
Abstract
Hepatocellular carcinoma (HCC) is a subtype of highly malignant carcinoma that occurs in the liver, improved understanding of the mechanisms behind HCC tumorigenesis and better clinical treatment options are urgently needed. Several pieces of evidence have implied that the tumorigenesis and progression of HCC are driven by various genomic mutations and alterations. In this review, we have provided an overview of driver mutations in different signaling pathways that dominate HCC tumorigenesis, as well as vital molecular events in HCC initiation. Meanwhile, we have also summarized different agents or tools that may be utilized for HCC treatment in patients with corresponding mutation events. These findings may expand our understanding of the inherent characteristics of HCC and provide new perspectives for the future clinical treatment of HCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Liver cancer is the sixth most common cancer and the third leading cause of cancer-related death worldwide, with 0.91 million newly diagnosed cases and 0.83 million deaths annually in recent years (Sung et al., 2021). Hepatocellular carcinoma (HCC) accounts for approximately 85–90% of all primary malignancies in liver. Epidemiologically, HCC can be caused by various risk factors, including alcohol abuse, metabolic syndrome, and chronic infection with hepatitis B virus (HBV) and hepatitis C virus (HCV). However, as our understanding of the molecular mechanisms behind HCC tumorigenesis improves, increasing attention has been paid to revealing the key mutations or molecular drivers for hepatocarcinogenesis (Forner et al., 2018). Specifically, accumulating studies based on large-scale, multi-omics analysis of HCC patients’ samples have provided a more and more comprehensive landscape of HCC genetic alterations (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research, 2017; Gao et al., 2019; Guichard et al., 2012). These efforts not only expand our knowledge of the key events dominating HCC development and progression but also pave the way for the development of new HCC treatment strategies, which may ultimately benefit HCC patients.
In this review, we mainly focused on the four most common driver mutations in HCC tumorigenesis and their corresponding therapeutic strategies: mutations in TP53 pathway to govern genomic integrity and regulate cell growth; mutations in WNT/β-catenin pathway to modulate cell growth and tissue development/homeostasis; dysregulation of NRF2/KEAP1 pathway in modulating cellular response to ROS stress; and abnormal expression of TERT in cell fate control. In addition, we also summarized two vital molecular events in HCC initiation, including HCC-related hepatitis viral infection and polyploid in hepatocytes. These recent findings may help understand the intrinsic biological nature of HCC and guide the development of better clinical treatments in the future.
Mutation of TP53 pathway in HCC
TP53, the most famous tumor suppressor, has been identified as having approximately 31%–58% mutation frequency in HCC (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research, 2017; Gao et al., 2019; Jiang et al., 2019), which makes it the most prominent driver mutation in HCC patients.
Normally, TP53 acts as a tumor suppressor, governing hepatocyte cell-cycle, apoptosis, and metabolism etc., and is expressed at very a low level in normal cells (Boutelle & Attardi, 2021). Canonical transcriptional targets of TP53 include Fas (Owen-Schaub et al., 1995), Bax (Miyashita & Reed, 1995), NOXA (Oda et al., 2000) and PUMA (Nakano & Vousden, 2001) in apoptosis pathway; CDKN1A (el-Deiry et al., 1994) and PAI-1 (Kunz et al., 1995) in cell cycle regulation; and GLS2 (Suzuki et al., 2010) and TIGAR (Bensaad et al., 2006) in cellular metabolism. Interestingly, TP53 also induces the transcription of its E3 ubiquitin ligase MDM2 by binding to the TP53-specific response elements in MDM2 promoter region. As the negative regulator of TP53, MDM2 not only suppresses the transcription of TP53 downstream targets by binding to the transactivation domain of TP53, but also mediates the polyubiquitination and proteasome-dependent degradation of TP53. Thus, the MDM2–TP53 axis strictly regulates the proliferation of hepatocyte (Moll & Petrenko, 2003).
The somatic mutations of TP53 in HCC exhibit several forms, such as splice site, missense mutation, nonsense mutation, multi hit, frame shift deletion, and in-frame deletion (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research, 2017; Gao et al., 2019). These mutations primarily exist in its DNA-binding domain, which leads to the dysregulation of its downstream target genes. Since MDM2 is also a TP53 target gene, the protein abundance of MDM2 is also reduced in TP53-mutated HCC cells, resulting in the accumulation of dysfunctional TP53 mutants and ultimately disrupted cellular homeostasis and tumorigenesis.
Studies have revealed that missense mutation is the predominant form of TP53 mutations in HCC, and the R249S amino acid substitution derived from a single base substitution is the dominant hotspot (Hsu et al., 1991; Hussain et al., 2007; Staib et al., 2003). According to the statistical data obtained from the COSMIC database (https://cancer.sanger.ac.uk/cosmic/gene/analysis?all_data=&coords=AA%3AAA&dr=&end=394&gd=&id=348585&ln=TP53&seqlen=394&sn=liver&start=1#ts), missense mutations account for 74.3% (1312/1767) of all the mutations observed in TP53 in HCC. Out of these, the R249S amino acid substitution is responsible for 30.1% (395/1312) of total missense mutations in TP53 associated with HCC. Mechanistically, the S249 residue of TP53-R249S mutant could be phosphorylated by CDK4/cyclin D1 during G1 phase of cell cycle in HCC cells, and then peptidyl-prolyl cis/trans isomerase PIN1 would facilitate the nuclear import of TP53-R249S by binding to the phosphorylated S249 site. The TP53-R249S might disrupt the interaction between FBXW7 and c-Myc in the nucleus, which helps c-Myc escape from FBXW7-mediated proteasome-dependent degradation. Consequently, stabilized c-Myc augments the survival and proliferation of HCC cells (Liao et al., 2017). Meanwhile, methyltransferase like SETDB1 has been reported to elevate the tumor-promoting capacity of TP53-R249S mutant by demethylating it at K370 (Fei et al., 2015).
High Aflatoxin B1 (AFB1) exposure and HBV infection are two risks identified highly associated with TP53-R249S mutation (Gouas et al., 2012; Weng et al., 2017). Several epidemiological studies reported that R249S mutation is frequently detected in HCC patients suffering from above risks (Coursaget et al., 1993; Soini et al., 1996; Stern et al., 2001). The metabolites produced from AFB1 could induce TP53-R249S mutation by forming covalent and premutagenic DNA adducts and resulting DNA damage (Besaratinia et al., 2009), while HBV-encoded x protein (HBx) might promote HCC by interplaying with TP53-R249S mutant (Gouas et al., 2010; Kew, 2011). Since TP53-R249S mutation predominately occurrs in carcinogenesis of liver than in other tissues, it may raise the potential of TP53-R249S as an HCC diagnostic marker for patients in areas with heavy AFB1 exposure or HBV infection risks.
Considering the potential diagnostic and therapeutic values of TP53 mutation in HCC, several treatment strategies by targeting TP53 mutant have been developed. CP-31398 is the first chemical identified that can restore the native conformation of TP53 to active downstream targets in tumor cells (Bykov et al., 2002; Rippin et al., 2002). CP-31398 has been found to inhibit the growth of liver cancer cells with mutated TP53 in a dose-dependent and TP53-dependent manner. Additionally, it has been found to block the growth of HCC xenograft tumors by activating TP53-responsive downstream molecules (He et al., 2016). Furthermore, CP-31398 has been shown to have a synergistic effect with CDK4 inhibitor PD-0332991, resulting in the suppression of HCC cell survival by blocking the TP53-R249S-c-Myc axis (Wang et al., 2020). Another compound, PRIMA-1, has been shown to induce apoptosis in TP53-R249S mutated HCC cells by reactivating the transactivation capacity of TP53-R249S mutant (Shi et al., 2008). Meanwhile, compounds purified from podophyllum derivatives have been reported to inhibit the growth of TP53-R249S mutated HCC cells by restoring the activity of wildtype TP53 (Chen et al., 2020). Recently, PK9318 has been reported to restore the TP53 transcriptional activity by binding to TP53-Y220C mutant in HCC cells harboring the corresponding mutation (Bauer et al., 2019). However, other TP53-mutant targeting drugs like COTI-2 (Guo et al., 2020; Lindemann et al., 2019; Synnott et al., 2020) and TAR1 (Orgad et al., 2010) that have been tested in other cancers, their capacity in treating TP53-mutated HCC has not been determined and is likely would be fully evaluated in the future.
Mutation of WNT/β-catenin pathway in HCC
The WNT/β-catenin pathway plays irreplaceable roles in regulating hepatocyte development and cellular homeostasis (Perugorria et al., 2019; Reya & Clevers, 2005). Different clinical studies have revealed that mutations in β-catenin exist in 17–27% HCC patients, while other 8–18% HCC patients harbor mutations in AXIN1, an important component of WNT/β-catenin signaling pathway (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research, 2017; Gao et al., 2019; Jiang et al., 2019).
Generally, there are two WNT/β-catenin pathways, namely canonical and non-canonical, functioning in cells. The canonical WNT/β-catenin cascade includes WNT, β-catenin, APC, AXIN, LRP5/6, FZD, DVL, GSK3β, and several downstream target genes. In the absence of WNT ligands, a portion of β-catenin is sequestered at the plasma membrane by E-cadherin (Niessen & Gottardi, 2008). The cytosolic β-catenin forms a complex with APC and AXIN1, in which the N-terminus of β-catenin is phosphorylated by CK1 at S45 site and GSK3β at Y41, S37 and S33 sites, respectively. This leads to the ubiquitination and proteasome degradation of β-catenin by E3 ubiquitin ligase SCFβ−TrCP (Liu et al., 2002). When WNT ligands are present, they bind to the FZD and LRP5/6 to form WNT-FZD-LRP complex. The complex then captures the AXIN1 and GSK3β by recruiting DVL at the plasma membrane, which finally stabilizes β-catenin by facilitating its escape from N-terminus phosphorylation and subsequent proteasome degradation, and the stabilized β-catenin then translocates into the nucleus (Tortelote et al., 2017). Nuclear β-catenin binds to transcription factors and transcription co-activators such as TCF/LEF(Korswagen & Clevers, 1999), HIF-1α (Tang et al., 2019), FOXO (Kamo et al., 2013) and SOX (Han et al., 2018) via its C-terminus, thereby initiating the transcription of downstream target genes responsible for cellular survival and proliferation. Apart from the canonical pathway, the non-canonical WNT signaling mainly functions in cell growth, fate determination and migration by interacting with Ca2+ and planar cell polarity pathways (Nishita et al., 2019).
Most somatic mutations of β-catenin in HCC are missense mutations and mainly exhibit gain-of-function (GOF) phenotypes. The phosphorylation sites or adjacent amino acids in exon 3 and armadillo repeat domains 5/6 are two mutational hotspots in β-catenin. Mechanistically, the former would prolong β-catenin half-life by avoiding phosphorylation and proteasome degradation (Cieply et al., 2009; Okabe et al., 2016), while the latter could reduce the binding of β-catenin to APC, thus activating the WNT/β-catenin cascade (Liu et al., 2020). AXIN1 and APC, as tumor suppressors and key components of β-catenin degradation complex, were also found to be mutated in HCC patients, while these mutations always present loss-of-function (LOF) phenotypes (Bugter et al., 2021). Interestingly, β-catenin, AXIN1 or APC rarely co-mutated in the same HCC patient (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research, 2017; Gao et al., 2019; Jiang et al., 2019), indicating that any single mutation in WNT/β-catenin pathway might be efficient to activate the downstream cascade.
The mutation or hyperactivation of WNT/β-catenin pathway is critically related to HCC tumorigenesis. Morphologically, WNT/β-catenin-mutated HCC tumors usually exhibit well-differentiated, micro-beam skeleton, pseudo-glandular architecture pattern, tumor cholestasis, and lack of inflammatory infiltration phenotypes (Calderaro et al., 2019). As a part of the underlying mechanism, several oncoproteins in HCC have been characterized as targets of β-catenin, such as c-Myc (He et al., 1998), Cyclin D1 (Shtutman et al., 1999), TBX3 (Liang et al., 2021), and KIF2C (Wei et al., 2021), which are implicated in HCC progression, metastasis, metabolism, and drug resistance (Khalaf et al., 2018). It is worthy noticing that β-catenin mutation also functions in HCC metabolism, as Glutamine synthetase (GS, or Glutamate-ammonia ligase, GLUL) in glutaminolysis is the target of β-catenin (Cadoret et al., 2002), and its expression is correlated with β-catenin GOF mutation in HCC (Lee et al., 2014). Meanwhile, our research in HBV-related HCC also revealed that β-catenin mutation could promote the glycolysis of tumor cells via enhancing ALDOA phosphorylation in HCC cells (Gao et al., 2019).
Different from worse outcomes caused by TP53 mutation, WNT/β-catenin mutation seems to be a neutral or even favorable factor for HCC patients. Unchanged or prolonged overall survival rates have been observed in different studies on HCC patients with WNT/β-catenin mutations (Ding et al., 2014b; Lu et al., 2014; Wang et al., 2015), which may be associated with different stages of HCC. However, targeting WNT/β-catenin also showed attractive potentials in HCC treatment. For examples, CGP049090 and PKF155-854 are two fungal derivatives refined for blocking the interaction between β-catenin and TCFs, both of which exhibit inhibitory effects on HCC cell growth (Lepourcelet et al., 2004; Wei et al., 2010). Meanwhile, TNKS inhibitor XAV939 could suppress HCC cell growth in vitro via destabilizing AXIN1 (Ma et al., 2015).
Dysregulation of NRF2/KEAP1 pathway in HCC
NRF2 and its E3 ubiquitin ligase KEAP1 are key players in the regulation of redox homeostasis in hepatocytes. Approximately 3–6% and 5–7% HCC patients have been found to carry NRF2 and KEAP1 mutations, respectively (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research, 2017; Gao et al., 2019; Guichard et al., 2012).
NRF2 is a transcriptional factor coded by NFE2L2 and mainly plays pivotal roles in the prevention or alleviation of redox imbalance in cells. The known regulatory targets of NRF2 include NQO1 in ROS resistance(V enugopal & Jaiswal, 1996), 53BP1 in DNA damage repair signaling (Kim et al., 2012), PSMA1 and POMP in proteostasis regulation (Kwak et al., 2003; Li et al., 2015), GSTA1, GSTM1 and AKR1C1 in multidrug resistance (MDR) pathways (Chanas et al., 2002; Lou et al., 2006), HMOX1 and FTH1 in Fe2+/heme metabolism pathway (Alam et al., 1999; Campbell et al., 2013), ME1 in NADPH synthesis (Lee et al., 2021), GCLM, GCLC and SLC11A7 in glutathione synthesis (Bea et al., 2003; Sasaki et al., 2002), as well as BCL-2 and BCL-xL in the apoptosis pathway (Niture & Jaiswal, 2012, 2013), etc. Therefore, NRF2 is recognized as a master regulator that orchestrates the NRF2-mediated oxidative stress response to maintain homeostasis in liver. KEAP1 is the adaptor protein for the KEAP1-CUL3-RBX1 complex (Zhang et al., 2004). Normally, the KELCH domain located in the C-terminus of KEAP1 interacts with the DLG and ETGE motifs in the NEH2 domain of NRF2 to mediate the ubiquitination and subsequent proteosome-mediated degradation of NRF2 in the cytosol (McMahon et al., 2006; Padmanabhan et al., 2008; Tong et al., 2006). When hepatocytes are under ROS stress, several sensor cysteines (especially C151) in KEAP1 would interact with electrophiles to change the molecular conformation of KEAP1, influencing the binding of KEAP1 to NRF2 and raising the abundance of NRF2 in the cytosol (Dayalan Naidu et al., 2018). Finally, NRF2 accumulated in the cytosol would be translocated into the nucleus to activate various downstream signaling pathways, protecting hepatocytes from ROS-induced damage. The KEAP1-NRF2 interaction would be restored once hepatocytes return to homeostasis statue (Baird & Yamamoto, 2020). Meanwhile, the autophagy-related protein SQSTM1/p62 also participates in NRF2 signaling by competitively interfering the interaction between KEAP1 and NRF2 (Inami et al., 2011; Komatsu et al., 2010).
Previous studies reported that NRF2 mutations in DLG and ETGE motifs located in exon 2 could activate the expression of NRF2 target genes in HCC cells, which is similar to the HCC cells bearing KEAP1 mutations (Goldstein et al., 2016). Generally, somatic mutations of NRF2 in HCC mainly occur in the DLG and ETGE motifs, while somatic mutations of KEAP1 are primarily located in BTB, IVR and KELCH domains, exhibiting GOF or LOF phenotypes respectively, both by disrupting KEAP1-NRF2 interaction (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research, 2017; Gao et al., 2019; Haque et al., 2020). In a mutational fashion similar to β-catenin/AXIN1/APC of WNT/β-catenin pathway in HCC, mutations of NRF2 and KEAP1 are also exclusive and seldom co-occur in the same HCC patient (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research, 2017).
GOF mutations of NRF2 have been reported as one of the driver mutations and potential inducers of HCC (Ngo et al., 2017; Orru et al., 2018; Zavattari et al., 2015). Actually, the mutation and dysregulation of NRF2/KEAP1 pathway could promote HCC occurrence and progression through different manners. For example, the upregulation of NQO1 and HMOX1 induced by constitutive NRF2 activation promotes the growth of HCC cells (Gan et al., 2010). Hyperactivation of NRF2 triggers the expression of MMP-9, promoting the migration and invasion of HCC cell (Endo et al., 2018; Zhang et al., 2015). Stabilized NRF2 increases BCL-xL expression to rescue HCC cells from chemotherapy-induced apoptosis (Niture & Jaiswal, 2013). Additionally, aberrant regulation of NRF2 also contributes to non-alcoholic fatty liver disease (NAFLD) by reprogramming lipid metabolism in the liver, which promotes hepatocarcinogenesis. FOXA1, as an important triglyceride synthesis suppressor, its expression could be reduced by NRF2 accumulation (Suzuki et al., 2019), which might be associated with the risk of NAFLD (Moya et al., 2012). Moreover, NRF2 activation by high fat diet (HFD) increases the protein abundance of the lipid metabolism regulator PPARγ, which ultimately triggers NAFLD by triglyceride accumulation (Li et al., 2020).
Due to numerous NRF2 targets are involved in MDR pathways, HCCs with NRF2 mutations or hyperactivation always exhibit a weak response to traditional chemotherapeutic agents such as cisplatin, sorafenib, and doxorubicin (Gao et al., 2013; Huang et al., 2021; Liu et al., 2022; Wu et al., 2019). However, both downregulating NRF2 expression and inhibiting NRF2-mediated transcription of target genes have exhibited promising outcomes for MDR avoidance. For instance, apigenin has been found to alleviate NRF2-induced MDR by blocking the PI3K/AKT/NRF2 pathway (Gao et al., 2013). Ursolic acid, a potential NRF2 inhibitor, has been reported to sensitize cisplatin-resistant HCC cells to cisplatin by inhibiting the NRF2 pathway (Wu et al., 2016). Metformin, the most famous agent for diabetic treatment, is prone to suppress HCC cell growth by inhibiting RAF/ERK/NRF2/HMOX1 signaling cascade (Do et al., 2013). Meanwhile, isoniazid could interfere with the translocation of NRF2 into the nucleus by reducing ERK1 phosphorylation, which benefits drug treatment in HCC (Verma et al., 2015).
Abnormal expression of TERT in HCC
TERT is the core catalytic enzyme of the telomerase complex coding by TERT (Dratwa et al., 2020; Gunes & Rudolph, 2013). Mutations in the TERT promoter have been observed in 30–60% of overall HCC patients, making it a remarkable somatic genetic alternation in HCC development (Cevik et al., 2015; Huang et al., 2015; Schulze et al., 2015; Totoki et al., 2014).
Telomeres are replicated DNA sequences (5′-TTAGGG-3′) located at the extremities of chromosomes. Under normal physiological circumstance, the telomeres are constantly shortened with each round of cell division until DNA damage protein is activated to initiate cell senescence. Thus, telomeres function as internal clocks to regulate cell proliferation and aging (Gunes & Rudolph, 2013). However, TERT maintains telomere length by adding repeat sequences to the extremities of chromosomes and protecting chromosomes from degradation (Rudolph et al., 2009).
Normally, telomerase is inactive and remains a quiescent stage in healthy adult liver (Mangnall et al., 2003), and the shortening of telomeres or insufficient telomerase activity has been found in chronic diseases or injury in liver (Miura et al., 1997; Wiemann et al., 2002). However, the re-expression and reactivation of TERT have been identified in liver cirrhosis and regenerative nodules staging early premalignant (Hytiroglou et al., 1998; Kotoula et al., 2002; Youssef et al., 2001). Furthermore, TERT overexpression by mutations in TRET promoter has been recognized as a hallmark of hepatocarcinogenesis (Kim et al., 1994; Miura et al., 1997). Generally, the hot spots of mutations in the TERT promoter in HCC are positions − 124 (G to A/T) and − 146 (G to A) before the ATG translation start site (Totoki et al., 2014), and the G to T substitution of − 124 is the most common. The new DNA sequences created by these mutations emerge as binding sites for transcription factors like ETS and TCF, which transcriptionally upregulate the expression of TERT and subsequently stabilizing the telomeres (Xu et al., 2008). Recently, a new mutation at − 297 (C to T) was reported to enhance TERT transcription by creating an AP2 consensus sequence (Lombardo et al., 2020). Interestingly, mutations in the TERT promoter have been reported to be frequently accompanied by mutations in β-catenin (Nault et al., 2013; Totoki et al., 2014), which hints at the potential synergistic carcinogenic effect between TERT promoter mutations and WNT/β-catenin pathway mutations (Park et al., 2009).
Apart from mutations in the TERT promoter, TERT expression in HCC can also be replenished via different ways. For example, HBV, rather than HCV, can be inserted into the promoter region of TERT to activate its expression (Paterlini-Brechot et al., 2003; Sung et al., 2012). A similar upregulation of TERT can also be observed in HCCs with adeno-associated virus type 2 (AAV2) infection, as AAV2 can also insert into the promoter region of TERT (Nault et al., 2015). The fusion of SLC7A2 and SLC12A7 with TERT has been identified in approximately 4% of HCCs. Since SLC7A2 and SLC12A7 are ubiquitously expressed in the liver, and the transcription of TERT is controlled by the promoters of SLC7A2 and SLC12A7, TRET abundance is upregulated in these fusion cases (Barthel et al., 2017). Meanwhile, amplification of TERT in chromosomes has been reported in 5% of HCCs, but the mechanisms behind remain unknown (Letouze et al., 2017; Schulze et al., 2015).
Considering that telomerases are only reactivated in HCC cells, not in normal hepatocytes, different therapeutic strategies targeting telomeres and telomerase have been used to treat HCCs with telomerase dysregulation. The TERT inhibitor BIBR1532 could suppress HCC cell growth by inhibiting TERT in vitro and in vivo (Tahtouh et al., 2015). Telomerase peptide vaccines like GV1001 and VX-001 have been developed to treat several human cancers, while only limited efficacy has been observed with these drugs on HCC treatment (Anguille et al., 2014; Brunsvig et al., 2011; Greten et al., 2010; Kotsakis et al., 2012).
HCC-related hepatitis viral infection (HBV/HCV)
Chronic infection with hepatitis viruses such as hepatitis B virus (HBV) and hepatitis C virus (HCV) has been identified as an etiological risk factor for the occurrence of HCC. Approximately 54% or 31% of HCCs have been reported to be associated with HBV or HCV infection, respectively (Ding et al., 2017; El-Serag, 2012; Yang et al., 2019), and HBV/HCV coinfection incidences have also been observed in HCC (Fattovich et al., 2004).
HBV belongs to the Hepadnaviridae family, which has a selective appetite for hepatocytes. The mature HBV possesses a 3.2 kb double-stranded relaxed circular DNA (rcDNA) genome and viral proteins. The viral proteins are composed of HBcAg (the core antigen), HBeAg (the excreted antigen e), L/M/S-HBsAg (three surface envelope proteins), viral polymerase (functions as RHAseH, DNA polymerase, and transcriptase) and HBx (functions in viral transcription and pathogenesis) (Coffin et al., 2011; Seeger & Mason, 2000; Tu et al., 2017). During the HBV lifecycle, the mature HBV enters the hepatocytes by binding of L-HBsAg to the HSPG (heparin sulfate proteoglycan receptor) on the surface of hepatocytes. Then, the nucleocapsid could be translocated into the nucleus after uncoating surface proteins, where rcDNA is transformed into covalently closed circular DNA (cccDNA). Pregenomic RNA (pgRNA) and subgenomic RNA are transcribed from cccDNA and are further be translated to produce viral proteins (HBx, L/M/S-HBsAg, etc.). Additionally, the pgRNA could also be encapsulated by HBcAg and reverse transcribed into rcDNA or double-stranded linear DNA (dslDNA). The newly assembled rcDNA would either go back to the nucleus to replenish the cccDNA pool or bud out of the current cell to infect other cells after being assembled with coat proteins, while dslDNA could integrate into the genome of host cells after being relocated into the nucleus (Nassal, 2015; Seeger & Mason, 2015; Summers & Mason, 1982; Turton et al., 2020).
Generally, HBV infection promotes HCC tumorigenesis via numerous mechanisms. Since HBV can integrate into the host hepatocyte genome, the genomic stability can be affected by HBV integration in certain areas such as CpG islands and telomeres, which leads to chromosomal rearrangements or gene copy number variation and contributes to HCC occurrence (Jiang et al., 2012; Yan et al., 2015). Meanwhile, insertional mutation of several oncoproteins and tumor suppressors can also be induced by HBV integration (Dong et al., 2015; Sung et al., 2012). Furthermore, dysregulated expression of viral proteins (especially HBx) by HBV genomic integration is associated with HCC tumorigenesis (Chisari et al., 1989). HBx, as key regulator for HBV oncogenicity, plays various significant roles in hepatocarcinogenesis by interacting with multiple cellular targets. Apart from enhancing HBV replication, HBx functions in chromatin and transcriptional control by interplaying with transcription factors like ATF/CREB, C/EBP, ETS, and TP53, chromatin-modifying enzymes such as PCAF and p300, as well as components for transcription (RPB5, TFIIB and TBP) (Levrero & Zucman-Rossi, 2016). HBx controls cell proliferation and death through regulating the expression of HIF-1α, ANG2 and TGF-1β (Sanz-Cameno et al., 2006; Yoo et al., 1996, 2003). As mentioned above, HBx can regulate senescence and telomeres via manipulating expression of TERT (Ozturk et al., 2009). Meanwhile, proto-oncogenic pathways like JAK-STAT, Src, survivin, WNT/β-catenin, and PI3K can also be modulated by HBx (Cha et al., 2004; Feitelson & Duan, 1997; Murakami, 2001; Shlomai et al., 2014).
HCV belongs to the Flaviviridae family and the Hepacivirus genus. Unlike HBV, HCV is an RNA virus with a 9.6 kb positive-sense, single-stranded RNA and receptor proteins (Lindenbach & Rice, 2005). During the HCV replication cycle, it enters hepatocytes via clathrin-dependent endocytosis after plenty of interactions between HCV particles and cellular receptors (Ding et al., 2014a). Then, the viral RNA is released into the cytoplasm, where it is further replicated and translated at the endoplasmic reticulum (ER) to produce a single polyprotein chain. The viral proteins are assembled from cleaved polyprotein chain and form premature HCV particles with viral RNA on lipid droplets, which are further processed at Golgi apparatus. Finally, the mature HCV particles bud out of the parental host cell via the secretory pathway and populate into the new hosts (Kim & Chang, 2013; Morozov & Lagaye, 2018). It is worthy noticing that HCV cannot integrate into host genome, which is different from HBV.
The mechanisms of HCV-mediated HCC occurrence are complex. In the same fashion with HBV infection, various cancer-related signaling pathways could be modulated by HCV. The relationship between HCV infection and TP53 signaling pathway remains vague. TP53 overexpression has been observed in the early stage of HCV-related HCC (Loguercio et al., 2003), while HCV viral protein NS5A has been reported to transcriptionally inactive TP53 by interacting with it, and another TP53 interactor hTAFII32 could suppress TP53 signaling by binding to NS5A (Lan et al., 2002). HCV infection actives the WNT/β-catenin signaling cascade by downregulating the expression of APC or AXIN2, which proceeds HCC tumorigenesis (Levrero, 2006). The RAS/RAF/MEK/MAPK pathway involved in HCC development is affected by HCV at different levels. Normally, HCV infection always accompanies with the hypermethylation at the promoter regions of many tumor suppressor genes (Quan et al., 2014; Wijetunga et al., 2017; Zekri Ael et al., 2014). RASSF1 and RASAL1, as two inhibitors for RAS, have their expressions downregulated by HCV infection via methylation on their gene promoter regions (Jin et al., 2007; Volodko et al., 2014). Moreover, the anti-HCV efficacy of IFN-γ is diminished by MEK1/2 activation, which implies the indispensable role of RAS/RAF/MEK/MAPK in HCV-induced HCC (Huang et al., 2006).
Currently, apart from vaccination strategy to prevent viral-induced HCC, the treatment of HBV-induced HCC patients mainly focuses on targeting the dysregulated signaling pathways influenced by viral infection. Alternatively, training hosts’ immune system to eliminate viral particles is a new strategy to treat viral-induced HCC patients. Cellular technology tools likes CAR-T have been used to precisely targeting HBV in HCC tumors and have exhibited promising performance (Tan et al., 2019). On the contrary, the direct-acting antiviral therapy on chronic HCV infection has shown promising success (Falade-Nwulia et al., 2017), which reflects the different biological attributes between HBV and HCV.
The polyploidy of hepatocytes in HCC
Polyploidy is a state in which cells possess more than two sets of homologous chromosomes, which is wildly presented in heart, bone marrow, pancreas, placenta, and liver (Anzi et al., 2018; Davoli & de Lange, 2011; Oates & Morgan, 1986; Pandit et al., 2013). Normally, diploid cells have only one single nucleus with 2n nuclear content, while polyploid cells could be mononucleate with a single 4n/8n/16n nucleus or binucleate with pairs of 2n/4n/8n nuclei. Hepatocytes exhibit different ploidy statues spatiotemporally. Hepatocytes are born to be diploid, and polyploid descendants are generated after rounds of mitosis. Polyploid hepatocytes account for 25–50% and more than 90% of all hepatocytes in humans and mice, respectively (Duncan, 2020), and the polyploid of hepatocytes is tightly associated with hepatocarcinogenesis.
Previous studies revealed that polyploid or quasi-polyploid phenotypes have been observed in nearly 30% of solid tumors (Bielski et al., 2018; Zack et al., 2013). However, the relationship between polyploid and HCC tumorigenesis is quit confusing. As polyploid is usually associated with hemostasis and normal functions of healthy liver, polyploid is now generally characterized as a gatekeeper of tumorigenesis in liver (Zhang et al., 2019). Polyploid hepatocytes have more copies of tumor suppressor genes than diploid cells, which makes them more resistant to oncogenic threats such as loss of heterozygosity (Lin et al., 2020; Zhang et al., 2018a, 2018b). Meanwhile, polyploid strictly limits the proliferation of hepatocytes via TP53-dependent “tetraploid checkpoint”, which avoids HCC occurrence (Kneissig et al., 2019; Tanaka et al., 2018). Consistently, HCC has been demonstrated to be derived from low polyploid hepatocytes, especially hepatocytes. However, polyploid seems to be a potential HCC tumorigenesis driver when TP53 is mutated or inactivated. It has been reported that binucleate polyploid was drastically reduced, while mononucleate polyploid was increased during liver tumorigenesis, and TP53 mutations are the key for increasing mononucleate polyploid hepatocytes (Bou-Nader et al., 2020). It is obvious that the “Jekyll and Hyde” roles of polyploid in HCC are largely dependent on the mutational state of TP53.
The PIDDosome is mainly responsible for the regulation of hepatic polyploidy, which is crucial for HCC development. The PIDDosome is a protein complex comprised of PIDD1, RAIDD, and CASP2, which functions as a sensor of supernumerary centrosomes to regulate TP53-mediated cell cycle modulation (Fava et al., 2017). PIDD1 is commonly expressed at the centriole during the cell cycle. When extra centrosomes are produced by the mitosis process in polyploid hepatocytes, RAIDD and CASP2 are recruited and form the PIDDosome with PIDD1. Once CASP2 is activated, MDM2 is cleaved to stabilize TP53 and activate p21-mediated cell cycle arrest, which keeps a moderate polyploidy level and restricts hyperpolyploidization of hepatocytes (Sladky et al., 2020a). It has been reported that the absence of the PIDDosome leads to hyperpolyploidy of hepatocytes and benefits the tumorigenesis resistance via a TP53-dependent manner (Sladky et al., 2020b), providing a new strategy for HCC treatment from a cell ploidy perspective.
Discussion and perspectives
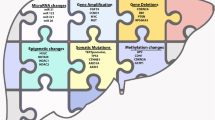
Hepatocellular carcinoma (HCC) is unique compared to other epithelium-derived solid tumors as it is mainly derived from healthy hepatocytes (Forner et al., 2018). In this review, we have presented the molecular and mutational landscape of HCC and briefly summarized the mechanisms behind HCC-promoting effects of these driver mutations and molecular events, as well as the potential therapeutic tools for HCC patients with different mutational landscapes (Fig. 1).

Driver mutations and molecular events in HCC. This diagram summarized the HCC-promoting mutations in pathways such as TP53, WNT/β-catenin, NRF2/KEAP1, and telomere regulation. Additionally, HBV/HCV infection and polyploidy of hepatocytes are significant molecular events in HCC tumorigenesis. Meanwhile, several potential therapeutic inhibitors and tools could be utilized to treat HCC patients with different mutational backgrounds.
Interestingly, HCC tumors are highly heterogeneous at both histological and molecular levels, and this phenotype seems to be a result of the different molecular events they undergo (Calderaro et al., 2019). It has been widely recognized that HCC could be physiologically and molecularly classified as “proliferative” and “non-proliferative” tumors (Zucman-Rossi et al., 2015). The “proliferative” HCC patients mainly harbor TP53 mutations and chromosomal instability, as well as high tumor grades and high-level of AFP, leading to worse survival outcomes. However, those HCC patients with “non-proliferative” tumors generally carry mutations in the WNT/β-catenin pathway and low level of immune cells infiltration. In contrast, they are always characterized with low-level tumor grades and possess better clinical outcomes (Ruiz de Galarreta et al., 2019). Mutations in TP53 and WNT/β-catenin, as two of the most prominent mutations in HCC, both play oncogenic roles in tumorigenesis. However, they seem to guide the development of HCC cells into different directions and lead to opposite outcomes, and the mechanisms behind this paradox remain to be further studied and summarized. Likewise, mutations in the TERT promoter always seem to be independent of the polyploid of tumor cells in HCC (Bielski et al., 2018). It has been found in both cirrhosis and non-cirrhosis derived HCCs and remains stable through different stages of HCC development (Farazi et al., 2003; Gunes & Rudolph, 2013; Nault et al., 2019). Mutations in the TERT promoter were also detected in dysplastic nodules that finally developed into malignancy tumors, while mutations in TP53 and WNT/β-catenin were absent in dysplastic nodules (Zucman-Rossi et al., 2015). This implies that TERT promoter mutations are the fundamental gatekeepers for malignant transformation of hepatocytes into HCC cells, and mutations of TP53 and WNT/β-catenin are HCC progressors rather than initiators. As emerging studies focus on the heterogeneity of HCC, the spatial and temporal evolution of HCC is being fully revealed, which would greatly broad our knowledge on HCC tumorigenesis.
Meanwhile, it is obvious that different mutations and molecular drivers never work independently during tumorigenesis. Instead, they usually interact with each other to synergistically function in HCC development and progress. As mentioned above, HBV-coded HBx and HCV viral protein NS5A could interact with TP53 mutant to promote HCC tumorigenesis. At the same time, hepatic viral infection constitutively activates NRF2 signaling by raising oxidative stress, which transcriptionally elevates the expression of MDM2, consequently leading to MDM2-mediated TP53 proteasomal degradation, and enhancing the proliferation of HCC cells (Aydin et al., 2017). HCV also promotes the WNT/β-catenin cascade by modulating the expression of APC and AXIN2. However, the tumor-promoting or tumor-suppressing roles of polyploid in HCC depend on the mutational status of TP53. Additionally, the mutations of TERT promoters in HCC are always accompanied by β-catenin mutations. The interactions among these molecular drivers, together with the HCC heterogeneity mentioned above, show us the complexity of signaling pathway networks regulating HCC tumorigenesis and the necessity to understand the mechanism behind HCC tumorigenesis from multiple dimensions. Apart from the molecular drivers discussed in this review, mutations in other cancer-associated genes such as KMT2C, ARID1A, and TSC have also been observed in patients with HCC (Gao et al., 2019; Jiang et al., 2019; Zucman-Rossi et al., 2015). It is essential to further investigate and summarize their distinct roles in the tumorigenesis of HCC.
For HCC clinical treatment, different inhibitors for HCC patients with different mutational landscapes show various performances in vivo and in vitro. Inhibitors against TP53 mutations and abnormal activation of NRF2 pathway seem to have promising effects in treatment for corresponding HCC patients, while inhibitors targeting mutations in WNT/β-catenin and TERT promoter had limited effects in HCC treatment. There are certainly diverse reasons behind this, and more in-depth research is needed to benefit HCC patients. Furthermore, the possibilities of combining mutational inhibitors with conventional chemotherapy drugs such as Gemcitabine, Sorafenib, and Lenvatinib, or immunotherapy agents like Opdivo and Keytruda, present promising avenues for research. These combination drug therapies have the potential to revolutionize HCC treatment and warrant further investigation.
References
Alam, J., Stewart, D., Touchard, C., Boinapally, S., Choi, A. M., & Cook, J. L. (1999). Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. Journal of Biological Chemistry, 274, 26071–26078.
Anguille, S., Smits, E. L., Lion, E., van Tendeloo, V. F., & Berneman, Z. N. (2014). Clinical use of dendritic cells for cancer therapy. The Lancet Oncology, 15, e257-267.
Anzi, S., Stolovich-Rain, M., Klochendler, A., Fridlich, O., Helman, A., Paz-Sonnenfeld, A., Avni-Magen, N., Kaufman, E., Ginzberg, M. B., Snider, D., et al. (2018). Postnatal exocrine pancreas growth by cellular hypertrophy correlates with a shorter lifespan in mammals. Developmental Cell, 45(726–737), e723.
Aydin, Y., Chedid, M., Chava, S., Danielle Williams, D., Liu, S., Hagedorn, C. H., Sumitran-Holgersson, S., Reiss, K., Moroz, K., Lu, H., et al. (2017). Activation of PERK-Nrf2 oncogenic signaling promotes Mdm2-mediated Rb degradation in persistently infected HCV culture. Science and Reports, 7, 9223.
Baird, L., & Yamamoto, M. (2020). The Molecular mechanisms regulating the KEAP1-NRF2 pathway. Molecular and Cellular Biology, 40(13), e00099-20.
Barthel, F. P., Wei, W., Tang, M., Martinez-Ledesma, E., Hu, X., Amin, S. B., Akdemir, K. C., Seth, S., Song, X., Wang, Q., et al. (2017). Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nature Genetics, 49, 349–357.
Bauer, M. R., Jones, R. N., Tareque, R. K., Springett, B., Dingler, F. A., Verduci, L., Patel, K. J., Fersht, A. R., Joerger, A. C., & Spencer, J. (2019). A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Medicinal Chemistry, 11, 2491–2504.
Bea, F., Hudson, F. N., Chait, A., Kavanagh, T. J., & Rosenfeld, M. E. (2003). Induction of glutathione synthesis in macrophages by oxidized low-density lipoproteins is mediated by consensus antioxidant response elements. Circulation Research, 92, 386–393.
Bensaad, K., Tsuruta, A., Selak, M. A., Vidal, M. N., Nakano, K., Bartrons, R., Gottlieb, E., & Vousden, K. H. (2006). TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell, 126, 107–120.
Besaratinia, A., Kim, S. I., Hainaut, P., & Pfeifer, G. P. (2009). In vitro recapitulating of TP53 mutagenesis in hepatocellular carcinoma associated with dietary aflatoxin B1 exposure. Gastroenterology, 137, 1127–1137.
Bielski, C. M., Zehir, A., Penson, A. V., Donoghue, M. T. A., Chatila, W., Armenia, J., Chang, M. T., Schram, A. M., Jonsson, P., Bandlamudi, C., et al. (2018). Genome doubling shapes the evolution and prognosis of advanced cancers. Nature Genetics, 50, 1189–1195.
Bou-Nader, M., Caruso, S., Donne, R., Celton-Morizur, S., Calderaro, J., Gentric, G., Cadoux, M., L’Hermitte, A., Klein, C., Guilbert, T., et al. (2020). Polyploidy spectrum: A new marker in HCC classification. Gut, 69, 355–364.
Boutelle, A. M., & Attardi, L. D. (2021). p53 and tumor suppression: It takes a network. Trends in Cell Biology, 31, 298–310.
Brunsvig, P. F., Kyte, J. A., Kersten, C., Sundstrom, S., Moller, M., Nyakas, M., Hansen, G. L., Gaudernack, G., & Aamdal, S. (2011). Telomerase peptide vaccination in NSCLC: A phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clinical Cancer Research, 17, 6847–6857.
Bugter, J. M., Fenderico, N., & Maurice, M. M. (2021). Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nature Reviews Cancer, 21, 5–21.
Bykov, V. J., Issaeva, N., Shilov, A., Hultcrantz, M., Pugacheva, E., Chumakov, P., Bergman, J., Wiman, K. G., & Selivanova, G. (2002). Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nature Medicine, 8, 282–288.
Cadoret, A., Ovejero, C., Terris, B., Souil, E., Levy, L., Lamers, W. H., Kitajewski, J., Kahn, A., & Perret, C. (2002). New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene, 21, 8293–8301.
Calderaro, J., Ziol, M., Paradis, V., & Zucman-Rossi, J. (2019). Molecular and histological correlations in liver cancer. Journal of Hepatology, 71, 616–630.
Campbell, M. R., Karaca, M., Adamski, K. N., Chorley, B. N., Wang, X., & Bell, D. A. (2013). Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxidative Medicine and Cellular Longevity, 2013, 120305.
Cancer Genome Atlas Research Network. Electronic address, w.b.e., and Cancer Genome Atlas Research, N. (2017). Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell, 169(1327–1341), e1323.
Cevik, D., Yildiz, G., & Ozturk, M. (2015). Common telomerase reverse transcriptase promoter mutations in hepatocellular carcinomas from different geographical locations. World Journal of Gastroenterology, 21, 311–317.
Cha, M. Y., Kim, C. M., Park, Y. M., & Ryu, W. S. (2004). Hepatitis B virus X protein is essential for the activation of Wnt/beta-catenin signaling in hepatoma cells. Hepatology, 39, 1683–1693.
Chanas, S. A., Jiang, Q., McMahon, M., McWalter, G. K., McLellan, L. I., Elcombe, C. R., Henderson, C. J., Wolf, C. R., Moffat, G. J., Itoh, K., et al. (2002). Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. The Biochemical Journal, 365, 405–416.
Chen, H., Zhang, M., Wang, Z., Li, L., Li, Q., & Wang, H. (2020). The effect of p53–R249S on the suppression of hepatocellular carcinoma cells survival induced by podophyllum derivatives. Anti-Cancer Agents in Medicinal Chemistry, 20, 865–874.
Chisari, F. V., Klopchin, K., Moriyama, T., Pasquinelli, C., Dunsford, H. A., Sell, S., Pinkert, C. A., Brinster, R. L., & Palmiter, R. D. (1989). Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell, 59, 1145–1156.
Cieply, B., Zeng, G., Proverbs-Singh, T., Geller, D. A., & Monga, S. P. (2009). Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology, 49, 821–831.
Coffin, C. S., Mulrooney-Cousins, P. M., van Marle, G., Roberts, J. P., Michalak, T. I., & Terrault, N. A. (2011). Hepatitis B virus quasispecies in hepatic and extrahepatic viral reservoirs in liver transplant recipients on prophylactic therapy. Liver Transplantation, 17, 955–962.
Coursaget, P., Depril, N., Chabaud, M., Nandi, R., Mayelo, V., LeCann, P., & Yvonnet, B. (1993). High prevalence of mutations at codon 249 of the p53 gene in hepatocellular carcinomas from Senegal. British Journal of Cancer, 67, 1395–1397.
Davoli, T., & de Lange, T. (2011). The causes and consequences of polyploidy in normal development and cancer. Annual Review of Cell and Developmental Biology, 27, 585–610.
Dayalan Naidu, S., Muramatsu, A., Saito, R., Asami, S., Honda, T., Hosoya, T., Itoh, K., Yamamoto, M., Suzuki, T., & Dinkova-Kostova, A. T. (2018). C151 in KEAP1 is the main cysteine sensor for the cyanoenone class of NRF2 activators, irrespective of molecular size or shape. Science and Reports, 8, 8037.
Ding, Q., von Schaewen, M., & Ploss, A. (2014a). The impact of hepatitis C virus entry on viral tropism. Cell Host & Microbe, 16, 562–568.
Ding, X., Yang, Y., Han, B., Du, C., Xu, N., Huang, H., Cai, T., Zhang, A., Han, Z. G., Zhou, W., et al. (2014b). Transcriptomic characterization of hepatocellular carcinoma with CTNNB1 mutation. PLoS ONE, 9, e95307.
Ding, X. X., Zhu, Q. G., Zhang, S. M., Guan, L., Li, T., Zhang, L., Wang, S. Y., Ren, W. L., Chen, X. M., Zhao, J., et al. (2017). Precision medicine for hepatocellular carcinoma: Driver mutations and targeted therapy. Oncotarget, 8, 55715–55730.
Do, M. T., Kim, H. G., Khanal, T., Choi, J. H., Kim, D. H., Jeong, T. C., & Jeong, H. G. (2013). Metformin inhibits heme oxygenase-1 expression in cancer cells through inactivation of Raf-ERK-Nrf2 signaling and AMPK-independent pathways. Toxicology and Applied Pharmacology, 271, 229–238.
Dong, H., Zhang, L., Qian, Z., Zhu, X., Zhu, G., Chen, Y., Xie, X., Ye, Q., Zang, J., Ren, Z., et al. (2015). Identification of HBV-MLL4 integration and its molecular basis in Chinese hepatocellular carcinoma. PLoS ONE, 10, e0123175.
Dratwa, M., Wysoczanska, B., Lacina, P., Kubik, T., & Bogunia-Kubik, K. (2020). TERT-regulation and roles in cancer formation. Frontiers in Immunology, 11, 589929.
Duncan, A. W. (2020). Hepatocyte ploidy modulation in liver cancer. EMBO Reports, 21, e51922.
El-Deiry, W. S., Harper, J. W., O’Connor, P. M., Velculescu, V. E., Canman, C. E., Jackman, J., Pietenpol, J. A., Burrell, M., Hill, D. E., Wang, Y., et al. (1994). WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res, 54, 1169–1174.
El-Serag, H. B. (2012). Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology, 142(1264–1273), e1261.
Endo, H., Owada, S., Inagaki, Y., Shida, Y., & Tatemichi, M. (2018). Glucose starvation induces LKB1-AMPK-mediated MMP-9 expression in cancer cells. Science and Reports, 8, 10122.
Falade-Nwulia, O., Suarez-Cuervo, C., Nelson, D. R., Fried, M. W., Segal, J. B., & Sulkowski, M. S. (2017). Oral direct-acting agent therapy for hepatitis C virus infection: A systematic review. Annals of Internal Medicine, 166, 637–648.
Farazi, P. A., Glickman, J., Jiang, S., Yu, A., Rudolph, K. L., & DePinho, R. A. (2003). Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Research, 63, 5021–5027.
Fattovich, G., Stroffolini, T., Zagni, I., & Donato, F. (2004). Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology, 127, S35-50.
Fava, L. L., Schuler, F., Sladky, V., Haschka, M. D., Soratroi, C., Eiterer, L., Demetz, E., Weiss, G., Geley, S., Nigg, E. A., et al. (2017). The PIDDosome activates p53 in response to supernumerary centrosomes. Genes & Development, 31, 34–45.
Fei, Q., Shang, K., Zhang, J., Chuai, S., Kong, D., Zhou, T., Fu, S., Liang, Y., Li, C., Chen, Z., et al. (2015). Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nature Communications, 6, 8651.
Feitelson, M. A., & Duan, L. X. (1997). Hepatitis B virus X antigen in the pathogenesis of chronic infections and the development of hepatocellular carcinoma. American Journal of Pathology, 150, 1141–1157.
Forner, A., Reig, M., & Bruix, J. (2018). Hepatocellular carcinoma. Lancet, 391, 1301–1314.
Gan, N., Sun, X., & Song, L. (2010). Activation of Nrf2 by microcystin-LR provides advantages for liver cancer cell growth. Chemical Research in Toxicology, 23, 1477–1484.
Gao, A. M., Ke, Z. P., Wang, J. N., Yang, J. Y., Chen, S. Y., & Chen, H. (2013). Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis, 34, 1806–1814.
Gao, Q., Zhu, H., Dong, L., Shi, W., Chen, R., Song, Z., Huang, C., Li, J., Dong, X., Zhou, Y., et al. (2019). Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell, 179(561–577), e522.
Goldstein, L. D., Lee, J., Gnad, F., Klijn, C., Schaub, A., Reeder, J., Daemen, A., Bakalarski, C. E., Holcomb, T., Shames, D. S., et al. (2016). Recurrent loss of NFE2L2 Exon 2 is a mechanism for Nrf2 pathway activation in human cancers. Cell Reports, 16, 2605–2617.
Gouas, D. A., Shi, H., Hautefeuille, A. H., Ortiz-Cuaran, S. L., Legros, P. C., Szymanska, K. J., Galy, O., Egevad, L. A., Abedi-Ardekani, B., Wiman, K. G., et al. (2010). Effects of the TP53 p. R249S mutant on proliferation and clonogenic properties in human hepatocellular carcinoma cell lines: Interaction with hepatitis B virus X protein. Carcinogenesis, 31, 1475–1482.
Gouas, D. A., Villar, S., Ortiz-Cuaran, S., Legros, P., Ferro, G., Kirk, G. D., Lesi, O. A., Mendy, M., Bah, E., Friesen, M. D., et al. (2012). TP53 R249S mutation, genetic variations in HBX and risk of hepatocellular carcinoma in The Gambia. Carcinogenesis, 33, 1219–1224.
Greten, T. F., Forner, A., Korangy, F., N’Kontchou, G., Barget, N., Ayuso, C., Ormandy, L. A., Manns, M. P., Beaugrand, M., & Bruix, J. (2010). A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer, 10, 209.
Guichard, C., Amaddeo, G., Imbeaud, S., Ladeiro, Y., Pelletier, L., Maad, I. B., Calderaro, J., Bioulac-Sage, P., Letexier, M., Degos, F., et al. (2012). Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nature Genetics, 44, 694–698.
Gunes, C., & Rudolph, K. L. (2013). The role of telomeres in stem cells and cancer. Cell, 152, 390–393.
Guo, Y., Zhu, X., & Sun, X. (2020). COTI-2 induces cell apoptosis in pediatric acute lymphoblastic leukemia via upregulation of miR-203. Bioengineered, 11, 201–208.
Han, F., Liu, W. B., Shi, X. Y., Yang, J. T., Zhang, X., Li, Z. M., Jiang, X., Yin, L., Li, J. J., Huang, C. S., et al. (2018). SOX30 inhibits tumor metastasis through attenuating Wnt-signaling via transcriptional and posttranslational regulation of beta-catenin in lung cancer. eBioMedicine, 31, 253–266.
Haque, E., Karim, M.R., Salam Teeli, A., Smiech, M., Leszczynski, P., Winiarczyk, D., Parvanov, E.D., Atanasov, A.G., & Taniguchi, H. (2020). Molecular mechanisms underlying hepatocellular carcinoma induction by aberrant NRF2 activation-mediated transcription networks: Interaction of NRF2-KEAP1 controls the fate of hepatocarcinogenesis. International Journal of Molecular Sciences, 21(5), e5378.
He, T. C., Sparks, A. B., Rago, C., Hermeking, H., Zawel, L., da Costa, L. T., Morin, P. J., Vogelstein, B., & Kinzler, K. W. (1998). Identification of c-MYC as a target of the APC pathway. Science, 281, 1509–1512.
He, X. X., Zhang, Y. N., Yan, J. W., Yan, J. J., Wu, Q., & Song, Y. H. (2016). CP-31398 inhibits the growth of p53-mutated liver cancer cells in vitro and in vivo. Tumour Biology, 37, 807–815.
Hsu, I. C., Metcalf, R. A., Sun, T., Welsh, J. A., Wang, N. J., & Harris, C. C. (1991). Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature, 350, 427–428.
Huang, D. S., Wang, Z., He, X. J., Diplas, B. H., Yang, R., Killela, P. J., Meng, Q., Ye, Z. Y., Wang, W., Jiang, X. T., et al. (2015). Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. European Journal of Cancer, 51, 969–976.
Huang, W., Chen, K., Lu, Y., Zhang, D., Cheng, Y., Li, L., Huang, W., He, G., Liao, H., Cai, L., et al. (2021). ABCC5 facilitates the acquired resistance of sorafenib through the inhibition of SLC7A11-induced ferroptosis in hepatocellular carcinoma. Neoplasia, 23, 1227–1239.
Huang, Y., Chen, X. C., Konduri, M., Fomina, N., Lu, J., Jin, L., Kolykhalov, A., & Tan, S. L. (2006). Mechanistic link between the anti-HCV effect of interferon gamma and control of viral replication by a Ras-MAPK signaling cascade. Hepatology, 43, 81–90.
Hussain, S. P., Schwank, J., Staib, F., Wang, X. W., & Harris, C. C. (2007). TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene, 26, 2166–2176.
Hytiroglou, P., Kotoula, V., Thung, S. N., Tsokos, M., Fiel, M. I., & Papadimitriou, C. S. (1998). Telomerase activity in precancerous hepatic nodules. Cancer, 82, 1831–1838.
Inami, Y., Waguri, S., Sakamoto, A., Kouno, T., Nakada, K., Hino, O., Watanabe, S., Ando, J., Iwadate, M., Yamamoto, M., et al. (2011). Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. Journal of Cell Biology, 193, 275–284.
Jiang, Y., Sun, A., Zhao, Y., Ying, W., Sun, H., Yang, X., Xing, B., Sun, W., Ren, L., Hu, B., et al. (2019). Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature, 567, 257–261.
Jiang, Z., Jhunjhunwala, S., Liu, J., Haverty, P. M., Kennemer, M. I., Guan, Y., Lee, W., Carnevali, P., Stinson, J., Johnson, S., et al. (2012). The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Research, 22, 593–601.
Jin, H., Wang, X., Ying, J., Wong, A. H., Cui, Y., Srivastava, G., Shen, Z. Y., Li, E. M., Zhang, Q., Jin, J., et al. (2007). Epigenetic silencing of a Ca(2+)-regulated Ras GTPase-activating protein RASAL defines a new mechanism of Ras activation in human cancers. Proc Natl Acad Sci U S A, 104, 12353–12358.
Kamo, N., Ke, B., Busuttil, R. W., & Kupiec-Weglinski, J. W. (2013). PTEN-mediated Akt/beta-catenin/Foxo1 signaling regulates innate immune responses in mouse liver ischemia/reperfusion injury. Hepatology, 57, 289–298.
Kew, M. C. (2011). Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. Journal of Gastroenterology and Hepatology, 26(Suppl 1), 144–152.
Khalaf, A. M., Fuentes, D., Morshid, A. I., Burke, M. R., Kaseb, A. O., Hassan, M., Hazle, J. D., & Elsayes, K. M. (2018). Role of Wnt/beta-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. Journal of Hepatocellular Carcinoma, 5, 61–73.
Kim, C. W., & Chang, K. M. (2013). Hepatitis C virus: Virology and life cycle. Clinical and Molecular Hepatology, 19, 17–25.
Kim, N. W., Piatyszek, M. A., Prowse, K. R., Harley, C. B., West, M. D., Ho, P. L., Coviello, G. M., Wright, W. E., Weinrich, S. L., & Shay, J. W. (1994). Specific association of human telomerase activity with immortal cells and cancer. Science, 266, 2011–2015.
Kim, S. B., Pandita, R. K., Eskiocak, U., Ly, P., Kaisani, A., Kumar, R., Cornelius, C., Wright, W. E., Pandita, T. K., & Shay, J. W. (2012). Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proceedings of the National Academy of Sciences of the United States of America, 109, E2949-2955.
Kneissig, M., Bernhard, S., & Storchova, Z. (2019). Modelling chromosome structural and copy number changes to understand cancer genomes. Current Opinion in Genetics & Development, 54, 25–32.
Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., Sou, Y. S., Ueno, I., Sakamoto, A., Tong, K. I., et al. (2010). The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature Cell Biology, 12, 213–223.
Korswagen, H. C., & Clevers, H. C. (1999). Activation and repression of wingless/Wnt target genes by the TCF/LEF-1 family of transcription factors. Cold Spring Harbor Symposia on Quantitative Biology, 64, 141–147.
Kotoula, V., Hytiroglou, P., Pyrpasopoulou, A., Saxena, R., Thung, S. N., & Papadimitriou, C. S. (2002). Expression of human telomerase reverse transcriptase in regenerative and precancerous lesions of cirrhotic livers. Liver, 22, 57–69.
Kotsakis, A., Vetsika, E. K., Christou, S., Hatzidaki, D., Vardakis, N., Aggouraki, D., Konsolakis, G., Georgoulias, V., Christophyllakis, C., Cordopatis, P., et al. (2012). Clinical outcome of patients with various advanced cancer types vaccinated with an optimized cryptic human telomerase reverse transcriptase (TERT) peptide: Results of an expanded phase II study. Annals of Oncology, 23, 442–449.
Kunz, C., Pebler, S., Otte, J., & von der Ahe, D. (1995). Differential regulation of plasminogen activator and inhibitor gene transcription by the tumor suppressor p53. Nucleic Acids Research, 23, 3710–3717.
Kwak, M. K., Wakabayashi, N., Greenlaw, J. L., Yamamoto, M., & Kensler, T. W. (2003). Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Molecular and Cellular Biology, 23, 8786–8794.
Lan, K. H., Sheu, M. L., Hwang, S. J., Yen, S. H., Chen, S. Y., Wu, J. C., Wang, Y. J., Kato, N., Omata, M., Chang, F. Y., et al. (2002). HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene, 21, 4801–4811.
Lee, D., Zhang, M. S., Tsang, F. H., Bao, M. H., Xu, I. M., Lai, R. K., Chiu, D. K., Tse, A. P., Law, C. T., Chan, C. Y., et al. (2021). Adaptive and constitutive activations of malic enzymes confer liver cancer multilayered protection against reactive oxygen species. Hepatology, 74, 776–796.
Lee, J. M., Yang, J., Newell, P., Singh, S., Parwani, A., Friedman, S. L., Nejak-Bowen, K. N., & Monga, S. P. (2014). beta-Catenin signaling in hepatocellular cancer: Implications in inflammation, fibrosis, and proliferation. Cancer Letters, 343, 90–97.
Lepourcelet, M., Chen, Y. N., France, D. S., Wang, H., Crews, P., Petersen, F., Bruseo, C., Wood, A. W., & Shivdasani, R. A. (2004). Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell, 5, 91–102.
Letouze, E., Shinde, J., Renault, V., Couchy, G., Blanc, J. F., Tubacher, E., Bayard, Q., Bacq, D., Meyer, V., Semhoun, J., et al. (2017). Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nature Communications, 8, 1315.
Levrero, M. (2006). Viral hepatitis and liver cancer: The case of hepatitis C. Oncogene, 25, 3834–3847.
Levrero, M., & Zucman-Rossi, J. (2016). Mechanisms of HBV-induced hepatocellular carcinoma. Journal of Hepatology, 64, S84–S101.
Li, B., Fu, J., Chen, P., Ge, X., Li, Y., Kuiatse, I., Wang, H., Wang, H., Zhang, X., & Orlowski, R. Z. (2015). The nuclear factor (Erythroid-derived 2)-like 2 and proteasome maturation protein axis mediate bortezomib resistance in multiple myeloma. Journal of Biological Chemistry, 290, 29854–29868.
Li, L., Fu, J., Liu, D., Sun, J., Hou, Y., Chen, C., Shao, J., Wang, L., Wang, X., Zhao, R., et al. (2020). Hepatocyte-specific Nrf2 deficiency mitigates high-fat diet-induced hepatic steatosis: Involvement of reduced PPARgamma expression. Redox Biology, 30, 101412.
Liang, B., Zhou, Y., Qian, M., Xu, M., Wang, J., Zhang, Y., Song, X., Wang, H., Lin, S., Ren, C., et al. (2021). TBX3 functions as a tumor suppressor downstream of activated CTNNB1 mutants during hepatocarcinogenesis. Journal of Hepatology, 75, 120–131.
Liao, P., Zeng, S. X., Zhou, X., Chen, T., Zhou, F., Cao, B., Jung, J. H., Del Sal, G., Luo, S., & Lu, H. (2017). Mutant p53 gains its function via c-Myc activation upon CDK4 phosphorylation at serine 249 and consequent PIN1 binding. Molecular Cell, 68(1134–1146), e1136.
Lin, Y. H., Zhang, S., Zhu, M., Lu, T., Chen, K., Wen, Z., Wang, S., Xiao, G., Luo, D., Jia, Y., et al. (2020). Mice with increased numbers of polyploid hepatocytes maintain regenerative capacity but develop fewer hepatocellular carcinomas following chronic liver injury. Gastroenterology, 158(1698–1712), e1614.
Lindemann, A., Patel, A. A., Silver, N. L., Tang, L., Liu, Z., Wang, L., Tanaka, N., Rao, X., Takahashi, H., Maduka, N. K., et al. (2019). COTI-2, a novel thiosemicarbazone derivative, exhibits antitumor activity in HNSCC through p53-dependent and -independent mechanisms. Clinical Cancer Research, 25, 5650–5662.
Lindenbach, B. D., & Rice, C. M. (2005). Unravelling hepatitis C virus replication from genome to function. Nature, 436, 933–938.
Liu, C., Li, Y., Semenov, M., Han, C., Baeg, G. H., Tan, Y., Zhang, Z., Lin, X., & He, X. (2002). Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell, 108, 837–847.
Liu, H., Zhao, L., Wang, M., Yang, K., Jin, Z., Zhao, C., & Shi, G. (2022). FNDC5 causes resistance to sorafenib by activating the PI3K/Akt/Nrf2 pathway in hepatocellular carcinoma cells. Frontiers in Oncology, 12, 852095.
Liu, P., Liang, B., Liu, M., Lebbink, J. H. G., Li, S., Qian, M., Lavrijsen, M., Peppelenbosch, M. P., Chen, X., & Smits, R. (2020). Oncogenic mutations in armadillo repeats 5 and 6 of beta-catenin reduce binding to APC, increasing signaling and transcription of target genes. Gastroenterology, 158(1029–1043), e1010.
Loguercio, C., Cuomo, A., Tuccillo, C., Gazzerro, P., Cioffi, M., Molinari, A. M., & Del Vecchio Blanco, C. (2003). Liver p53 expression in patients with HCV-related chronic hepatitis. Journal of Viral Hepatitis, 10, 266–270.
Lombardo, D., Saitta, C., Giosa, D., Di Tocco, F. C., Musolino, C., Caminiti, G., Chines, V., Franze, M. S., Alibrandi, A., Navarra, G., et al. (2020). Frequency of somatic mutations in TERT promoter, TP53 and CTNNB1 genes in patients with hepatocellular carcinoma from Southern Italy. Oncology Letters, 19, 2368–2374.
Lou, H., Du, S., Ji, Q., & Stolz, A. (2006). Induction of AKR1C2 by phase II inducers: Identification of a distal consensus antioxidant response element regulated by NRF2. Molecular Pharmacology, 69, 1662–1672.
Lu, L. C., Shao, Y. Y., Lee, Y. H., Hsieh, M. S., Hsiao, C. H., Lin, H. H., Kao, H. F., Ma, Y. Y., Yen, F. C., Cheng, A. L., et al. (2014). beta-catenin (CTNNB1) mutations are not associated with prognosis in advanced hepatocellular carcinoma. Oncology, 87, 159–166.
Ma, L., Wang, X., Jia, T., Wei, W., Chua, M. S., & So, S. (2015). Tankyrase inhibitors attenuate WNT/beta-catenin signaling and inhibit growth of hepatocellular carcinoma cells. Oncotarget, 6, 25390–25401.
Mangnall, D., Bird, N. C., & Majeed, A. W. (2003). The molecular physiology of liver regeneration following partial hepatectomy. Liver International, 23, 124–138.
McMahon, M., Thomas, N., Itoh, K., Yamamoto, M., & Hayes, J. D. (2006). Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the Nrf2-Keap1 complex. Journal of Biological Chemistry, 281, 24756–24768.
Miura, N., Horikawa, I., Nishimoto, A., Ohmura, H., Ito, H., Hirohashi, S., Shay, J. W., & Oshimura, M. (1997). Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genetics and Cytogenetics, 93, 56–62.
Miyashita, T., & Reed, J. C. (1995). Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell, 80, 293–299.
Moll, U. M., & Petrenko, O. (2003). The MDM2-p53 interaction. Molecular Cancer Research, 1, 1001–1008.
Morozov, V. A., & Lagaye, S. (2018). Hepatitis C virus: Morphogenesis, infection and therapy. World Journal of Hepatology, 10, 186–212.
Moya, M., Benet, M., Guzman, C., Tolosa, L., Garcia-Monzon, C., Pareja, E., Castell, J. V., & Jover, R. (2012). Foxa1 reduces lipid accumulation in human hepatocytes and is down-regulated in nonalcoholic fatty liver. PLoS ONE, 7, e30014.
Murakami, S. (2001). Hepatitis B virus X protein: A multifunctional viral regulator. Journal of Gastroenterology, 36, 651–660.
Nakano, K., & Vousden, K. H. (2001). PUMA, a novel proapoptotic gene, is induced by p53. Molecular Cell, 7, 683–694.
Nassal, M. (2015). HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut, 64, 1972–1984.
Nault, J. C., Datta, S., Imbeaud, S., Franconi, A., Mallet, M., Couchy, G., Letouze, E., Pilati, C., Verret, B., Blanc, J. F., et al. (2015). Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nature Genetics, 47, 1187–1193.
Nault, J. C., Mallet, M., Pilati, C., Calderaro, J., Bioulac-Sage, P., Laurent, C., Laurent, A., Cherqui, D., Balabaud, C., & Zucman-Rossi, J. (2013). High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nature Communications, 4, 2218.
Nault, J. C., Ningarhari, M., Rebouissou, S., & Zucman-Rossi, J. (2019). The role of telomeres and telomerase in cirrhosis and liver cancer. Nature Reviews. Gastroenterology & Hepatology, 16, 544–558.
Ngo, H. K. C., Kim, D. H., Cha, Y. N., Na, H. K., & Surh, Y. J. (2017). Nrf2 Mutagenic Activation Drives Hepatocarcinogenesis. Cancer Research, 77, 4797–4808.
Niessen, C. M., & Gottardi, C. J. (2008). Molecular components of the adherens junction. Biochimica Et Biophysica Acta, 1778, 562–571.
Nishita, M., Saji, T., & Minami, Y. (2019). Non-canonical Wnt signaling and cellular responses. Clinical Calcium, 29, 291–297.
Niture, S. K., & Jaiswal, A. K. (2012). Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. Journal of Biological Chemistry, 287, 9873–9886.
Niture, S. K., & Jaiswal, A. K. (2013). Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radical Biology & Medicine, 57, 119–131.
Oates, P. S., & Morgan, R. G. (1986). Changes in pancreatic acinar cell nuclear number and DNA content during aging in the rat. The American Journal of Anatomy, 177, 547–554.
Oda, E., Ohki, R., Murasawa, H., Nemoto, J., Shibue, T., Yamashita, T., Tokino, T., Taniguchi, T., & Tanaka, N. (2000). Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science, 288, 1053–1058.
Okabe, H., Kinoshita, H., Imai, K., Nakagawa, S., Higashi, T., Arima, K., Uchiyama, H., Ikegami, T., Harimoto, N., Itoh, S., et al. (2016). Diverse basis of beta-catenin activation in human hepatocellular carcinoma: implications in biology and prognosis. PLoS ONE, 11, e0152695.
Orgad, S., Dimant, H., Dor-On, E., Azriel-Rosenfeld, R., Benhar, I., & Solomon, B. (2010). TAR1, a human anti-p53 single-chain antibody, restores tumor suppressor function to mutant p53 variants. Journal of Immunotherapy, 33, 146–154.
Orru, C., Szydlowska, M., Taguchi, K., Zavattari, P., Perra, A., Yamamoto, M., & Columbano, A. (2018). Genetic inactivation of Nrf2 prevents clonal expansion of initiated cells in a nutritional model of rat hepatocarcinogenesis. Journal of Hepatology, 69, 635–643.
Owen-Schaub, L. B., Zhang, W., Cusack, J. C., Angelo, L. S., Santee, S. M., Fujiwara, T., Roth, J. A., Deisseroth, A. B., Zhang, W. W., Kruzel, E., et al. (1995). Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Molecular and Cellular Biology, 15, 3032–3040.
Ozturk, M., Arslan-Ergul, A., Bagislar, S., Senturk, S., & Yuzugullu, H. (2009). Senescence and immortality in hepatocellular carcinoma. Cancer Letters, 286, 103–113.
Padmanabhan, B., Tong, K. I., Kobayashi, A., Yamamoto, M., & Yokoyama, S. (2008). Structural insights into the similar modes of Nrf2 transcription factor recognition by the cytoplasmic repressor Keap1. Journal of Synchrotron Radiat., 15, 273–276.
Pandit, S. K., Westendorp, B., & de Bruin, A. (2013). Physiological significance of polyploidization in mammalian cells. Trends in Cell Biology, 23, 556–566.
Park, J. I., Venteicher, A. S., Hong, J. Y., Choi, J., Jun, S., Shkreli, M., Chang, W., Meng, Z., Cheung, P., Ji, H., et al. (2009). Telomerase modulates Wnt signalling by association with target gene chromatin. Nature, 460, 66–72.
Paterlini-Brechot, P., Saigo, K., Murakami, Y., Chami, M., Gozuacik, D., Mugnier, C., Lagorce, D., & Brechot, C. (2003). Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene, 22, 3911–3916.
Perugorria, M. J., Olaizola, P., Labiano, I., Esparza-Baquer, A., Marzioni, M., Marin, J. J. G., Bujanda, L., & Banales, J. M. (2019). Wnt-beta-catenin signalling in liver development, health and disease. Nature Reviews Gastroenterology & Hepatology, 16, 121–136.
Quan, H., Zhou, F., Nie, D., Chen, Q., Cai, X., Shan, X., Zhou, Z., Chen, K., Huang, A., Li, S., et al. (2014). Hepatitis C virus core protein epigenetically silences SFRP1 and enhances HCC aggressiveness by inducing epithelial-mesenchymal transition. Oncogene, 33, 2826–2835.
Reya, T., & Clevers, H. (2005). Wnt signalling in stem cells and cancer. Nature, 434, 843–850.
Rippin, T. M., Bykov, V. J., Freund, S. M., Selivanova, G., Wiman, K. G., & Fersht, A. R. (2002). Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene, 21, 2119–2129.
Rudolph, K. L., Hartmann, D., & Opitz, O. G. (2009). Telomere dysfunction and DNA damage checkpoints in diseases and cancer of the gastrointestinal tract. Gastroenterology, 137, 754–762.
Ruiz de Galarreta, M., Bresnahan, E., Molina-Sanchez, P., Lindblad, K. E., Maier, B., Sia, D., Puigvehi, M., Miguela, V., Casanova-Acebes, M., Dhainaut, M., et al. (2019). beta-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discovery, 9, 1124–1141.
Sanz-Cameno, P., Martin-Vilchez, S., Lara-Pezzi, E., Borque, M. J., Salmeron, J., Munoz de Rueda, P., Solis, J. A., Lopez-Cabrera, M., & Moreno-Otero, R. (2006). Hepatitis B virus promotes angiopoietin-2 expression in liver tissue: Role of HBV x protein. American Journal of Pathology, 169, 1215–1222.
Sasaki, H., Sato, H., Kuriyama-Matsumura, K., Sato, K., Maebara, K., Wang, H., Tamba, M., Itoh, K., Yamamoto, M., & Bannai, S. (2002). Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. Journal of Biological Chemistry, 277, 44765–44771.
Schulze, K., Imbeaud, S., Letouze, E., Alexandrov, L. B., Calderaro, J., Rebouissou, S., Couchy, G., Meiller, C., Shinde, J., Soysouvanh, F., et al. (2015). Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nature Genetics, 47, 505–511.
Seeger, C., & Mason, W. S. (2000). Hepatitis B virus biology. Microbiology and Molecular Biology Reviews, 64, 51–68.
Seeger, C., & Mason, W. S. (2015). Molecular biology of hepatitis B virus infection. Virology, 479–480, 672–686.
Shi, H., Lambert, J. M., Hautefeuille, A., Bykov, V. J., Wiman, K. G., Hainaut, P., & Caron de Fromentel, C. (2008). In vitro and in vivo cytotoxic effects of PRIMA-1 on hepatocellular carcinoma cells expressing mutant p53ser249. Carcinogenesis, 29, 1428–1434.
Shlomai, A., de Jong, Y. P., & Rice, C. M. (2014). Virus associated malignancies: The role of viral hepatitis in hepatocellular carcinoma. Seminars in Cancer Biology, 26, 78–88.
Shtutman, M., Zhurinsky, J., Simcha, I., Albanese, C., D’Amico, M., Pestell, R., & Ben-Ze’ev, A. (1999). The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A, 96, 5522–5527.
Sladky, V. C., Knapp, K., Soratroi, C., Heppke, J., Eichin, F., Rocamora-Reverte, L., Szabo, T. G., Bongiovanni, L., Westendorp, B., Moreno, E., et al. (2020a). E2F-family members engage the PIDDosome to limit hepatocyte ploidy in liver development and regeneration. Developmental Cell, 52(335–349), e337.
Sladky, V. C., Knapp, K., Szabo, T. G., Braun, V. Z., Bongiovanni, L., van den Bos, H., Spierings, D. C., Westendorp, B., Curinha, A., Stojakovic, T., et al. (2020b). PIDDosome-induced p53-dependent ploidy restriction facilitates hepatocarcinogenesis. EMBO Reports, 21, e50893.
Soini, Y., Chia, S. C., Bennett, W. P., Groopman, J. D., Wang, J. S., DeBenedetti, V. M., Cawley, H., Welsh, J. A., Hansen, C., Bergasa, N. V., et al. (1996). An aflatoxin-associated mutational hotspot at codon 249 in the p53 tumor suppressor gene occurs in hepatocellular carcinomas from Mexico. Carcinogenesis, 17, 1007–1012.
Staib, F., Hussain, S. P., Hofseth, L. J., Wang, X. W., & Harris, C. C. (2003). TP53 and liver carcinogenesis. Human Mutation, 21, 201–216.
Stern, M. C., Umbach, D. M., Yu, M. C., London, S. J., Zhang, Z. Q., & Taylor, J. A. (2001). Hepatitis B, aflatoxin B(1), and p53 codon 249 mutation in hepatocellular carcinomas from Guangxi, People’s Republic of China, and a meta-analysis of existing studies. Cancer Epidemiology, Biomarkers & Prevention, 10, 617–625.
Summers, J., & Mason, W. S. (1982). Replication of the genome of a hepatitis B–like virus by reverse transcription of an RNA intermediate. Cell, 29, 403–415.
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., & Bray, F. (2021). Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians, 71, 209–249.
Sung, W. K., Zheng, H., Li, S., Chen, R., Liu, X., Li, Y., Lee, N. P., Lee, W. H., Ariyaratne, P. N., Tennakoon, C., et al. (2012). Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nature Genetics, 44, 765–769.
Suzuki, A., Shim, J., Ogata, K., Yoshioka, H., & Iwata, J. (2019). Cholesterol metabolism plays a crucial role in the regulation of autophagy for cell differentiation of granular convoluted tubules in male mouse submandibular glands. Development, 146.
Suzuki, S., Tanaka, T., Poyurovsky, M. V., Nagano, H., Mayama, T., Ohkubo, S., Lokshin, M., Hosokawa, H., Nakayama, T., Suzuki, Y., et al. (2010). Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proceedings of the National Academy of Sciences of the United States of America, 107, 7461–7466.
Synnott, N. C., O’Connell, D., Crown, J., & Duffy, M. J. (2020). COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Research and Treatment, 179, 47–56.
Tahtouh, R., Azzi, A. S., Alaaeddine, N., Chamat, S., Bouharoun-Tayoun, H., Wardi, L., Raad, I., Sarkis, R., Antoun, N. A., & Hilal, G. (2015). Telomerase inhibition decreases alpha-fetoprotein expression and secretion by hepatocellular carcinoma cell lines: In vitro and in vivo study. PLoS ONE, 10, e0119512.
Tan, A. T., Yang, N., Lee Krishnamoorthy, T., Oei, V., Chua, A., Zhao, X., Tan, H. S., Chia, A., Le Bert, N., Low, D., et al. (2019). Use of expression profiles of HBV-DNA integrated into genomes of hepatocellular carcinoma cells to select T cells for immunotherapy. Gastroenterology, 156(1862–1876), e1869.
Tanaka, K., Goto, H., Nishimura, Y., Kasahara, K., Mizoguchi, A., & Inagaki, M. (2018). Tetraploidy in cancer and its possible link to aging. Cancer Science, 109, 2632–2640.
Tang, E., Wang, Y., Liu, T., & Yan, B. (2019). Gastrin promotes angiogenesis by activating HIF-1alpha/beta-catenin/VEGF signaling in gastric cancer. Gene, 704, 42–48.
Tong, K. I., Katoh, Y., Kusunoki, H., Itoh, K., Tanaka, T., & Yamamoto, M. (2006). Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Molecular and Cellular Biology, 26, 2887–2900.
Tortelote, G. G., Reis, R. R., de Almeida Mendes, F., & Abreu, J. G. (2017). Complexity of the Wnt/betacatenin pathway: Searching for an activation model. Cellular Signalling, 40, 30–43.
Totoki, Y., Tatsuno, K., Covington, K. R., Ueda, H., Creighton, C. J., Kato, M., Tsuji, S., Donehower, L. A., Slagle, B. L., Nakamura, H., et al. (2014). Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nature Genetics, 46, 1267–1273.
Tu, T., Budzinska, M.A., Shackel, N.A., & Urban, S. (2017). HBV DNA integration: molecular mechanisms and clinical implications. Viruses, 9(4), 75.
Turton, K.L., Meier-Stephenson, V., Badmalia, M.D., Coffin, C.S., & Patel, T.R. (2020). Host transcription factors in hepatitis B virus RNA synthesis. Viruses, 12(2), 160.
Venugopal, R., & Jaiswal, A. K. (1996). Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:Quinone oxidoreductase1 gene. Proceedings of the National Academy of Sciences of the United States of America, 93, 14960–14965.
Verma, A. K., Yadav, A., Dewangan, J., Singh, S. V., Mishra, M., Singh, P. K., & Rath, S. K. (2015). Isoniazid prevents Nrf2 translocation by inhibiting ERK1 phosphorylation and induces oxidative stress and apoptosis. Redox Biology, 6, 80–92.
Volodko, N., Gordon, M., Salla, M., Ghazaleh, H. A., & Baksh, S. (2014). RASSF tumor suppressor gene family: Biological functions and regulation. FEBS Letters, 588, 2671–2684.
Wang, H., Liao, P., Zeng, S. X., & Lu, H. (2020). Co-targeting p53–R249S and CDK4 synergistically suppresses survival of hepatocellular carcinoma cells. Cancer Biology & Therapy, 21, 269–277.
Wang, Z., Sheng, Y. Y., Gao, X. M., Wang, C. Q., Wang, X. Y., Lu, X. U., Wei, J. W., Zhang, K. L., Dong, Q. Z., & Qin, L. X. (2015). beta-catenin mutation is correlated with a favorable prognosis in patients with hepatocellular carcinoma. Molecular and Clinical Oncology, 3, 936–940.
Wei, S., Dai, M., Zhang, C., Teng, K., Wang, F., Li, H., Sun, W., Feng, Z., Kang, T., Guan, X., et al. (2021). KIF2C: A novel link between Wnt/beta-catenin and mTORC1 signaling in the pathogenesis of hepatocellular carcinoma. Protein & Cell, 12, 788–809.
Wei, W., Chua, M. S., Grepper, S., & So, S. (2010). Small molecule antagonists of Tcf4/beta-catenin complex inhibit the growth of HCC cells in vitro and in vivo. International Journal of Cancer, 126, 2426–2436.
Weng, M. W., Lee, H. W., Choi, B., Wang, H. T., Hu, Y., Mehta, M., Desai, D., Amin, S., Zheng, Y., & Tang, M. S. (2017). AFB1 hepatocarcinogenesis is via lipid peroxidation that inhibits DNA repair, sensitizes mutation susceptibility and induces aldehyde-DNA adducts at p53 mutational hotspot codon 249. Oncotarget, 8, 18213–18226.
Wiemann, S. U., Satyanarayana, A., Tsahuridu, M., Tillmann, H. L., Zender, L., Klempnauer, J., Flemming, P., Franco, S., Blasco, M. A., Manns, M. P., et al. (2002). Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. The FASEB Journal, 16, 935–942.
Wijetunga, N. A., Pascual, M., Tozour, J., Delahaye, F., Alani, M., Adeyeye, M., Wolkoff, A. W., Verma, A., & Greally, J. M. (2017). A pre-neoplastic epigenetic field defect in HCV-infected liver at transcription factor binding sites and polycomb targets. Oncogene, 36, 2030–2044.
Wu, L. L., Cai, W. P., Lei, X., Shi, K. Q., Lin, X. Y., & Shi, L. (2019). NRAL mediates cisplatin resistance in hepatocellular carcinoma via miR-340-5p/Nrf2 axis. J Cell Commun Signal, 13, 99–112.
Wu, S., Zhang, T., & Du, J. (2016). Ursolic acid sensitizes cisplatin-resistant HepG2/DDP cells to cisplatin via inhibiting Nrf2/ARE pathway. Drug Design, Development and Therapy, 10, 3471–3481.
Xu, D., Dwyer, J., Li, H., Duan, W., & Liu, J. P. (2008). Ets2 maintains hTERT gene expression and breast cancer cell proliferation by interacting with c-Myc. Journal of Biological Chemistry, 283, 23567–23580.
Yan, H., Yang, Y., Zhang, L., Tang, G., Wang, Y., Xue, G., Zhou, W., & Sun, S. (2015). Characterization of the genotype and integration patterns of hepatitis B virus in early- and late-onset hepatocellular carcinoma. Hepatology, 61, 1821–1831.
Yang, J. D., Hainaut, P., Gores, G. J., Amadou, A., Plymoth, A., & Roberts, L. R. (2019). A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nature Reviews Gastroenterology & Hepatology, 16, 589–604.
Yoo, Y. G., Oh, S. H., Park, E. S., Cho, H., Lee, N., Park, H., Kim, D. K., Yu, D. Y., Seong, J. K., & Lee, M. O. (2003). Hepatitis B virus X protein enhances transcriptional activity of hypoxia-inducible factor-1alpha through activation of mitogen-activated protein kinase pathway. Journal of Biological Chemistry, 278, 39076–39084.
Yoo, Y. D., Ueda, H., Park, K., Flanders, K. C., Lee, Y. I., Jay, G., & Kim, S. J. (1996). Regulation of transforming growth factor-beta 1 expression by the hepatitis B virus (HBV) X transactivator. Role in HBV pathogenesis. The Journal of Clinical Investigation, 97, 388–395.
Youssef, N., Paradis, V., Ferlicot, S., & Bedossa, P. (2001). In situ detection of telomerase enzymatic activity in human hepatocellular carcinogenesis. The Journal of Pathology, 194, 459–465.
Zack, T. I., Schumacher, S. E., Carter, S. L., Cherniack, A. D., Saksena, G., Tabak, B., Lawrence, M. S., Zhsng, C. Z., Wala, J., Mermel, C. H., et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nature Genetics, 45, 1134–1140.
Zavattari, P., Perra, A., Menegon, S., Kowalik, M. A., Petrelli, A., Angioni, M. M., Follenzi, A., Quagliata, L., Ledda-Columbano, G. M., Terracciano, L., et al. (2015). Nrf2, but not beta-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology, 62, 851–862.
Zekri Ael, R., Nassar, A. A., El-Din El-Rouby, M. N., Shousha, H. I., Barakat, A. B., El-Desouky, E. D., Zayed, N. A., Ahmed, O. S., El-Din Youssef, A. S., Kaseb, A. O., et al. (2014). Disease progression from chronic hepatitis C to cirrhosis and hepatocellular carcinoma is associated with increasing DNA promoter methylation. Asian Pacific Journal of Cancer Prevention, 14, 6721–6726.
Zhang, D. D., Lo, S. C., Cross, J. V., Templeton, D. J., & Hannink, M. (2004). Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Molecular and Cellular Biology, 24, 10941–10953.
Zhang, M., Zhang, C., Zhang, L., Yang, Q., Zhou, S., Wen, Q., & Wang, J. (2015). Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer, 15, 531.
Zhang, S., Lin, Y. H., Tarlow, B., & Zhu, H. (2019). The origins and functions of hepatic polyploidy. Cell Cycle, 18, 1302–1315.
Zhang, S., Nguyen, L. H., Zhou, K., Tu, H. C., Sehgal, A., Nassour, I., Li, L., Gopal, P., Goodman, J., Singal, A. G., et al. (2018a). Knockdown of anillin actin binding protein blocks cytokinesis in hepatocytes and reduces liver tumor development in mice without affecting regeneration. Gastroenterology, 154, 1421–1434.
Zhang, S., Zhou, K., Luo, X., Li, L., Tu, H. C., Sehgal, A., Nguyen, L. H., Zhang, Y., Gopal, P., Tarlow, B. D., et al. (2018b). The polyploid state plays a tumor-suppressive role in the liver. Developmental Cell, 47, 390.
Zucman-Rossi, J., Villanueva, A., Nault, J. C., & Llovet, J. M. (2015). Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology, 149(1226–1239), e1224.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (grants 2020YFA0803203, 2019YFA0802102), grants from National Natural Science Foundation of China (81925029, 81790253, 82230098, 32221002), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB19020203), and the CAS project for young scientists in basic research (YSBR-014) and Shanghai Municipal Science and Technology Major Project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Author Daming Gao is a member of the Editorial Board for Genome Instability & Disease.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, R., Lin, M. & Gao, D. Major genomic mutations driving hepatocellular carcinoma. GENOME INSTAB. DIS. 4, 239–253 (2023). https://doi.org/10.1007/s42764-023-00103-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42764-023-00103-7