
Abstract
Humans frequently contract urinary tract infections (UTIs), which can be brought on by uropathogens (UPs) that are multi-drug resistant. Treatment for UTIs brought on by pathogenic UPs that produce extended-spectrum lactamases (ESBLs) is more costly and potentially fatal. As a result, the objective of this study was to use culture, biochemical, and 16S rRNA sequencing to identify and characterize UPs isolated from outpatients in Noakhali, Bangladesh, who had symptoms of UTIs. ESBL gene identification and quinolone resistance gene typing were then performed on the isolates using polymerase chain reaction (PCR). Throughout the trial’s 8-month duration, 152 (76%) of 200 urine samples were positive for the presence of UPs. The overall number of UPs recovered was 210, with 39 individuals having multiple UPs present in their samples. Among all of the isolates, Escherichia coli (45.24%, 95/210; 95% confidence interval (CI): 35.15–57.60%), Enterobacter spp. (24.76%, 52/210; CI: 19.15–35.77%), Klebsiella spp. (20.95%; 44/210; CI: 15.15–30.20%), and Providencia spp. (9.05%; 19/210; CI: 4.95–19.25%) were the four most prevalent bacteria found in the isolates. The UPs displayed a very high level of resistance to piperacillin 96.92% (126/130), ampicillin 90% (117/130), nalidixic acid 77.69% (101/130), cefazolin 70% (91/130), amoxicillin 50% (55/130), cefazolin 42.31% (55/130), nitrofurantoin 43.08% (56/130), and ciprofloxacin 33.08% (43/130), whereas resistance to netilmicin (3.85%), amikacin (4.62%), and imipenem (9.23%) was low. Individually, every species of E. coli and Providencia spp. showed greater ampicillin, amikacin, cefazolin, cefazolin, and nalidixic acid resistance than the others. The bivariate results indicate several antibiotic pairings, and isolates had meaningful associations. All MDR isolates were subjected to PCR, which revealed that blaCTX-M-15 genes predominated among the isolates, followed by the blaTEM class (37%). Isolates also had the qnrS, aac-6´-Ib-cr, and gyrA genes. The findings provide worrying indications of a major expansion of MDR isolates in the study locations, particularly the epidemiological balCTX-M 15, with the potential for the transmission of multi-drug-resistant UP strains in the population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introductions
Every year, 150 to 250 million individuals worldwide are affected by urinary tract infections (UTIs), one of the recognized bacterial illnesses [1]. Age and gender have different impacts on the severity of UTIs. Due to the difference in their anatomy from men’s, it is more prevalent in the female population [2]. With their detrimental effects on health, UTI raises some serious issues. Dysuria, frequency, urgency, suprapubic pain, costovertebral soreness, chills, and fever are typical symptoms [3, 4]. Around 75% of UTI infections are caused by UPs Escherichia coli (UPEC), making it the most common bacteria. Other species that may cause the illness include Klebsiella pneumoniae, Proteus, Acinetobacter, Staphylococcus spp., group B Streptococcus (GBS), Citrobacter spp., Enterobacter aerogenes, Salmonella spp., and Pseudomonas aeruginosa [5,6,7].
Additionally, contamination jointly by many bacterial populations would result in more severe circumstances than the others [8, 9]. Antibiotic administration is the traditional strategy for treating UTIs [9]. Due to increased antibiotic resistance, many UPs cannot be treated today [10]. An error rate of more than 30% may occur when a diagnosis is made based on clinical symptoms rather than laboratory tests [11]. However, many underdeveloped nations do so to cut costs and increase antimicrobial resistance [8]. Antibiotic misuse and self-medication worsened the situation, especially in the case of UP treatment [12]. Up to 60% of the antimicrobial medications administered in American ICUs are nonsensical, inappropriate, or subpar [13]. In the UK, 69% of all prescriptions were for antibiotics, and more than 20% of them were judged unsuitable [11].
Moreover, Asia, Africa, the Middle East, and Europe continue to face an increasing issue with multi-drug-resistant bacteria [14,15,16]. Bangladesh has a high prevalence of UTI illnesses and is struggling with the serious issue of antibiotic resistance UPs. According to a recent UK survey, antibiotic prescriptions in primary care are declining; however, this is not the situation in Bangladesh [17]. DNA from both chromosomal and plasmid sources is crucial in developing antibiotic resistance. ESBL-producing bacteria are increasingly more prevalent among clinical isolates, as it has been observed. Producing lactamase makes microorganisms resistant to lactam antibiotics, particularly Gram-negative bacteria. These plasmid-borne enzymes confer MDR, making it harder to treat these infections [18, 19]. Extended-spectrum lactamase (ESBL)–producing microbes have become more prevalent recently, limiting the range of available treatments. Due to the synthesis of several beta-lactamases, including CTX-M, TEM, and SHV, which are encoded by genes for blaCTX-M, blaTEM, and blaSHV, respectively, they become resistant to third-generation cephalosporins, aminoglycosides, and fluoroquinolones [20, 21]. The medicine of choice for resistant strains is now carbapenem due to the rise of 3rd generation antibiotic resistance. But during the past 20 years, rising carbapenem usage revealed increased carbapenem resistance [22]. Moreover, the development of resistance against one antibiotic may also influence the outcome of another antibiotic resistance [17]. Healthcare authorizations will run out of treatment alternatives, increasing hospitalization if this pattern of antibiotic overuse persists [15]. When administering antibiotics, healthcare providers should exercise deliberate decision-making. In light of the localized way of antibiotic resistance, they ought to recommend antibiotics [13]. We require a ton of data regarding the resistance pattern to do this. The antibiotic susceptibility data of UPs are distributed differently from time to time and location to location. Regular monitoring in a specific location is required due to rising antibiotic resistance in UTIs. The current study aims to identify probable MDR UPs in both culture-dependent and independent ways, followed by quantifying the frequency of UTIs among outpatients in Noakhali, Bangladesh, and identifying the genes responsible for medication resistance.
Materials and methods
Study sites, patient selection, sampling, and processing of urine samples
All urine samples were taken between June 2021 and January 2022 at several neighborhood hospitals in Maijdhee (22.87091° N, 91.0938° E), Noakhali, Bangladesh (Fig. 1; Supplementary Fig. 1, obtained from the Google Map). All patients who visited the indoor and outdoor departments of the several community hospitals in Maijdhee, Noakhali, for a confirmed diagnosis of UTI were taken into consideration for the study. Patients with UTI symptoms and signs who had received a clinical diagnosis of a UTI from nearby physicians and who voluntarily consented to participate, signed a permission form, and consented to submit a midstream specimen of urine (MSU) were evaluated for inclusion. So, after obtaining consent, the sampling process was described and each participant was given a sterile urine collection cup (BD, Woodmead, South Africa). Sedation, cremation, or animal sacrifice was not employed in this experiment. Two hundred fresh urine samples were obtained from the study locations (approximately 20–30 ml). The samples were collected with the method previously mentioned [23]. After collection, samples were immediately maintained at 4 °C and shipped on ice to the department of microbiology’s laboratory at NSTU for examination within 8 h. The total heterotrophic count was performed by serially diluting samples in normal saline (up to a dilution factor of 103), then plating 100 µl of each dilution on nutrient agar medium. The plates were then incubated at 37 °C for one night.

UPs investigations of UTI infections in Bangladesh are shown graphically. Studies characterize UPs AMR properties in UTI samples from sick people by using both phenotypic (selected culture and biochemical testing) and molecular methods (16S rRNA gene sequencing, marker-specific PCR). The majority of UTI cases in Bangladesh are caused by phylogenetically related, highly infectious, multi-drug-resistant (MDR) UP strains
Culture-dependent identifications of the UPs
For the potential detection of Enterobacteriaceae, midstream specimen of urine (MSU) was inoculated onto Nutrient Agar (NA) (Oxoid™, Thermo Scientific, UK), MacConkey Agar (MAC) (Oxoid™, Thermo Scientific, UK), and Eosin Methylene Blue (EMB) agar (Oxoid™, Thermo Scientific, UK). The presence of two different colony types in equal numbers was viewed as a possible co-infection, whereas the growth of 105 colony forming units (CFU)/ml of colonies with comparable shape was considered positive for UTIs [24]. Pure colonies were obtained by choosing morphologically different colonies from each plate and streaking them further on EMB and MAC plates. Single colonies were again selected and subcultured on NA plates. All plates were incubated at 37 °C for 18 h. These microorganisms were identified based on their colony morphology, and Gram-staining [25]. Gram-positive bacteria were further confirmed based on their biochemical characteristics in triple sugar iron, indole, methyl-red, Voges-Proskauer, citrate, catalase, and oxidase tests. The 16S rRNA gene sequencing method was used for further culture-independent (not based on cultivation to study microorganisms in a specific ecosystem) identification of the selected UPs.
Detection of multi-drug resistant profile of UPs
Seventy identified bacterial populations were examined for the purpose of detecting the multi-drug resistance (MDR) impact. The in vitro antibiogram profile of UP isolates was determined using the disk diffusion method following the Clinical Laboratory Standards Institute (CLSI) 2022 guidelines. Using the traditional disk diffusion approach, the isolated bacteria’s pattern of antibiotic resistance to commonly used antimicrobial medicines was identified. We employed 16 wide spectrum conventional antibiotics, including AMP = Ampicillin (10 µg), AX = Amoxicillin (10 µg), CFM = Cefepime (30 µg), CTX = Cefotaxime (30 µg), CAZ = Cefazolin (30 µg), CTR = Ceftazidime (30 µg), CZ = Cefazolin (30 µg), CIP = Ciprofloxacin (30 µg), LE = Levofloxacin (5 µg), AK = Amikacin (30 µg), NET = Netilmicin (30 µg), PC = Piperacillin (100 µg), F = Nitrofurantoin (300 µg), NA = Nalidixic Acid (30 µg), IPM = Imipenem (10 µg), and TE = Tetracycline (30 µg).The antibiotic disks were then placed on the surface of the seeded plates using sterile forceps in the proper spatial arrangement after each isolate was scattered across the surface of Mueller–Hinton plates that had been manufactured. After 24 h of infection at 37 °C, the distinct zone of inhibition was visible on the plates. After incubation, the zones of total inhibition were assessed. The sensitivity patterns were established using a calibrated ruler and then assessed using typical Clinical Laboratory Standards Institute (CLSI) standards [26] with slight modifications where E. coli DH5α ATCC 53868 was used as a quality control strain of antibiotics [16].
Molecular identification of the UPs and phylogenetic analyses
According to [27, 28], bacterial DNA was extracted from biochemically characterized UP isolates and the 16S rRNA gene was sequenced. The genomic DNA of the isolates was first extracted using the boiling method [29]. Using the universal primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-CTACGGCTACCTTGTTACGA-3′), the 16S rRNA genes of representative randomly chosen isolates were amplified by PCR and sequenced [30]. Agarose gel electrophoresis (1.2% wt/vol) was used to validate the presence of PCR products. To see the DNA, 0.5 g/ml ethidium bromide was utilized. To assemble forward and reverse sequences, the SeqMan Genome Assembler [31] was used. The forward and reverse sequences were then compared to the partial 16S rRNA sequences in the GenBank database of the National Center for Biotechnology Information (NCBI) using the Basic Local Alignment Search Tool (BLAST). Molecular Evolutionary Genetics Analysis (MEGA) version 7.0 was used to align the 16S rRNA sequences (Sanger sequencing), which had been amplified from each unique bacterial isolate, with each other and with pertinent reference sequences retrieved from the NCBI database [32]. The phylogenetic tree was constructed using the maximum-likelihood method, and the evolutionary distances were calculated using the Kimura-Nei method (Kimura MA, 1980).
Molecular characterization of antibiotics resistance genes
PCR was used to find patterns in the resistance genes. Using particular primers, PCR amplification was used to detect class A β-lactamase genes (blaTEM, blaCTX-M-15, blaSIM-1), carbapenemase genes (blaSIM-1, blaIMP-4), aminoglycoside and ciprofloxacin resistance genes (aac-6'-Ib-cr), and plasmid-mediated quinolone resistance genes (gyrA, qnrS). The same primers were utilized, and Supplementary table 1 [33,34,35,36,37] displays the anticipated PCR product lengths. Using a Bio-Rad ChemiDoc™ Imaging System, amplicons were separated using electrophoresis in 1.5% agarose gel [28].
Statistical analysis
All the data was entered into an Excel sheet and uploaded onto the SPSS software (version 23.0 IBM, Armonk, NY). Multidrug-resistant (MDR) UP prevalence was calculated and reported as a percentage.
Data avail ability statement
All data are provided within the text, tables, and figures. The sequences of 15 bacterial 16S rRNA genomes were submitted to the NCBI database under the identifiers. The raw reads were submitted to the NCBI accession numbers OP514787–OP514801.
Results
Prevalence of concerning UPs in urine samples
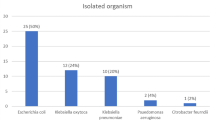
In urine samples, the prevalence of positive sample sources was 152 (76%) (p = 0.007). At first, a total heterotrophic plate count was performed to provide a snapshot of the overall bacterial population in samples. The heterotrophic plate count showed that the patient samples contained anything from 1.211 × 106 CFU/ml to 2.68 × 107 CFU/ml of viable bacteria. Finally, 210 UP isolates covering E. coli, Enterobacter spp., Klebsiella spp., and Providencia spp. were obtained from these 152 patient urine samples through cultural assay and finally confirmed by Gram-staining and biochemical assays. Out of 152 samples, 73 (48.03%) were confirmed to be positive for E. coli which was significantly higher (chi-square test, 95% CI, p = 0.008) compared to Enterobacter spp. 20 (13.16%), Klebsiella spp. 14 (9.21%), and Providencia spp. 13 (8.55%). Interestingly, in 39/152 (25.66%) (p = 0.004), samples harbored mixed UPs, in which 20 (13.16%) (p = 0.002) samples contained a combination of three UP strains (e.g., E. coli, Klebsiella spp., and Providencia spp.) where 19 (12.5%) (p = 0.001) samples contain a combination of two UP strains (e.g., E. coli, Klebsiella spp.). Under standard growth conditions, bacteria were identified as follows: E. coli (95/210; 45.24%; 95% confidence interval (CI): 35.15–57.60%), Enterobacter spp. (52/210; 24.76%; 95% confidence interval (CI): 19.15–35.77%), Klebsiella spp. (44/210; 20.95%; 95% confidence interval (CI): 15.15–30.20%), and Providencia spp. (19/210; 9.05%; 95% confidence interval (CI): 4.95–19.25%) from urine samples (Table 1).
UP identification by different biochemical tests
The indole test yielded positive results for Providencia spp. but negative results for Enterobacter. The gelatin hydrolysis and H2S test was the additional identification test that was carried out particularly for Providencia spp. Both responses were shown to be adverse. The indole test was found to be positive for every E. coli isolate, but the citrate and urease tests yielded negative results. The urease test yielded positive results for Klebsiella spp.
Molecular identification and phylogenetic analysis of UP strains
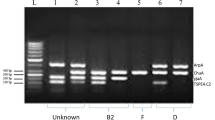
With the use of 16S rRNA sequencing, potential UP strains were discovered at random. The PCR amplification of 16S rRNA gene was done for 15 randomly UP isolates to detect the species of UPs. In this study, the PCR product appeared as a single-band DNA with a size equal to 1500 bp fragment corresponding to the 16S rRNA amplicon (Fig. 2). After being blasted, the 16S rRNA sequences were eventually submitted in GenBank with accession numbers (https://www.ncbi.nlm.nih.gov/genbank/). Additionally, figure depicts the phylogenic tree of probable UP strains based on 16S rRNA gene sequences in comparison to isolates and type strains. The 16S DNA sequences from the GenBank database and the sequences from the UP strains under study were aligned. Klebsiella pneumoniae, Enterobacter asburiae, Enterobacter cloacae, Escherichia coli, and Providencia stuartii were among the 15 UP isolates that were discovered. As can be shown, Providencia spp. isolate ASB5 has a 16S rRNA sequence that is most closely related to Providencia stuartii (97%) that was previously isolated from the patient's urine samples in China and the USA. Furthermore, Klebsiella isolates ASB160 and ASB22 from patient blood and stool samples in Tanzania and China are most closely related to Klebsiella pneumoniae (98%) than any other isolates. Additionally, the Enterobacter isolates ASB110 and ASB113 are most closely related to the previously isolated Enterobacter asburiae (99%) and Enterobacter cloacae (96%), which were both mostly recovered from water samples. In this tree, two groups of Escherichia coli subspecies strains were identified. In the first isolate cluster, ASBX16, ASB2, and ASBY05 had a 95–99% similarity to recovered Escherichia coli from non-human sources. While this is happening, second cluster isolates ASB102, ASB115, ASB137c, ASB63, ASBU20, and ASBU9 show a high degree of similarity to human urine-isolated E. coli (99%) (Fig. 3).

Following the amplification of 16S rRNA, PCR products were run on an agarose gel; lane 1 represents a 1-kb marker (PROMEGA, USA), lane 3 represents a positive control, lane 2 represents a negative control, and lanes 4, 5,6, 7, 8, 9, and 10 represent the positive UP strains ASB22, ASB10, ASB20, ASB11, ASB115, ASB37, and ASB2 at 1500 bp. Red box indicating the position of gene

Based on the nucleotide sequences of the truncated 16S rRNA gene, phylogenetic inference of the UPs (blue mark). A variety of microorganisms were represented by the selection of bacterial reference sequences. As percentages, 40% bootstrap values (n = 1000 replicates) are given. The scale bar displays the number of modifications per nucleotide site. Staphylococcus aureus served as the out-group. The accession numbers, sources, and isolated countries are indicated after each sequence. Clusters contained closely linked genomic sequences with a low branch deviation (cut of 0.0005). In the UPB from Noakhali, Bangladesh, 16S rRNA sequences are split into five clades designated as group I to group V. Here, fuchsia, blue, green, and red colors indicate the various group represented by Klebsiella spp., Enterobacter spp., E. coli, Providencia spp. respectively. The best-fitting substitution model (Kimura 2-parameter distance) was used to construct the tree using the MEGA 7 program
Antimicrobial susceptibility profile
The antimicrobial treatment response of each of the isolates of E. coli, Enterobacter spp., Klebsiella spp., and Providencia spp. was examined as indicated in (Table 2). Antibiotic resistances in E. coli isolates were detectable for each of the 16 tested antibiotics. 100%, 100%, 90.91%, 84.09%, 72.72%, 68.18%, 59.09%, 59.09%, 50%, 45.45%, 45.45%, 40.91%, and 31.82% of all isolated E. coli from urine were resistant to TE, PC, AMP, CZ, NA, AX, CFM, CTR, CTX, CAZ, LE, F, and CIP respectively (Fig. 4; Table 2). UTI E. coli isolates had lower resistance in AK, IPM, and NET (Table 2). Bivariate analysis showed high positive associations between NA and AX (p = 0.035), LE and CAZ (p = 0.006), CTR and AX (p = 0.001), CTR and CAZ (p = 0.044), and NA and AMP (p = 0.008) resistance profiles (Fig. 5). In a similar fashion, Klebsiella spp. isolated from UTI samples were found to be inhibited at the same level by AX, CZ, LE, and F (50%) (Fig. 4; Table 2). To make matters worse, Klebsiella spp. showed substantial increases in antibiotic resistance in response to all (AMP 100%, TE 100%, NA 83.33%, and CIP 62.5%) except for CTX (16.67%) and CAZ (33.33%) of the antibiotics assayed (Fig. 4; Table 2). Most interestingly, antibiotics namely CFM, CTR, AK, and NET are still very effective drugs in the case of UTI Klebsiella spp. Bivariate analysis revealed a strong positive significant association between F and AX (p = 0.004), CZ and AX (p = 0.008), LE and AX (p = 0.033), CZ and CAZ (p = 0.002), F and CZ (p = 0.047), LE and CZ (p = 0.0053), F and CIP (p = 0.081), F and LE (p = 0.013), LE and CIP (p = 0.002), and CAZ and CTX (p = 0.022) (Fig. 5; Supplementary table 2). We also looked for moderately significant correlations (both positive and negative) between a number of antibiotic combinations and their ability to kill Klebsiella spp. (Fig. 5). Isolates of Providencia spp. from urinary tract infections showed variable (between 53 and 91%) resistance to AMP, TE, CZ, PC, NA, and CAZ (Table 2). CTX and AX (p = 0.003), CAZ and AX (p = 0.025), IPM and CFM (p = 0.017), CIP and CTR (p = 0.008), TE and PC (p = 0.001), LE and CIP (p = 0.005), and AK and NET (p = 0.004) all showed high to moderately significant positive relationships (Fig. 5; Supplementary table 2). Additionally, strong negative connections were also seen (Fig. 5). Resistance rates to TE, AMP, NA, PC, and CZ were 100% (30/30), 83.33% (25/30), 93.33% (28/30), 80% (22/30), 66.67% (20/22), and 66.67% (20/20) among the 30 Enterobacter spp. isolates tested (Table 2). CTX and CFM had a very significant positive relationship (p = 0.004), as did LE and AX (p = 0.002), F and CZ (p = 0.0035), and CZ and AX (p = 0.016), as determined by bivariate analysis (Fig. 5; Supplementary table 2). We also looked for moderately significant relationships (both positive and negative) between antibiotic combinations and Enterobacter spp. resistance (Fig. 4).

Antibiotic resistance pattern of UPs by disk diffusion method. The antimicrobial resistance (AMR) patterns of the four UPs obtained from 152 UTI samples (E. coli, 44; Klebsiella spp., 24; Enterobacter spp., 30; Providencia spp., 32) for sixteen commonly used. Here, AMP, Ampicillin; AX, Amoxicillin; CFM, Cefepime; CTX, Cefotaxime; CAZ, Cefazolin; CTR, Ceftazidime; CZ, Cefazolin; CIP, Ciprofloxacin; LE, Levofloxacin; AK, Amikacin; NET, Netilmicin; PC, Piperacillin; F, Nitrofurantoin; NA, Nalidixic Acid; IPM, Imipenem; TE, Tetracycline. More details about AMR profiles can be found in the text and in Table 2

Pearson correlation coefficients in between any of two antibiotics resistance to bacterial isolates (A Providencia; B E. coli; C Enterobacter; D Klebsiella). A p-value < 0.05 was deemed as statistically significant; orange, highly significant; yellow, moderately significant; light blue, non-significant; AMP, Ampicillin; AX, Amoxicillin; CFM, Cefepime; CTX, Cefotaxime; CAZ, Cefazolin; CTR, Ceftazidime; CZ, Cefazolin; CIP, Ciprofloxacin; LE, Levofloxacin; AK, Amikacin; NET, Netilmicin; PC, Piperacillin; F, Nitrofurantoin; NA, Nalidixic Acid; IPM, Imipenem; TE, Tetracycline
Genotypes of antimicrobial resistant genes
AMR genes blaCTX-M-15, blaTEM, qnrS, aac-6'-Ib-cr, and gyrA were detected by gene-specific PCR in 70 out of 85 resistant isolates, accounting for 96% of the total. Fourteen of these 70 isolates (87.5%) contained several ESBL genes as opposed to only one. blaCTX-M-15 (29 out of 70 isolates, 41%) and blaTEM (26 out of 70 isolates, 37%) were the two most common ESBL genes found, while all isolates lack the other three genes blaSIM-1, blaSHV, and blaIMP-4. PCR amplification for genes product is shown in Fig. 5. Additionally, electrophoresis revealed the presence of the qnrS, aac-6'-Ib-cr, and gyrA genes in 40% (28/70), 48% (34/70), and 70% (49/70) of the UP isolates, respectively (Fig. 5).
Discussion
One of the most prevalent bacterial infections in hospitals is urinary tract infection (UTIs), which is challenging to treat [38]. Antibiotic usage is on the rise, which increases the number of germs that are resistant to numerous drugs and makes the situation more precarious. Therefore, intensifying efforts to solve this issue is essential for delivering better healthcare. The purpose of this study was to describe the ESBL genes in UTI isolates obtained from patients who attended the neighborhood hospitals in the Noakhali district, with a particular emphasis on the frequency of gram-negative bacteria and their pattern of antibiotic resistance (Fig. 1). We have also concentrated on MDR co-infection in UTI patients. In this study, we found that the total number of positive samples was 152 (76%), of which 19 (12.5%) were composed of prevalent unimicrobial organisms and 20 (13.16%) of which were composed of polymicrobial organisms. This number was higher than that reported in the study by Haque et al. [39] which found that 59.67% of samples were positive, with 94.41% of those consisting of unimicrobial and 5.59% of those composed of polymicrobial microorganism growths. Another research conducted in Bangladesh found that 2% of samples had polymicrobial growth and that 31.67% of samples had unimicrobial growth [40]. Patient difficulties during UTI may be attributable to the presence of numerous UPs in the same sample, albeit this has not been validated. This coinfection has the potential to significantly impact the difficulty of treating UTIs. According to our research, E. coli, Enterobacter spp., Klebsiella spp., and Providencia spp. were the most common microorganisms. The only difference between [40] and [41] is that they did not locate Providencia spp. However, E. coli, Klebsiella spp., Providencia spp., and many more species were discovered in prior research by [42]. This variance might be due to the temporal, geographical, and environmental factors of the patients under investigation. Compared to the others, we recorded a larger number of E. coli cases. Given the biological characteristics of women, it is possible that a sizable colony of E. coli is already flourishing in female feces. Importantly, the etiology of UTI may have other causes due to its ascending character [43, 44]. Numerous investigations have noted the increased prevalence of E. coli in UTI samples [45,46,47]. Another major public health worry, along with the co-infection of bacteria in UTI samples, is the development of AMR. The vulnerability of the patient’s treatment and life has increased as a result of rising antibiotic resistance among UPs in Bangladesh [48]. MDR UPs emerge as antibiotics are used more often in therapeutic settings. In this investigation, we found that AMP, NA, PC, and TE resistance was present in more than 72% of isolated E. coli, Klebsiella spp., Enterobacter spp., and Providencia spp. A similar report was made for AX and NA resistance patterns in research by [39]. The resistance rate of UPEC for fluoroquinolone and third-generation cephalosporin (3GC) was greater but lower in nitrofurantoin demonstrated, according to several other types of research by [39, 43, 49]. However, we discovered lesser resistance to CIP and increased resistance to 3GC and nitrofurantoin in the current investigation for E. coli, which is entirely consistent with the findings of [17]. Our research showed that resistance to 3GC is much lower in Klebsiella spp., Enterobacter spp., and Providencia spp. than it is in E. coli, in contradiction to the results of a prior experiment by Haque et al. (2015) [39]. When compared to Klebsiella spp. in this study, the resistance patterns of Enterobacter spp. and E. coli to CIP are higher in 3GC and lower. However, according to a different research [50], CIP resistance among K. pneumoniae isolates was low (10%), whereas AMP and F resistance were both prevalent. In the past, 3GC and nitrofurantoin [17] medications have been utilized as options for treating bacterial infections for many years. However, the incidence of greater resistance to such treatments suggests that these medications are no longer useful for treating UTI patients [43]. CIP is the medicine of choice now in Bangladesh for treating both severe and simple UTIs caused by bacteria [39]. According to Akram et al. and Rani et al. [4, 51], E. coli in India was significantly resistant to NET, AK and IPM. In contrast, we discovered in the current investigation that NET, IPM, and AK exhibit lesser resistance to E. coli, which was completely consistent with the earlier studies by [4, 43]. Intriguingly, Soleimani et al. [52] reported a similar finding to our findings in the cases of Klebsiella spp., Enterobacter spp., and Providencia spp. in one of the earlier research carried out in Saudi Arabia. IPM is hence a potent antibacterial drug that is being utilized to treat a variety of illnesses.
A significant obstacle to the fight against infections is the proliferation of ESBL among Enterobacteriaceae and other Gram-negative pathogens in the community and hospitals. All around the world, the prevalence of ESBL-producing microbes is rising. When it comes to β-lactam antibiotics like penicillins, cephalosporins, monobactams, and even carbapenems, the incidence of ESBL-producing Enterobacteriaceae strains differs substantially between Asian nations [53]. Our research revealed that blaCTX-M-15 was the most often seen ESBL gene (41% or 29 of 70 isolates), followed by blaTEM (37% or 26 of 70 isolates) (Fig. 6). Our results corroborate those of previous and current studies on hospital and community-acquired UTI [14, 16] and are in line with those of studies conducted in the same setting that also found CTX-M-15 to be the most common ESBL genotype[17]. It is widely known that CTX-M-15 and other ESBLs of the CTX-M type propagate rapidly among members of the family Enterobacteriaceae. Some researchers have speculated that CTX-M enzymes might have originated from the extensive usage of ceftriaxone and cefotaxime [18]. Because blaTEM was the second most prevalent ESBL gene, it is possible that TEM-type ESBLs are present in at least some community strains. We also found differences in antibiotic resistance among blaTEMonly isolates, which may point to the presence of ESBLs that are inhibited by clavulanic acid but were not uncovered in our analysis, or to the presence of additional genes of the blaTEM type. In this work, strains of quinolone-resistant bacteria containing the plasmid-mediated quinolone resistance (PMQR) genes qnrS and aac-6'-Ib-cr were shown to be resistant to antibiotics. aac-6'-Ib-cr was the most often found PMQR gene (48%), which is consistent with other research where aac-6'-Ib-cr gene detection was more prevalent in MDR UP isolates [19, 21, 54]. Even while PMQR genes only produce modest amounts of FQ resistance, it has been discovered that these genes promote the selection of further chromosome-encoded resistance mechanisms [22]. In this investigation, almost 70% of isolates had quinolone resistance-determining regions (QRDR) mutations in the gene gyrA. Previous investigations carried out in Jordan [19, 21] also revealed a presence of gyrA gene strongly correlated with the resistant of fluoroquinolones groups of antibiotics. Although they have not done sequencing for the conformation of mutation as like our study. The widespread and ineffective use of quinolones as empirical therapy for UTIs may help to explain these high rates. According to Paiva et al. [55], plasmid DNA contained the ciprofloxacin resistance gene in UPs E. coli. Fluoroquinolone resistance led to the evolution of several mutations in several genes, which are finally reflected in the phenotypic traits of the resistant organism. In our research, we discovered that UTI-causing organisms that have the ESBL and quinolone resistance genes exhibit a high level of antibiotic resistance (AR). AR UPs appear to be extensively distributed in both community and hospital settings. The high levels of bacterial antibiotic resistance may be due to a variety of factors. One of them is that the pace of rising resistance frequency varies from organism to organism, possibly as a result of regional differences. Another potential cause of antibiotic abuse in our nation is the inappropriate use of antibiotics, such as self-medication.

ARG analysis of representative UPs. Red box indicates the appropriate position of the bands. Here, A blaCTX-M-15 and blaTEM gene product, B blaTEM gene, and C qnrS and aac-6´-Ib-cr gene product as well as gyrA gene product. In figure A, lane 1 represents a 1-kB marker (PROMEGA, USA), lanes 2 and 9 represent negative controls, lanes 5 and 10 represent a positive control, and other lanes represent the positive gene products at 688 bp and 861 bp, respectively. In figure B, lane 1 represents a 100-bp marker (PROMEGA, USA), lanes 3 and 7 represents positive controls, lanes 2 and 6 represent a negative control, and other lanes represent the positive gene products at 428 bp and 519 bp, respectively. In figure C, lane 1 represents a 100-bp marker (PROMEGA, USA), lane 2 represents a negative control, lane 3 represents a positive control, and other lanes represent the positive gene products at 490 bp
Conclusions
In occurrences of UTI in Bangladesh, UPs caused by E. coli, Enterobacter spp., Klebsiella spp., and Providencia spp. remain a major cause for concern. The fact that E. coli has the highest incidence in Bangladesh (> 45.0%), followed by the other three, is a major public health concern. Urinary pathogens (UPs) recovered from patients are related to numerous harmful bacterial strains reported from throughout the globe, as shown by phylogenetic research. UPs were shown to be an MDR pathogen due to the presence of blaCTX-M-15 and blaTEM genes, as well as three other AMR genes that are roughly connected to aminoglycoside and ciprofloxacin, as determined by the antibiotic sensitivity testing. Based on our findings, the relatively high prevalence of MDR across all β-lactamase genes and plasmid-mediated quinolone resistance genes with UP phenotypes in Bangladesh is a cause for worry and may have far-reaching consequences for the environment. However, more work has to be done in the future to comprehend the circulation of antimicrobial use and to monitor the spread of ARGs with greater population numbers in various ecosystems of the medical sectors of Bangladesh.
Data availability
The data relating to this manuscript are available upon request.
References
Masajtis-Zagajewska A, Nowicki M (2017) New markers of urinary tract infection. Clin Chim Acta 471:286–291
Mishra MP, Sarangi R, Padhy RN (2016) Prevalence of multidrug resistant uropathogenic bacteria in pediatric patients of a tertiary care hospital in eastern India. J Infect Public Health 9(3):308–314
Ahmed N, Khalid H, Mushtaq M, Basha S, Rabaan AA, Garout M, Yean CY (2022) The molecular characterization of virulence determinants and antibiotic resistance patterns in human bacterial uropathogens. Antibiotics 11(4):516
Akram M, Shahid M, Khan AU (2007) Etiology and antibiotic resistance patterns of community-acquired urinary tract infections in JNMC Hospital Aligarh, India. Ann Clin Microbiol Antimicrob 6(1):1–7
Balachandran L, Jacob L, Al Awadhi R, Yahya LO, Catroon KM, Soundararajan LP,... Hussein YA (2022) Urinary tract infection in pregnancy and its effects on maternal and perinatal outcome: a retrospective study. Cureus 14(1)
Call ZD, Jang I, Geiss BJ, Dandy DS, Henry CS (2022) Progress toward a simplified UTI diagnostic: pump-free magnetophoresis for E. coli detection. Anal Chem 94(21):7545–7550
Gililland JM, Anderson LA, Erickson J, Pelt CE & Peters CL (2013) Mean 5-year clinical and radiographic outcomes of cementless total hip arthroplasty in patients under the age of 30. BioMed research international 2013
Khoshnood S, Heidary M, Mirnejad R, Bahramian A, Sedighi M, Mirzaei H (2017) Drug-resistant gram-negative uropathogens: a review. Biomed Pharmacother 94:982–994
Mollick S, Dasgupta T, Hasnain MJ, Ahmed M (2016) Isolation and characterization of pathogens responsible for urinary tract infection in Bangladesh and determination of their antibiotic susceptibility pattern. J Appl Pharmaceut Sci 6(4):072–076
Ahmed OB, Omar AO, Asghar AH, Elhassan MM, Al-Munawwarah AM, Arabia S (2013) Prevalence of TEM, SHV and CTX-M genes in Escherichia coli and Klebsiella spp urinary isolates from Sudan with confirmed ESBL phenotype. Life Sci J 10(2):191–195
Al-Naqshbandi AA, Chawsheen MA, Abdulqader HH (2019) Prevalence and antimicrobial susceptibility of bacterial pathogens isolated from urine specimens received in rizgary hospital—Erbil. J Infect Public Health 12(3):330–336
Jassim A-M (2010) In-home drug storage and self-medication with antimicrobial drugs in Basrah. Iraq Oman Med J 25(2):79
Paul R (2018) State of the globe: rising antimicrobial resistance of pathogens in urinary tract infection. J Global Infect Dis 10(3):117
Alizade H (2018) Escherichia coli in Iran: an overview of antibiotic resistance: a review article. Iran J Public Health 47(1):1
Novelli A, Rosi E (2017) Pharmacological properties of oral antibiotics for the treatment of uncomplicated urinary tract infections. J Chemother 29(1):10–18
Hasan MS, Sultana M, Hossain MA (2019) Complete genome arrangement revealed the emergence of a poultry origin superbug Citrobacter portucalensis strain NR-12. J Global Antimicrobial Resistance 18:126–129
Hossain A, Hossain SA, Fatema AN, Wahab A, Alam MM, Islam MN, Ahsan GU (2020) Age and gender-specific antibiotic resistance patterns among Bangladeshi patients with urinary tract infection caused by Escherichia coli. Heliyon 6(6):e04161
Sultana M, Parvez AK, Sultana KF, Mukharje SK, Hossain MA (2019) Characterization of extended spectrum β-Lactamase producing bactieria isolated from urinary tract infections. Bangladesh Med Res Counc Bull 45(1):23–33
Rajan S, Prabavathy JV (2012) Antibiotic sensitivity and phenotypic detection of ESBL producing E. coli strains causing urinary tract infection in a community hospital, Chennai, Tamil Nadu India. WebmedCentral Pharmaceut Sci 3(11):003840
Gundran RS, Cardenio PA, Villanueva MA, Sison FB, Benigno CC, Kreausukon K, Punyapornwithaya V (2019) Prevalence and distribution of blaCTX-M, blaSHV, blaTEM genes in extended-spectrum β-lactamase-producing E. coli isolates from broiler farms in the Philippines. BMC Vet Res 15(1):1–8
Shakya P, Shrestha D, Maharjan E, Sharma VK, Paudyal R (2017) ESBL production among E coli and Klebsiella spp causing urinary tract infection: a hospital based study. Open Microbiol J 11:23
Zowawi HM, Harris PN, Roberts MJ, Tambyah PA, Schembri MA, Pezzani MD, Paterson DI (2015) The emerging threat of multidrug-resistant Gram-negative bacteria in urology. Nat Rev Urol 12(10):570–584
Kubone PZ, Mlisana KP, Govinden U, Abia ALK, Essack SY (2020) Antibiotic susceptibility and molecular characterization of uropathogenic Escherichia coli associated with community-acquired urinary tract infections in urban and rural settings in South Africa. Tropical Med Infectious Dis 5(4):176
Ramachandran G, Rajivgandhi GN, Chackaravarthi G, Kanisha CC, Siddiqi MZ, Alharbi NS, Manoharan N (2021) Isolation and molecular identification of extended spectrum beta-lactamase producing bacteria from urinary tract infection. J Infection Public Health 14(12):1911–1916
Hoque MN, Talukder AK, Saha O, Hasan MM, Sultana M, Rahman AA, Das ZC (2022) Antibiogram and virulence profiling reveals multidrug resistant Staphylococcus aureus as the predominant aetiology of subclinical mastitis in riverine buffaloes. Vet Med Sci 8:2631–45
Clinical and Laboratory Standards Institut (2022) Performance standards for antimicrobial susceptibility testing, M100, 32nd ed. Clinical and Laboratory Standards Institute, Wayne, PA
Saha O, Rakhi NN, Hoque MN, Sultana M, Hossain MA (2021) Genome-wide genetic marker analysis and genotyping of Escherichia fergusonii strain OTSVEF-60. Braz J Microbiol 52(2):989–1004
Saha O, Hoque MN, Islam OK, Rahaman MM, Sultana M, Hossain MA (2020) Multidrug-resistant avian pathogenic Escherichia coli strains and association of their virulence genes in Bangladesh. Microorganisms 8(8):1135
Momtaz S, Saha O, Usha MK, Sultana M, Hossain MA (2018) Occurrence of pathogenic and multidrug resistant Salmonella spp in poultry slaughter-house in Bangladesh. Biores Commu (BRC) 4(2):506–515
Hoque MN, Istiaq A, Clement RA, Gibson KM, Saha O, Islam OK, Hossain MA (2020) Insights into the resistome of bovine clinical mastitis microbiome, a key factor in disease complication. Front Microbiol 11:860
Swindell SR & Plasterer TN (1997) Seqman. In Sequence data analysis guidebook (75–89). Springer, Totowa, NJ
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mole Biol Evol 33(7):1870–1874
Doi Y, Adams-Haduch JM, Endimiani A, Sidjabat HE, Gaddad SM, Paterson DL (2008) High prevalence of CTX-M-15-producing Klebsiella pneumoniae among inpatients and outpatients with urinary tract infection in Southern India. J Antimicrob Chemother 61(6):1393–1394
Hoşoğlu S, Gündeş S, Kolaylõ F, Karadenizli A, Demirdağ K, Günaydõn M, Ucmak H (2007) Extended-spectrum beta-lactamases in ceftazidime-resistant Escherichia coli and Klebsiella pneumoniae isolates in Turkish hospitals. Indian J Med Microbiol 25(4):346–350
Cattoir V, Poirel L, Rotimi V, Soussy CJ, Nordmann P (2007) Multiplex PCR for detection of plasmid-mediated quinolone resistance qnr genes in ESBL-producing enterobacterial isolates. J Antimicrob Chemother 60(2):394–397
Platell JL, Cobbold RN, Johnson JR, Heisig A, Heisig P, Clabots C, Trott DJ (2011) Commonality among fluoroquinolone-resistant sequence type ST131 extraintestinal Escherichia coli isolates from humans and companion animals in Australia. Antimicrobial Agents Chemother 55(8):3782–3787
Zhao L, Zhang J, Zheng B, Wei Z, Shen P, Li S, Xiao Y (2015) Molecular epidemiology and genetic diversity of fluoroquinolone-resistant Escherichia coli isolates from patients with community-onset infections in 30 Chinese county hospitals. J Clin Microbiol 53(3):766–770
Asare KK, Amoah S, Coomson CA Jr, Banson C, Yaro D, Mbata J, Opoku YK (2022) Antibiotic-resistant pathogenic bacterial isolates from patients attending the outpatient department of university of Cape Coast hospital, Ghana a retrospective study between 2013–2015. PLoS Global Public Health 2(5):e0000417
Haque R, Akter M, Salam M (2015) Prevalence and susceptibility of uropathogens: a recent report from a teaching hospital in Bangladesh. BMC Res Notes 8(1):1–5
Parveen R, Saha SK, Shamshuzzaman SM, Rashid AL, Chowdhury A, Muazzam N (2011) Detection of uropathogens by using chromogenic media (Hicrome UTI agar), CLED agar and other conventional media. Faridpur Med Coll J 6(1):46–50
Rahman SR, Ahmed MF, Begum A (2014) Occurrence of urinary tract infection in adolescent and adult women of shanty town in Dhaka City. Bangladesh Ethiopian J Health Sci 24(2):145–152
Niranjan V, Malini A (2014) Antimicrobial resistance pattern in Escherichia coli causing urinary tract infection among inpatients. Indian J Med Res 139(6):945
Ahmed M, Moremi N, Mirambo MM, Hokororo A, Mushi MF, Seni J, Mshana SE (2015) Multi-resistant gram negative enteric bacteria causing urinary tract infection among malnourished underfives admitted at a tertiary hospital, northwestern Tanzania. Italian J Pediat 41(1):1–5
Parveen R, Yusuf MA, Sharmin I, Islam MS, Rahim I (2015) Antibiotic sensitivity of bacteria causing urinary tract infection. Bangladesh J Infect Dis 2(1):13–18
Yousefi A, Torkan S (2017) Uropathogenic Escherichia coli in the urine samples of Iranian dogs: antimicrobial resistance pattern and distribution of antibiotic resistance genes. Biomed Res Int 2017:10
Singhal A, Sharma R, Jain M, Vyas L (2014) Hospital and community isolates of uropathogens and their antibiotic sensitivity pattern from a tertiary care hospital in North West India. Ann Med Health Sci Res 4(1):51–56
Beyene G, Tsegaye W (2011) Bacterial uropathogens in urinary tract infection and antibiotic susceptibility pattern in Jimma University Specialized Hospital, Southwest Ethiopia. Ethiop J Health Sci 21(2):141–146
Rahman MM, Haq JA, Hossain MA, Sultana R, Islam F, Islam AS (2004) Prevalence of extended-spectrum β-lactamase-producing Escherichia coli and Klebsiella pneumoniae in an urban hospital in Dhaka. Bangladesh Int J Antimicrobial Agents 24(5):508–510
Sharmin S, Alamgir F, Fahmida M, Saleh AA (2009) Antimicrobial sensitivity pattern of uropathogens in children. Bangladesh J Med Microbiol 3(2):18–22
Al Wutayd O, Al Nafeesah A, Adam I, Babikir I (2018) The antibiotic susceptibility patterns of uropathogens isolated in Qassim, Saudi Arabia. J Infect Dev Countries 12(11):946–952
Rani L, Pinnelli VBK, Hemavathi BS, Rajendran R (2012) Utility Urinary of HiChrome Tract Infection (UTI) agar medium for identification of uropathogens: a comparative study with other conventional media. J Chem Pharm Res 1(4):95–105
Soleimani N, Aganj M, Ali L, Shokoohizadeh L, Sakinc T (2014) Frequency distribution of genes encoding aminoglycoside modifying enzymes in uropathogenic E coli isolated from Iranian hospital. BMC Res Notes 7(1):1–5
Khaleque M, Akter S, Mondal DK, Akhter H, Khan SI, Begum A (2017) Molecular characterization of extended spectrum β-lactamase producing bacteria isolated from urinary tract infected patients. Bangladesh Tropical Biomed 34(3):512–523
Chervet D, Lortholary O, Zahar JR, Dufougeray A, Pilmis B, Partouche H (2018) Antimicrobial resistance in community-acquired urinary tract infections in Paris in 2015. Medecine et maladies infectieuses 48(3):188–192
Paiva MC, Nascimento AMA, Camargo ILBC, Lima-Bittencourt CI, Nardi RMD (2012) The first report of the qnrB19, qnrS1 and aac (6)-Ib-cr genes in urinary isolates of ciprofloxacin-resistant Escherichia coli in Brazil. Mem Inst Oswaldo Cruz 107:687–689
Acknowledgements
The authors acknowledge the Department of Microbiology, Noakhali Science and Technology University and Research Cell for providing the research facilities with funding.
Author information
Authors and Affiliations
Contributions
KFS, AA, SA, TJ, and OS carried out the studies (sampling, sequencing, molecular, and data analysis) and participated in drafting the manuscript. SRS and AA critically reviewed and drafted the manuscript. OS visualized figures, interpreted data and results, critically reviewed, and edited the manuscript. KFS and FA supervised the sampling and supervised the whole work. KFS and OS developed the hypothesis, supervised the whole work, and helped to prepare and critically revise the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study obtained ethical clearance from the appropriate authorities. Moreover, verbal and written consent forms were also obtained from all of the participants.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Responsible Editor: Fernando R. Pavan
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sultana, K.F., Akter, A., Saha, S.R. et al. Bacterial profile, antimicrobial resistance, and molecular detection of ESBL and quinolone resistance gene of uropathogens causing urinary tract infection in the southeastern part of Bangladesh. Braz J Microbiol 54, 803–815 (2023). https://doi.org/10.1007/s42770-023-00942-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-023-00942-2