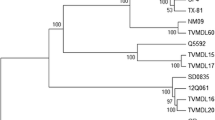
The second step of the phylogenetic analysis was performed using a 438 nt fragment, encompassing the HVR region of the subunit S1 of BCoV. This analysis aimed to establish phylogenetic relationships with other BCoV isolates from Brazil and globally that could not be included in the first analysis because of limited nt sequences available in public databases. The percent identities of nt among Brazilian strains collected from different locations and years ranged from 95.3 to 99.1%. The BOV19-NS sample identified in this study showed higher (98.4%) nt identity with a Brazilian BRCoV isolate PD1 (GenBank accession number KT381471.1), and lower (95.5%) nt identity with the prototype Mebus strain (GenBank accession number U00735.2). The phylogenetic tree revealed the formation of two major clades: Clade I, containing the classical BCoV strains, and Clade II, containing American, European, and Asian isolates. Clade II was subdivided into two monophyletic groups, according to Suzuki's classification [13], namely the American and European clades. The Brazilian BOV19-NS strain grouped in an American-type clade, formed a distinct subclade separate from the Asian and Cuban isolates (Fig. 2).

Phylogenetic analysis based on 438 nt of the polymorphic region HVR of the S gene (nt 1,339–1,777 of Mebus strain) from different BCoV strains. The phylogenetic tree was constructed using the maximum likelihood method, Tamura–Nei evolutionary model, with Gamma 5 distribution (ML + TN93 + G) and 1,000 bootstrap replicates. Bootstrap values > 70 are indicated next to nodes. The isolates included in the analysis are identified using the GenBank accession number / strain name / country of origin / year of isolation. Respiratory isolates are indicated with (R), and the enteric isolates with (E). The sequence obtained in this study is indicated by the symbol ■
Isolation of BCoV in cell culture
BOV19-NS was successfully isolated from HRT-18 cells. From the second to the ninth passages, characteristic cytopathic effects were observed, including granular, rounded, and swollen cells. BCoV was detected in all cell passages via partial amplification of the N gene from the supernatant. The S gene was successfully sequenced from three contiguous genomic segments (1,448 nt) of the BOV19-NS isolates at the third, fourth, seventh, eighth, and ninth passages.
Molecular characterization of BOV19-NS before and after passage in cell culture
The 1,451 nt fragment (nt 1,258–2,709, relative to the Mebus prototype strain) of BOV19-NS was analyzed before and after successive passages in cell culture (third, fourth, seventh, eighth, and ninth passages). The results showed that a single nt substitution was identified at position 2,428 starting from the seventh passage, resulting in a non-synonymous transversion mutation that caused a substitution of asparagine for tyrosine at aa residue 810 (N810Y), as shown in Fig. 3. No insertions or deletions were observed. In silico analysis revealed that among the 15 aa changes observed, three (P501S, P546S, and A769S) resulted in a loss of hydrophobicity, and one (S543A) gained hydrophobicity. In addition, four polar (hydrophilic) aa underwent charge changes (H470D changed from positive to negative, N531D from neutral to negative, and T540K and Y571H from neutral to positive).

Amino acid deduced sequences alignment (aa 420–903 of Mebus strain) of the S gene. Comparison between the direct sequencing of the biological sample (MT346358 BOV19-NS), its third, fourth, seventh, eighth, and ninth passages (MT346360 BOV19-NS-3P, MT346361 BOV19-NS-4P, MT346362 BOV19-NS-7P, MT346363 BOV19-NS-8P, and MT346364 BOV19-NS-9P) and prototype strains of BCoV. The dots (.) indicate identical aa; the red frame encompassing aa 452–593 indicates the HVR region; the green frame encompassing aa 763–768 indicates the proteolytic cleavage site; the smaller black frames encompassing three aa indicate the potential glycosylation sites; the black arrows indicate the cysteine residues
Among the 29 cysteine residues detected in the studied region, 15 were concentrated in the polymorphic HVR region and were conserved compared to those of other prototype strains. In addition, we identified nine putative glycosylation sites that were highly conserved among the BCoV strains (Fig. 3).
Discussion
snRT-PCR targeting the N gene was efficient for diagnosing BCoV in calves with respiratory clinical signs, as evidenced by the detection of BCoV in the nasal secretions of the 11 evaluated dairy heifer calves. The choice of the target region for genomic amplification is crucial for optimizing the diagnosis of BCoV. The N gene is highly conserved among BCoV isolates and is abundant in infected cells as subgenomic RNA, making it an ideal target for molecular diagnostic techniques [20, 21].
According to Suzuki et al. [13], the evolution of BCoVs may be closely associated with variations in the HE, S, and N genes and the ORF1 region, as the clustering patterns observed in the phylogenetic analysis of these genes were similar to those found in the analysis of the whole genome. Using 153 complete sequences of the S gene of BCoV, these authors divided the European and American strains into 11 (G1–G3 and G5–G12) and three (G4, G13, and G14) genotypes, respectively. However, the genotypic characterization of Brazilian BCoV isolates was not possible in the study by Suzuki et al. [13] owing to the unavailability of complete nt sequences of the S gene from the Brazilian strains. In our phylogenetic analysis, the use of a partial nt sequence (1,448 nt) of the S gene led to the same grouping of genotypes. This was possible because the analyzed fragment corresponded to the most variable region of the S gene, specifically the 3' half of the S1 gene. BOV19-NS strain was classified within the American-type clade; however, it was grouped into a subclade composed exclusively of Brazilian strains and separated from the 14 genotypes previously determined by Suzuki et al. [13]. The results of our analysis, in conjunction with the existing classification, provide additional support for the existence of a putative G15 genotype among Brazilian BCoV strains [22].
Phylogenetic analysis of the HVR region (438 nt) of the S gene revealed a distinct phylogeographic pattern compared to that of the larger fragment. Specifically, the analysis generated two large clades, separating the prototype strains (Clade I) from the European and American types (Clade II). In this analysis, we expanded our sample size to include five additional BCoV isolates from other regions of Brazil. Again, all Brazilian strains clustered together, forming a distinct subclade separate from the Asian and Cuban strains (Fig. 2). Differences in the percentage of nt identity (95.7 to 99.1%) among Brazilian BCoV strains indicated that multiple strains of the virus may be circulating within the country. It is believed that the commerce and the intense movement of livestock between different regions of a country favor the diffusion of distinct strains and the emergence of new genetic variants of BCoV.
Discriminating between enteric and respiratory BCoV isolates remains a challenge, with conflicting findings reported in the literature [23]. Some studies have reported the absence of consistent genetic or antigenic markers to distinguish BCoVs in different clinical syndromes [24,25,26,27], whereas others have suggested genetic, pathogenic, and antigenic differences between the BRCoV and BECoV strains [28,29,30,31,32,33]. However, our study revealed a tendency to segregate the strains based on their geographic and temporal origins rather than their clinical origins (enteric or respiratory), which is consistent with recent findings reported by Suzuki et al. [13], Temizkan and Alkan [34], and Frucchi et al. [22].
When analyzing the respiratory sample BOV19-NS and its respective passages after isolation in HRT-18 cell culture, no alterations were observed, except for a non-synonymous mutation detected at residue 810, which was only present from the seventh passage onwards. Enteric strains were more prone to changes during adaptation to cell culture and passages than BCoV respiratory strains. For example, in a study conducted by Zhang et al. [32] using the complete genome, an enteric isolate showed 104 nt mutations, whereas the respiratory isolate of the same animal presented only eight alterations after 15 and 14 passages in cell culture, respectively. Therefore, certain BCoV strains may require additional cumulative passages to exhibit substantial genetic mutations or changes in tropism or virulence.
Based on the in silico analysis, we found that changes may have occurred in the aa charges covering the HVR and RBD regions of the S gene in the analyzed strains. These regions contain important neutralizing epitopes responsible for interactions with host cell receptors. Substitutions of a hydrophobic aa for a polar/charged aa or vice versa, in addition to polar aa charge replacements, can alter the conformation of the protein. This structural change may alter viral host tropism and viral fitness and lead to the emergence of escape virus mutants [35, 36].
The observed aa changes were consistent with those previously reported. For example, specific mutations in antigenic regions such as P501S and P546S can induce changes in antigenicity or virulence among strains [37]. Yoo and Deregt [36] demonstrated that a single aa substitution from alanine to valine at aa residue 528 (A528V) in the S1 subunit of the S gene induced by in vitro selection pressure leads to resistance against monoclonal antibody neutralization. Our isolate BCoV strain harboured the same mutation. This is the first report of this mutation in a wild-type BCoV isolate.
Some authors have suggested that specific changes in residues 510 (S510T), 531 (N531D), and 578 (T578S) may serve as markers for respiratory strains [28, 36, 38, 39]. Our isolate also showed mutations suggested as BRCoV marker candidates at residues 510 and 531. Hasoksuz et al. [11] demonstrated that aa changes in S510T and N531D led to a reduction in hydrophilicity and alteration to a negative charge, respectively, which could affect the folding of the protein and its physical and chemical properties. Another factor that could be characteristic of respiratory isolates is the substitution of glycine at residue 531 with aspartic acid or asparagine in enteric isolates [36]. However, contrary to the speculation of Yoo and Deregt [36], the BOV19-NS strain had an aspartic acid substitution at residue 531 instead of glycine. Other studies have also observed aspartic acid in respiratory isolates, such as glycine in enteric isolates; however, it is unclear whether this residue constitutes a respiratory marker [11, 40, 41]. Molecular characterization of Cuban isolates from fecal samples of cows with clinical signs of WD also showed that changes in residues 510, 531, and 578 were not exclusive to BRCoV [39].
Based on these analyses, we believe that there are no specific markers for BRCoV or BECoV infection. The co-detection of BCoV in both nasal and fecal samples has been reported by other authors, suggesting that animals may be primarily infected via the oronasal route. After viral replication in the respiratory tract, the virions are swallowed and migrate to the gastrointestinal tract [11, 39, 42]. An experimental study conducted on gnotobiotic calves deprived of colostrum showed that diarrheal disease occurs after inoculation with BRCoV isolates [43].
The cleavage site KRRSRR (764–768) was preserved in the analyzed respiratory sample and its passages in cell culture, with only one change occurring immediately after the cleavage site, A769S. Chouljenko et al. [28] proposed that such replacement would be specific to respiratory isolates. However, studies on respiratory and enteric isolates have shown that both these can have the aa serine, indicating that this alteration is not a reliable marker for respiratory tropism [11, 39,40,41].
Consistent with the findings of Yoo et al. [44], of the 29 cysteine residues detected in the studied portion, 15 residues were in the HVR region. In addition, they were conserved compared to other strains. The high concentration of this aa in the HVR region may be involved in the formation of antigenic determinants via disulfide bonds [36]. These residues confer stability to the structure and function of proteins by forming disulfide bonds and maintaining adequate maturation and localization via intermolecular protein–protein interactions [45, 46].
In conclusion, the present study demonstrated that BCoV strains variants circulating within Brazil. Possibly the Brazilian strains, constitute a new genotype distinct from all other known BCoVs circulating worldwide. This is the first report of BCoV isolated in cell culture associated with respiratory disease in Brazil. Comparison of the nt sequences of the wild-type and cell-culture-adapted BCoV strain revealed a single aa substitution from the seventh passage onwards that did not affect the topology of the phylogenetic tree. Further studies with other Brazilian BCoV strains should be conducted to determine whether this mutation is a potential marker of adaptation and attenuation in HRT-18 cells.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
Change history
07 March 2024
A Correction to this paper has been published: https://doi.org/10.1007/s42770-024-01296-z
References
ICTV (2022) International committee on taxonomy of viruses-virus taxonomy: release. https://talk.ictvonline.org/taxonomy/. Accessed 25 Jan 2023.
Boileau MJ, Kapil S (2010) Bovine coronavirus associated syndromes. Vet Clin North Am Food Anim Pract 26:123–146. https://doi.org/10.1016/j.cvfa.2009.10.003
Park SJ, Kim GY, Choy HE, Hong YJ, Saif LJ, Jeong JH, Park SI, Kim HH, Kim SK, Shin SS, Kang MI, Cho KO (2007) Dual enteric and respiratory tropisms of winter dysentery bovine coronavirus in calves. Arch Virol 152:1885–1900. https://doi.org/10.1007/s00705-007-1005-2
Cavanagh D (1995) The coronavirus surface glycoprotein. In: Siddell SG (ed) The coronaviridae. Springer US, Boston, pp 73–113
Bidokhti MR, Traven M, Krishna NK, Munir M, Belak S, Alenius S, Cortey M (2013) Evolutionary dynamics of bovine coronaviruses: natural selection pattern of the spike gene implies adaptive evolution of the strains. J Gen Virol 94:2036–2049. https://doi.org/10.1006/viro.1996.8344
Peng G, Xu L, Lin YL, Chen L, Pasquarella JR, Holmes KV, Li F (2012) Crystal structure of bovine coronavirus spike protein lectin domain. J Biol Chem 287:41931–41938. https://doi.org/10.1074/jbc.M112.418210
Alekseev KP, Vlasova AN, Jung K, Hasoksuz M, Zhang X, Halpin R, Wang S, Ghedin E, Spiro D, Saif LJ (2008) Bovine-like coronaviruses isolated from four species of captive wild ruminants are homologous to bovine coronaviruses, based on complete genomic sequences. J Virol 82:12422–12431. https://doi.org/10.1128/jvi.01586-08
Saif LJ, Jung K (2020) Comparative Pathogenesis of Bovine and Porcine Respiratory Coronaviruses in the Animal Host Species and SARS-CoV-2 in Humans. J Clin Microbiol 58(8):e01355-e1420. https://doi.org/10.1128/JCM.01355-20
Smith FL, Heller MC, Crossley BM, Clothier KA, Anderson ML, Barnum SS, Pusterla N, Rowe JD (2022) Diarrhea outbreak associated with coronavirus infection in adult dairy goats. J Vet Intern Med 36:805–811. https://doi.org/10.1111/jvim.16354
Ballesteros ML, Sanchez CM, Enjuanes L (1997) Two amino acid changes at the N-terminus of transmissible gastroenteritis coronavirus spike protein result in the loss of enteric tropism. Virology 227:378–388. https://doi.org/10.1006/viro.1996.8344
Hasoksuz M, Sreevatsan S, Cho KO, Hoet AE, Saif LJ (2002) Molecular analysis of the S1 subunit of the spike glycoprotein of respiratory and enteric bovine coronavirus isolates. Virus Res 84:101–109. https://doi.org/10.1016/s0168-1702(02)00004-7
Rekik MR, Dea S (1994) Comparative sequence analysis of a polymorphic region of the spike glycoprotein S1 subunit of enteric bovine coronavirus isolates. Arch Virol 135:319–331. https://doi.org/10.1007/BF01310017
Suzuki T, Otake Y, Uchimoto S, Hasebe A, Goto Y (2020) Genomic characterization and phylogenetic classification of bovine coronaviruses through whole genome sequence analysis. Viruses 12(2):183. https://doi.org/10.3390/v12020183
Zhu Q, Li B, Sun D (2022) Advances in bovine coronavirus epidemiology. Viruses 14(5):1109. https://doi.org/10.3390/v14051109
Malpica JM, Fraile A, Moreno I, Obies CI, Drake JW, Garcia-Arenal F (2002) The rate and character of spontaneous mutation in an RNA virus. Genetics 162:1505–1511. https://doi.org/10.1093/genetics/162.4.1505
Chung H, Noh JY, Koo BS, Hong JJ, Kim HK (2022) SARS-CoV-2 mutations acquired during serial passage in human cell lines are consistent with several of those found in recent natural SARS-CoV-2 variants. Comput Struct Biotechnol J 20:1925–1934. https://doi.org/10.1016/j.csbj.2022.04.022
Borucki MK, Allen JE, Chen-Harris H, Zemla A, Vanier G, Mabery S, Torres C, Hullinger P, Slezak T (2013) The role of viral population diversity in adaptation of bovine coronavirus to new host environments. PLoS One 8:e52752. https://doi.org/10.1371/journal.pone.0052752
Takiuchi E, Alfieri AF, Alfieri AA (2008) Molecular analysis of the bovine coronavirus S1 gene by direct sequencing of diarrheic fecal specimens. Braz J Med Biol Res 41:277–282. https://doi.org/10.1590/s0100-879x2008000400004
Alfieri AA, Parazzi ME, Takiuchi E, Medici KC, Alfieri AF (2006) Frequency of group A rotavirus in diarrhoeic calves in Brazilian cattle herds, 1998–2002. Trop Anim Health Prod 38:521–526. https://doi.org/10.1007/s11250-006-4349-9
Takiuchi E, Stipp DT, Alfieri AF, Alfieri AA (2006) Improved detection of bovine coronavirus N gene in faeces of calves infected naturally by a semi-nested PCR assay and an internal control. J Virol Methods 131:148–154. https://doi.org/10.1016/j.jviromet.2005.08.005
Hiscox JA, Cavanagh D, Britton P (1995) Quantification of individual subgenomic mRNA species during replication of the coronavirus transmissible gastroenteritis virus. Virus Res 36:119–130. https://doi.org/10.1016/0168-1702(94)00108-o
Frucchi APS, Dall Agnol AM, Bronkhorst DE, Beuttemmuller EA, Alfieri AA, Alfieri AF (2022) Bovine coronavirus co-infection and molecular characterization in dairy calves with or without clinical respiratory disease. Front Vet Sci 9:895492. https://doi.org/10.3389/fvets.2022.895492
Vilcek S, Jackova A, Kolesarova M, Vlasakova M (2017) Genetic variability of the S1 subunit of enteric and respiratory bovine coronavirus isolates. Acta Virol 61:212–216. https://doi.org/10.4149/av_2017_02_12
Hasoksuz M, Lathrop SL, Gadfield KL, Saif LJ (1999) Isolation of bovine respiratory coronaviruses from feedlot cattle and comparison of their biological and antigenic properties with bovine enteric coronaviruses. Am J Vet Res 60:1227–1233
Reynolds DJ, Debney TG, Hall GA, Thomas LH, Parsons KR (1985) Studies on the relationship between coronaviruses from the intestinal and respiratory tracts of calves. Arch Virol 85:71–83. https://doi.org/10.1007/BF01317007
Tsunemitsu H, Yonemichi H, Hirai T, Kudo T, Onoe S, Mori K, Shimizu M (1991) Isolation of bovine coronavirus from feces and nasal swabs of calves with diarrhea. J Vet Med Sci 53:433–437. https://doi.org/10.1292/jvms.53.433
Zhang X, Herbst W, Kousoulas KG, Storz J (1994) Comparison of the S genes and the biological properties of respiratory and enteropathogenic bovine coronaviruses. Arch Virol 134:421–426. https://doi.org/10.1007/BF01310579
Chouljenko VN, Kousoulas KG, Lin X, Storz J (1998) Nucleotide and predicted amino acid sequences of all genes encoded by the 3’ genomic portion (9.5 kb) of respiratory bovine coronaviruses and comparisons among respiratory and enteric coronaviruses. Virus Genes 17:33–42. https://doi.org/10.1023/A:1008048916808
Chouljenko VN, Lin XQ, Storz J, Kousoulas KG, Gorbalenya AE (2001) Comparison of genomic and predicted amino acid sequences of respiratory and enteric bovine coronaviruses isolated from the same animal with fatal shipping pneumonia. J Gen Virol 82:2927–2933. https://doi.org/10.1099/0022-1317-82-12-2927
Hasoksuz M, Lathrop S, Al-dubaib MA, Lewis P, Saif LJ (1999) Antigenic variation among bovine enteric coronaviruses (BECV) and bovine respiratory coronaviruses (BRCV) detected using monoclonal antibodies. Arch Virol 144:2441–2447. https://doi.org/10.1007/s007050050656
Lin XQ, O’Reilly KL, Storz J, Purdy CW, Loan RW (2000) Antibody responses to respiratory coronavirus infections of cattle during shipping fever pathogenesis. Arch Virol 145:2335–2349. https://doi.org/10.1007/s007050070024
Lin XQ, O’Reilly KL, Storz J (2002) Antibody responses of cattle with respiratory coronavirus infections during pathogenesis of shipping fever pneumonia are lower with antigens of enteric strains than with those of a respiratory strain. Clin Diagn Lab Immunol 9:1010–1013. https://doi.org/10.1128/cdli.9.5.1010-1013.2002
Zhang X, Hasoksuz M, Spiro D, Halpin R, Wang S, Vlasova A, Janies D, Jones LR, Ghedin E, Saif LJ (2007) Quasispecies of bovine enteric and respiratory coronaviruses based on complete genome sequences and genetic changes after tissue culture adaptation. Virology 363:1–10. https://doi.org/10.1016/j.virol.2007.03.018
Temizkan SS, Alkan F (2021) Bovine coronavirus infections in Turkey: molecular analysis of the full-length spike gene sequences of viruses from digestive and respiratory infections. Arch Virol 166:2461–2468. https://doi.org/10.1007/s00705-021-05147-2
Schaefer C, Rost B (2012) Predict impact of single amino acid change upon protein structure. BMC genomics 13(Suppl 4):S4. https://doi.org/10.1186/1471-2164-13-S4-S4
Yoo D, Deregt D (2001) A single amino acid change within antigenic domain II of the spike protein of bovine coronavirus confers resistance to virus neutralization. Clin Diagn Lab Immunol 8:297–302. https://doi.org/10.1128/CDLI.8.2.297-302.2001
Zhang XM, Kousoulas KG, Storz J (1991) Comparison of the nucleotide and deduced amino acid sequences of the S genes specified by virulent and avirulent strains of bovine coronaviruses. Virology 183:397–404. https://doi.org/10.1016/0042-6822(91)90154-4
Gelinas AM, Boutin M, Sasseville AM, Dea S (2001) Bovine coronaviruses associated with enteric and respiratory diseases in Canadian dairy cattle display different reactivities to anti-HE monoclonal antibodies and distinct amino acid changes in their HE, S and ns4.9 protein. Virus Res 76:43–57. https://doi.org/10.1016/s0168-1702(01)00243-x
Martínez N, Brandão PE, de Souza SP, Barrera M, Santana N, de Arce HD, Pérez LJ (2012) Molecular and phylogenetic analysis of bovine coronavirus based on the spike glycoprotein gene. Infect Genet Evol 12:1870–1878. https://doi.org/10.1016/j.meegid.2012.05.007
Jeong JH, Kim GY, Yoon SS, Park SJ, Kim YJ, Sung CM, Jang OJ, Shin SS, Koh HB, Lee BJ, Lee CY, Kang MI, Kim HJ, Park NY, Cho KO (2005) Detection and isolation of winter dysentery bovine coronavirus circulated in Korea during 2002–2004. J Vet Med Sci 67:187–189. https://doi.org/10.1292/jvms.67.187
Kanno T, Hatama S, Ishihara R, Uchida I (2007) Molecular analysis of the S glycoprotein gene of bovine coronaviruses isolated in Japan from 1999 to 2006. J Gen Virol 88:1218–1224. https://doi.org/10.1099/vir.0.82635-0
Thomas CJ, Hoet AE, Sreevatsan S, Wittum TE, Briggs RE, Duff GC, Saif LJ (2006) Transmission of bovine coronavirus and serologic responses in feedlot calves under field conditions. Am J Vet Res 67:1412–1420. https://doi.org/10.2460/ajvr.67.8.1412
Cho KO, Hasoksuz M, Nielsen PR (2001) Cross-protection studies between respiratory and calf diarrhea and winter dysentery coronavirus strains in calves and RT-PCR and nested PCR for their detection. Arch Virol 146:2401–2419. https://doi.org/10.1007/s007050170011
Yoo D, Parker MD, Song J, Cov GJ, Deregt D, Babiuk LA (1991) Structural analysis of the conformational domains involved in neutralization of bovine coronavirus using deletion mutants of the spike glycoprotein S1 subunit expressed by recombinant baculoviruses. Virology 183:91–98. https://doi.org/10.1016/0042-6822(91)90121-q
Meitzler JL, Hinde S, Banfi B, Nauseef WM, Ortiz de Montellano PR (2013) Conserved cysteine residues provide a protein-protein interaction surface in dual oxidase (DUOX) proteins. J Biol Chem 288:7147–7157. https://doi.org/10.1074/jbc.M112.414797
Morand S, Agnandji D, Noel-Hudson MS, Nicolas V, Buisson S, Macon-Lemaitre L, Gnidehou S, Kaniewski J, Ohayon R, Virion A, Dupuy C (2004) Targeting of the dual oxidase 2 N-terminal region to the plasma membrane. J Biol Chem 279:30244–30251. https://doi.org/10.1074/jbc.M405406200
Acknowledgements
The authors would like to thank the Brazilian Federal Agency for Support and Evaluation of Graduate Education (CAPES/MEC) for granting a scholarship to the first author.
Funding
This work was supported by Universidade Federal do Paraná-UFPR, Universidade Estadual de Londrina-UEL, CAPES/MEC-Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, and National Institute of Science and Technology of Dairy Production Chain (INCT-Leite / CNPq, CAPES, and Araucaria Foundation-FAP/PR).
Author information
Authors and Affiliations
Contributions
Janaina Lustosa de Mello: Conceptualization, Methodology, Investigation, Data curation, Formal analysis, Writing-original draft. Daniela Lorencena: Investigation. Ruana Renostro Delai: Investigation, Data curation. Andressa Fernanda Kunz: Investigation, Validation. Flávia Possatti: Investigation, Data curation. Amauri Alcindo Alfieri: Visualization, Writing-review & editing. Elisabete Takiuchi: Conceptualization, Formal analysis, Supervision, Visualization, Writing-review & editing.
Corresponding author
Ethics declarations
Ethics approval
This study was approved by the Ethics Committee for the Use of Animals of the Federal University of Paraná, under protocol nº. 34/2016.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Wrong supplementary file 1 had been published.
Responsible Editor: Fernando R. Spilki
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
de Mello, J.L., Lorencena, D., Delai, R.R. et al. A comprehensive molecular analysis of bovine coronavirus strains isolated from Brazil and comparison of a wild-type and cell culture-adapted strain associated with respiratory disease. Braz J Microbiol 55, 1967–1977 (2024). https://doi.org/10.1007/s42770-024-01287-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-024-01287-0