
Abstract
Poly(ADP-ribosyl)ation (PARylation), a type of post-translational modification catalyzed by poly(ADP-ribose) polymerase (PARP), is implicated in numerous biological processes including DNA repair, chromatin remodeling, programmed cell death, RNA regulation, and PAR-dependent ubiquitination. The advent of PARP inhibitors represents a new synthetic lethality paradigm for killing tumors bearing BRCA mutations in which tumor-specific defects are exploited to create a vulnerability that causes tumor cell death. To date, four PARP inhibitors have been approved by the US Food and Drug Administration for treatment of several types of cancer. In this review, we summarize the current knowledge of the molecular functions of PARP1 and highlight the recent advances in the use of PARP inhibitors in cancer treatment and the problem of drug resistance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Since Chambon et al. first reported poly(ADP-ribosyl)ation (PARylation) in 1963, researchers have continued to make new discoveries about this post-translational modification (PTM) (Chambon et al., 1963). Over the past 60 years, PARylation has been found to play an important role in DNA repair, chromatin remodeling, programmed cell death, RNA regulation, and PAR-dependent ubiquitination (Anderson & Kedersha, 2008; Bock et al., 2015; Eustermann et al., 2011; Li et al., 2015; Smith et al., 2019). In parallel, the discovery of the synthetic lethality between poly(ADP-ribose) polymerase (PARP) inhibitors (PARPis) and breast cancer susceptibility gene (BRCA) mutations has also made PARPis a research hotspot in the anti-cancer field (Bryant et al., 2005; Farmer et al., 2005). To date, PARPis are the most successful example of using synthetic lethality to kill tumor cells. In this paper, after providing a brief review of PARylation and the PARP family, we focus on the current status and progress in understanding PARP molecular functions. In addition, the most recent findings regarding the clinical application of PARPis and the mechanisms of drug resistance are also described to enlighten future research.
PARylation and PARP
What is PARylation?
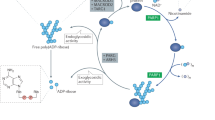
PARylation is a reversible post-translational modification that alters protein function. Specifically, (ADP-ribosyl)ation transferases, also known as PARPs, catalyze the covalent attachment of ADP-ribose to target proteins (Gibson & Kraus, 2012) (Fig. 1). ADP-ribose is predominantly attached to the serine residues of the target substrates, which include PARP itself and other proteins, such as histones, DNA repair proteins, transcription factors, and chromatin modulators (Palazzo et al., 2018). PARylation consists of three processes: first, an ADP-ribose monomer is attached to an amino acid residue (initiation), and then a (2’-1’’) or (2’’-2’’’) ribose-ribose glycosidic bond is formed between ADP-ribose units (elongation or branching) (Rolli et al., 1997). These catalytic processes form linear or branched PAR structures on target proteins, the size of which depends on the accessibility of the substrate (Alvarez-Gonzalez & Mendoza-Alvarez, 1995; Ruf et al., 1998).

Dynamic synthesis and degradation of PAR chain. The dynamic PARylation process involves a complex interplay between PARPs and PAR-degrading enzymes. Specifically, PARylation is initiated when PARPs recognize specific signals such as DNA damage or other signaling events. PARP then be recruited to the target protein and begin to break down NAD + into ADP-ribose units, leading to the attachment of ADP-ribose units to the target protein to create a linear or branched PAR chain. Once DNA damage is repaired, PARG and ARH3 cleave the bonds between ADP-ribose subunits, while TARG and MACROD1/2 remove the terminal ADP-ribose attachments, which results in the complete removal of PARylation from target proteins. PAR, poly(ADP-ribose); MAR, mono(ADP-ribose)
Excessive PARylation, which disrupts chromatin modification and DNA synthesis, is highly cytotoxic (Prokhorova et al., 2021). To avoid these cytotoxic effects, PAR glycohydrolase (PARG), ADP-ribosylhydrolase 3 (ARH3), terminal ADP-ribose protein glycohydrolase (TARG), and mono-ADP ribosylhydrolase 1/2 (MACROD1/2) catabolize PAR chains to maintain PARylation levels within a dynamic harmless range (Brochu et al., 1994; Jankevicius et al., 2013; Oka et al., 2006; Sharifi et al., 2013) (Fig. 1). PARG and ARH3 are the main enzymes that cleave the bonds between ADP-ribose subunits, and PARG is much more efficient than ARH3 in the clearance of PAR chains (Oka et al., 2006; Slade et al., 2011). TARG and MACROD1/2 remove the terminal serine-ADP-ribose attachments, which results in the complete removal of PARylation from target proteins (Peterson et al., 2011; Rosenthal et al., 2013). ARH3 is also responsible for the degradation of mitochondrial matrix-associated PAR (Niere et al., 2012). The release of ADP-ribose subunits can generate ATP for DNA repair, which guarantees rapid repair of DNA damage even under energy depletion (Maruta et al., 2007). In addition, PARG is also involved in regulating chromatin structure during DNA damage (Rack et al., 2016).
The PARP family
Seventeen PARP family members have been discovered in mammalian species; these proteins are named PARP1–16 with PARP5 having two subtypes, PARP5a and PARP5b (Langelier et al., 2012). However, not all PARP members have the same ability to synthesize PAR. PARP1, PARP2, and PARP5 have the highest activity, PARP9 and PARP13 lack catalytic activity, and other PARPs only synthesize mono(ADP-ribose) (Vyas et al., 2014). The different catalytic activities of the PARPs correspond to their different intracellular localizations. PARP1 is only detected in the nucleus, while PARP6, PARP8, PARP12, PARP13, PARP15, and PARP16 are more likely to be localized in the cytoplasm. Other PARPs have been found in the nucleus and cytoplasm (Vyas et al., 2013).
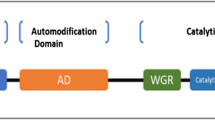
Since PARPs were identified, most research has focused on PARP1, the founding member of the PARP family. To date, six domains have been identified in PARP1, namely three zinc-binding domains (Zn1, Zn2, Zn3), an auto modification domain (AD), a Trp-Gly-Arg domain (WGR), and a catalytic domain (CAT) (Alemasova & Lavrik, 2019). Zn1 and Zn2 are responsible for recognizing particular DNA structures and single-strand breaks (SSBs) and promoting allosteric activation of other domains, while Zn3 is associated with DNA-dependent catalytic activity and chromatin compaction (Langelier et al., 2010, 2011). The WGR domain is the central component interacting with DNA when DNA is damaged (Langelier et al., 2012). The CAT consists of a helical subdomain and ADP-ribosyl transferase (ART) domain, which is the catalytic core and responsible for the binding of NAD+ and acceptor (Ruf et al., 1996). ART is highly conserved in all PARP family members, and its His-Tyr-Glu triad is regarded as the ART signature (Alemasova & Lavrik, 2019). When a DNA lesion occurs, Zn1, Zn3, and WGR bind to the damaged DNA, which induces helical domain unfolding and binding to NAD+, thereby activating PARP1 (Langelier et al., 2012).
Molecular functions of PARylation and PARP
DNA repair
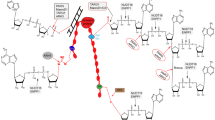
A wide variety of endogenous or exogenous noxious stimuli such as radiation and chemical mutagens can induce genomic DNA damage, including SSBs and double-strand breaks (DSBs) (Huang & Kraus, 2022). SSBs occur with a high frequency in every cell, once every 1–10 s, but they do not cause mutations in all cases (Demple & Harrison, 1994). The main reason for this is the rapid DNA damage repair mediated by PARP1 (Fig. 2). As a DNA lesion sensor, PARP1 recognizes SSBs, becomes activated, and PARylates itself or other proteins (Eustermann et al., 2011). The PARylated proteins then recruit DNA repair factors that repair DNA damage by interacting with histone PARylation factor 1 (HPF1), a coregulator of PARP1-dependent histone PARylation (Eustermann et al., 2011; Gibbs-Seymour et al., 2016). HPF1 prevents excessive auto-modification of PARP1 and promotes histone PARylation to ensure an efficient cellular response to DNA damage (Gibbs-Seymour et al., 2016).

The role of PARP1 in DNA repair and synthetic lethality. Endogenous or exogenous noxious stimuli induce genomic DNA SSBs. Under normal physiological conditions, PARP1 acts as a DNA lesion sensor to recognize SSBs and undergoes moderate auto-PARylation by interacting with HPF1. Activated PARP1 recruits DNA repair factors including XRCC1, LIG3, POLB, and PNKP to repair DNA damage. In the presence of PARPis, PARP1 is “trapped” on nicked DNA and fails to be auto-PARylated. The “trapped” PARP1 then causes replication fork collapse, which potentially upgrades the SSB to a DSB. With wild-type BRCA, the MRN complex recognizes DSBs and promotes HR, while with mutant BRCA, DSBs either remain unrepaired or are repaired through NHEJ, which leads to cell death
X-ray repair cross-complementing 1 (XRCC1) is another effector playing a leading role in DNA damage repair (Hanzlikova et al., 2017). In response to the PARylation of PARP1, XRCC1 is rapidly recruited to DNA damage sites and attracts other SSB repair proteins such as DNA ligase 3 (LIG3), DNA polymerase β (POLB), and bifocal polynucleotide kinase 3ʹ-phosphatase (PNKP) (Caldecott et al., 1994, 1996; Whitehouse et al., 2001). Notably, XRCC1 acts as a regulator of PARP1 activity. Recent studies have found that XRCC1-deficient cells cannot rapidly resume transcription after DNA base damage because of abnormal PARP1 activity (Adamowicz et al., 2021). If not repaired in time, accumulated SSBs lead to more severe DNA damage—DSBs. In that case, PARP1 recognizes DSBs and participates in the early recruitment of mitotic recombination 11 (MRE11) to promote homologous recombination (HR) (Schlacher et al., 2011).
In eukaryotic cells, HR is a high-fidelity repair pathway, in which homologous sequences of the sister chromatid are used as a template to repair damage during the S phase (Ray Chaudhuri & Nussenzweig, 2017). Specifically, the MRN complex, consisting of MRE11, RAD50, and NBS1, recognizes DSBs and binds to the damaged DNA end, and promotes the end resection to produce 3’ single-strand DNA (Haince et al., 2008). BRCA1, BRCA2 and partner and localizer of BRCA2 (PALB2) are then recruited to regulate the binding of RAD51 to DNA damage sites(C.-C. ). Next, RAD51 catalyzes DNA extension after the pairing of 3’ single-stranded DNA with homologous sequences on the sister chromatid (Yang et al., 2017). Finally, the copied double-strand DNA is connected to the nicked DNA by DNA ligase to complete repair.
Although PARP1 is the most active member of the PARP family, other members have also been reported to play a role in DNA damage repair (Amé et al., 1999; Beck et al., 2014; Q. ). Hanzlikova et al. found overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin (Hanzlikova et al., 2017). As mentioned earlier, the rapid recruitment of XRCC1 is a critical step in repairing SSBs. Although the total ADP-ribosylation level is visibly reduced in the lack of PARP1, the individual PARP2 protein is sufficient to maintain near-normal XRCC1 recruitment (Hanzlikova et al., 2017). Moreover, PARP3 and PARP1 act synergistically in response to X-irradiation in human and mouse cells (Boehler et al., 2011). In the absence of PARP3, cells become more sensitive to antitumoral drugs generating DSBs and show a significant delay in the repair of radio-induced DSBs (Beck et al., 2014). Besides, Day et al. found that PARP3 regulates G quadruplex DNA in response to DNA damage, which suppresses repair by nonhomologous end-joining (NHEJ) and HR (Day et al., 2017).
Chromatin remodeling
Once DNA damage occurs, cells initiate chromatin remodeling promptly to increase DNA accessibility, facilitating the entry of repair proteins to repair the damaged DNA strand. Smith et al. found that PAR-dependent chromatin remodeling promotes the formation of a particular chromatin conformation rather than simply unfolding chromatin (Smith et al., 2019). This exposes the DNA damage and makes it more accessible to binding repair proteins to improve the efficacy of DNA repair (Smith et al., 2019). The PARylated PARP1 recruits chromatin remodeling enzymes namely HPF1, amplified-in-liver-cancer 1, aprataxin and PNKP-like factor, and chromodomain helicase DNA binding protein 6 (CHD6) to induce chromatin remodeling and pull repair factors close to DNA damage (Bilokapic et al., 2020). In addition, PARP1-mediated PARylation can modulate chromatin conformation independent of DNA damage. H1 histone, a major substrate of PARP1, is released from chromatin during DNA replication. Then PARP1 takes the place of H1 histone and binds with linker DNA, facilitating the formation of a chromatin conformation conducive to transcription (Krishnakumar et al., 2008). Of note, PARP1 mediates the condensation of chromatin when it is not bound to DNA, but promotes decondensation when it binds DNA (Bock et al., 2015). The bidirectional interaction between PARP1 and histone indicates that histone can also regulate PARP1 activity. Consistent with this, a study in Drosophila found that histone H2A and H2B can suppress the catalytic activity of PARP1, while histone H4 continually activates PARP1 by interacting with the CAT domain, which facilitates chromatin remodeling through the accumulation of PARylation modifications (Boamah et al., 2012).
Cell death
Excessive PARylation is cytotoxic. Overactivation of PARP1 leads to cell death through the following three pathways. First, the process of PARylation consumes a large amount of NAD+, which impacts glycolysis and oxidative phosphorylation, ultimately resulting in metabolic catastrophe and cell death (Alano et al., 2010). Second, PAR can mediate a form of programmed cell death named parthanatos, which is an important cell death pathway in neurons. PAR-mediated parthanatos is associated with Parkinson's disease, brain damage following ischemia/reperfusion, and other neurological diseases (Park et al., 2020). The proposed mechanism underlying PAR-mediated parthanatos is related to apoptosis-inducing factor (AIF), a caspase-independent apoptosis-inducing factor, which is located in mitochondria under physiological conditions and participates in oxidative phosphorylation (Daugas et al., 2000). The release of a large amount of PAR into the cytoplasm triggers the translocation of AIF from the cytoplasm to the nucleus, which results in DNA fragmentation and chromatin condensation, and ultimately induces parthanatos both in vivo and in vitro (Mashimo et al., 2021; S.-W. Yu et al., 2002, 2006). It is argued that AIF binds PAR with high affinity, promoting AIF release from mitochondria (Gagné et al., 2008). In addition, mitochondrial membrane depolarization caused by the depletion of NAD+ may promote AIF release (S.-W. Yu et al., 2002). The third way in which overactivation of PARP1 leads to cell death is through PARP1-mediated necrosis, another type of programmed cell death distinct from apoptosis (Xu et al., 2006). Exposure to high levels of reactive oxygen species/reactive nitrogen species can over-activate PARP1 and induce necrosis, which is involved in c-Jun N-terminal kinase activation and mitochondrial dysfunction (Chiu et al., 2011; Xu et al., 2006).
Other functions
Early studies on PARP and PARylation mainly revealed their role in the regulation of DNA, and later studies gradually revealed their role in the regulation of RNA. In eukaryotes, the DNA sequence is first transcribed into pre-mRNA, which undergoes alternative splicing, capping, and polyadenylation before being exported from the nucleus (Bock et al., 2015). PARP1 and PARylation modulate the alternative splicing process in two ways. On the one hand, PARP1 directly affects alternative splicing by binding with the chromatin structure that regulates RNA polymerase elongation (Bock et al., 2015). On the other hand, PARP1 participates in the PARylation of splicing factors to regulate spliceosome protein activity or expression, which indirectly affects alternative splicing (Isabelle et al., 2012; Matveeva et al., 2016). In addition, PARP1-mediated PARylation of poly(A) polymerase, the enzyme catalyzing poly-adenylation, blocks mRNA maturation (Manco et al., 2022). Heterogeneous nuclear-ribonucleoproteins, a subgroup of RNA-binding proteins (RBPs) and PAR-binding proteins, affect mRNA trafficking and splicing by binding to PAR (Gagné et al., 2003; Ji & Tulin, 2013).
PARylation is also involved in RNA regulation during stress. When cells encounter external stimuli and stress, initiation of mRNA translation is reduced or inhibited. mRNAs and the corresponding RBPs aggregate to form membrane-less organelles—stress granules. In the interphase cytoplasm, PAR catalyzes stress granule assembly to recruit RBPs to specific locations, thereby regulating mRNA translation and stability under stress (Anderson & Kedersha, 2008). Studies have found that the RNA decay factor G3BP1, translational suppressor TIA1, and miRNA-binding argonaute proteins (Ago1–4) are modified by PARylation in an unstressed state (Isabelle et al., 2012; Leung et al., 2011). The level of PARylation of these proteins increases during stress, which relieves miRNA-mediated translation inhibition and miRNA-directed mRNA cleavage (Leung et al., 2011).
Recent studies have found cross-talk between PARylation and other protein PTMs such as phosphorylation, acetylation, methylation, and ubiquitination, to fine-tune protein functions and create a complex signaling network (Kassner et al., 2013; Li et al., 2015; Messner et al., 2010; Zhou et al., 2020). For example, PARylation of the target protein may serve as a signal for its subsequent ubiquitylation and induce ubiquitin–proteasome degradation (Li et al., 2015). Ubiquitin E3 ligase binds to PAR through the PAR-binding motif and is activated to promote substrate ubiquitylation (DaRosa et al., 2015). The PAR-dependent E3 ligases identified so far include BRCA1–BARD, RNF146, CHFR, and Iduna, which regulate the abundance of substrate proteins, such as Axin1/2, 3BP2, PTEN, AMOT-family proteins, PARP1, RNF146, and PARP5a/b (Hu et al., 2021; Kang et al., 2011; Kashima et al., 2012; Zhang et al., 2011). However, there are still questions to be answered, such as what factors determine the specificity of PAR-dependent ubiquitylation targets and whether other E3 ligases are also PAR-dependent. In addition, PARylation and other PTMs can be mutually exclusive events, and the presence of one modification can influence the other. Acetylation and PARylation compete for the same lysine. Indeed it has been demonstrated that histone H4 lysine 16 acetylation impairs the PARylation of the histone H4, which is essential for regulating transcription and chromatin remodeling (Messner et al., 2010).Zhou et al. found that enhanced STAT3 PARylation can significantly inhibit its phosphorylation at TRY705 in hepatoma cells (Zhou et al., 2020). Similarly, PARylation of histone H3 inhibits SET7/9-dependent methylation of H3 (Kassner et al., 2013). In conclusion, the crosstalk between PARylation and other PTMs is a dynamic and complex network that regulates various cellular processes. Understanding the interplay between these modifications will provide insights into the regulation of protein function in health and disease.
PARP inhibitor (PARPi)
Mechanism of PARPi action
Two studies in 2005 both observed that BRCA1/2-deficient cells were highly sensitive to PARPis, which resulted in apoptosis of cancer cells (Bryant et al., 2005; Farmer et al., 2005). Based on this phenomenon and the fact that SSB repair predominantly depends on PARP, scientists proposed the synthetic lethality theory (Fig. 2). PARPi binds to catalytic sites in PARP, which “traps” it on nicked DNA and prevents its auto-PARylation. The “trapped” PARP1 then causes replication fork collapse, which potentially upgrades SSBs to the more severe type of damage, DSBs. However, HR, the preferred DSB repair pathway, is defective in BRCA1/2 mutant cells. Consequently, DSBs remain unrepaired, which ultimately leads to cell death. Other models involving synthetic lethality have been proposed to explain the antitumor effect of PARPis. In BRCA1-deficient cells, PARPis derepress the error-prone NHEJ pathway by inhibiting PARylation of Ku70/Ku80; the use of this rapid but low-fidelity repair pathway increases the probability of cell death (Hochegger et al., 2006; Patel et al., 2011). In addition, PARP1-dependent microhomology-mediated end-joining, a compensatory DNA repair pathway, which is more active when HR is deficient, is also inhibited by PARPis, facilitating DSB accumulation (Ceccaldi et al., 2015).
Clinical approval of PARPis
Considerable research has focused on the use of PARPis as single agents to exploit tumor-specific defects (Drew et al., 2016; Gelmon et al., 2011; Litton et al., 2018; Mirza et al., 2016; Mukhopadhyay et al., 2010). To date, the FDA has approved four PARPis, namely olaparib, rucaparib, niraparib, and talazoparib. Olaparib was the first PARPi approved by the US Food and Drug Administration (FDA) (in 2014) for the treatment of relapsed platinum-sensitive ovarian cancer with germline BRCA mutations. The efficacy of this treatment was supported by data from a phase II randomized trial showing that patients with BRCA mutations had a longer median progression-free survival (PFS) with olaparib therapy than with placebo therapy (Ledermann et al., 2014). Subsequent clinical trials supported the efficacy of olaparib as a treatment for BRCA-mutated platinum-sensitive relapsed serous ovarian cancer and BRCA-mutated ovarian cancer after platinum-based therapy, and it was approved as a first-line maintenance therapy in ovarian cancer (Golan et al., 2019; Poveda et al., 2021; Robson et al., 2017).
The second PARPi, rucaparib was approved by the FDA in 2016 for the treatment of patients with advanced ovarian cancer with BRCA mutations who had received two or more previous chemotherapy regimens, combined with an FDA-approved next-generation sequencing-based companion diagnosis (Kristeleit et al., 2017). In 2018, the FDA approved rucaparib as a second-line maintenance therapy for adult patients with recurrent ovarian cancer that were in complete or partial remission prior to being treated with platinum-containing chemotherapy, without the need for a companion diagnosis (Coleman et al., 2017).
Niraparib was the third approved PARPi (in 2017) and the first PARPi used as maintenance therapy for platinum-sensitive ovarian cancer; it was also approved as maintenance therapy for ovarian cancer in 2019 (Mirza et al., 2016; Moore et al., 2019). In 2018, the FDA approved the most recent PARPi—talazoparib—for germline BRCA-mutant HER2-negative locally advanced or metastatic breast cancer (Litton et al., 2018). However, despite the approval of olaparib, rucaparib, and niraparib as maintenance therapies, the FDA is preparing to withdraw marketing approval for their use as third-line or higher treatments because of their potential to increase the risk of death.
In vitro studies have shown that different PARPis vary in cytotoxicity, which is significantly correlated with trapping capacity, despite having an equal ability to inhibit the catalytic activity of PARP (Murai et al., 2014). Thus, the dosage of PARPi used for clinical applications depends on the trapping capacity. For example, talazoparib shows the greatest PARP trapping ability and the highest cytotoxicity among these PARPis, which means that a lower dose is generally selected in the clinic (Lord & Ashworth, 2017).
Unanswered questions about synthetic lethality
While PARPis have resulted in the expected clinical success for certain tumor types, subsequent research and clinical trials have reported unanticipated results. For example, several clinical trials have shown that PARPis not only significantly improve the survival of ovarian cancer patients harboring BRCA mutations but also have high efficacy in patients with wild-type BRCA (Coleman et al., 2019; González-Martín et al., 2019; Ray-Coquard et al., 2019). In addition, in some tumors with a smaller probability of carrying BRCA mutations than ovarian and breast cancer, such as prostate cancer and pancreatic ductal adenocarcinoma, PARPi showed unexpectedly high efficacy, leading to its approval for clinical application (Clarke et al., 2018; Raphael et al., 2017; Robinson et al., 2015). These findings show that the underlying mechanism of synthetic lethality has not been fully elucidated.
Although many models have been proposed, there are still some issues regarding the interaction between PARPis and BRCA mutations. A controversial point is the hypothesis that most cancer-associated BRCA1 mutations are located in the domains involved in the core DNA damage response function because BRCA-deficient tumor cells fail to repair DSBs. However, abundant BRCA1 mutations have instead been found in or near the middle region of the protein (Mallery et al., 2002; Manke et al., 2003; Rebbeck et al., 2015; Solmaz et al., 2020). The mechanism by which PARPis destroy cells with intact DNA DSB repair function must still be addressed. Moreover, PARPis should have similar efficacies for HR-deficient patients with non-BRCA mutations, but this is not the case. It was found that tumors bearing certain non-BRCA HR factor mutants, such as ataxia telangiectasia mutated (ATM), are not as sensitive to PARPis as tumors with BRCA mutants (Rafiei et al., 2020).
At present, BRCA and HR status are important factors affecting the selection of drugs for ovarian and breast cancer patients. However, even if BRCA mutation testing or HR deficiency (HRD) testing is performed according to the guidelines before drug administration, the clinical effect is not fully in line with expectations (Del Campo et al., 2019). Thus, uncovering the molecular mechanism of PARPi in synthetic lethality will guide clinical trials toward outcomes of antitumor efficacy beyond HRD status.
Acquired resistance to PARPis
PARPi treatment often has good efficacy in the initial application, but acquired resistance emerges gradually, which is a serious clinical problem. The following section provides a brief overview of the mechanisms of PARPi resistance, including restoration of HR, stabilization of the replication fork, overexpression of efflux pumps, and inactivation of PARG (Fig. 3).

Mechanisms of PARPi resistance. a, b Restoration of HR. a The frameshift mutation of an HR-related protein produces a nonfunctional truncated protein, while a reversion mutation near the original mutated region restores the reading frame, enabling the expression of a partially functional protein. b When DNA damage is generated, resected DNA ends (in red) initiate HR to repair the DSB while unresected DNA ends (in green) promote the initiation of NHEJ. TP53BP1 is the initiator of NHEJ and is involved in inhibition of end resection. In BRCA1-deficient tumors, deletion of TP53BP1, as well as its downstream proteins RIF1, MAD2L2, and the shieldin complex, leads to restoration of DNA end resection and HR. c Stabilization of the replication fork. In BRCA-deficient cells, nucleases including MRE11 and MUS81 are recruited to the stalled replication fork and mediate its degradation due to the loss of BRCA1/2 protection. Depletion of CHD4 as well as PTIP and EZH2 inhibits the recruitment of nucleases and stabilizes the replication fork, conferring resistance to PARPis. In parallel, fork collapse could be prevented by RADX. d Overexpression of efflux pumps. Overexpression of efflux pumps contributes to decreasing the intracellular concentration of drugs, giving rise to multidrug resistance. e Inactivation of PARG. PARG and PARPi together act as barriers to the formation of PAR chains; thus, PARG downregulation rescues PARylation upon PARPi treatment
Restoration of HR
The restoration of HR is the most well-described mechanism by which BRCA-deficient cells acquire chemotherapeutic resistance. Among the several ways to achieve restoration of HR, the chief one is reversion mutations of BRCA1/2 (Fig. 3a) (Ganesan, 2018). Most mutations in BRCA1/2 are frameshifts, producing non-functional truncated proteins and subsequently resulting in BRCA1/2-dependent HRD (Gorodetska et al., 2019). However, insertions/deletions may reoccur near the original mutated regions, which restores the reading frame and gives rise to partially functional proteins (Barber et al., 2013; Kondrashova et al., 2017; Norquist et al., 2011). Multiple studies have found secondary mutations of BRCA1/2 in PARPi-resistant tumors, resulting in the restoration of HR (Barber et al., 2013; Loehr et al., 2022; Norquist et al., 2011; Patch et al., 2015). This “sneaky” mechanism supports both tumorigenesis and the development of drug resistance. It is unclear whether reversion mutations are induced by DNA damage reagents such as PARPis or are pre-existing and selected by PARPi treatment. Restoration of HR also occurs in BRCA-independent HRD cells and results in the development of resistance to PARPi and platinum; in these cells restoration is caused by reversion mutations in other HR genes such as RAD51C, RAD51D, and PALB2 (Goodall et al., 2017; Kondrashova et al., 2017).
Another mechanism underlying the restoration of HR is the release from DNA end resection inhibition (Fig. 3b). When DNA damage is generated, resected DNA ends initiate HR to repair the DSB while the unresected DNA ends promote the initiation of NHEJ (Ceccaldi et al., 2016). Tumor protein p53 binding protein 1 (TP53BP1) is the initiator of NHEJ, and it is involved in inhibition of end resection. Deletion of TP53BP1, as well as its downstream proteins replication timing regulatory factor 1 (RIF1), mitotic arrest deficient 2 like 2 (MAD2L2), and the shieldin complex, leads to restoration of DNA end resection and HR in BRCA1-deficient mouse models (Bunting et al., 2010). Similarly, it was reported that the CTC1-STN1-TEN1 complex could protect DSBs from end resection to inhibit the initiation of HR (Barazas et al., 2018). In line with the above observations, clinical data indicate that patients with BRCA1-deficient breast cancer acquire PARPi and platinum resistance due to mutations in TP53BP1 (Waks et al., 2020). Of note, the restoration of HR due to the deletion of TP53BP1 and its downstream proteins was exclusively observed in BRCA1-deficient tumors (Dias et al., 2021). Unfortunately, this resistance mechanism is difficult to overcome because of the restoration of DNA repair function, and in this case immunotherapy or anti-angiogenic drugs may be promising options.
Stabilization of the replication fork
Apart from HR, BRCA1/2 is also involved in maintaining replication fork stability (Liptay et al., 2020). In BRCA-deficient cells, nucleases including MRE11 are recruited to stalled replication forks and mediate their degradation due to the loss of BRCA1/2 protection (Fig. 3c) (Liptay et al., 2020). Several proteins have been identified as regulators of replication fork protection in the context of BRCA deficiency. Depletion of CHD4 as well as PAX interacting protein 1 (PTIP) inhibits MRE11 recruitment and stabilizes the replication fork, conferring resistance to PARPis (Guillemette et al., 2015; Ray Chaudhuri et al., 2016). In line with this, clinical survival analysis showed a significant correlation between reduced CHD4 expression and a shorter PFS (Guillemette et al., 2015). MUS81 structure-specific endonuclease subunit (MUS81) is another nuclease participating in the degradation of the replication fork. The deficiency of enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) prevents MUS81 recruitment at the stalled fork and maintains fork stability. A low level of EZH2 is considered to be a predictor of chemotherapy resistance and poor prognosis in cancer patients bearing BRCA2 mutations (Rondinelli et al., 2017). In addition, the single-strand DNA binding protein RADX prevents fork collapse by regulating RAD51. Inactivation of RADX drives chemotherapeutic resistance in BRCA2-deficient cells through fork collapse mediated by over-activated RAD51 (Dungrawala et al., 2017). Notably, the mechanisms of replication fork stabilization stated here are independent of the restoration of HR, which indicates that the replication fork may serve as a target for preventing the emergence of drug resistance in the future (Dungrawala et al., 2017; Ray Chaudhuri et al., 2016).
Overexpression of efflux pumps
Overexpression of efflux pumps contributes to decreasing the intracellular concentration of drugs, giving rise to multidrug resistance (Fig. 3d) (Andrei et al., 2020; Lepeltier et al., 2020). Research in mouse models of BRCA1-deficient breast cancer showed that long-term exposure to olaparib upregulates ABCB1, an ATP-binding cassette transporter family member, and confers resistance to PARPis; this resistance can be overcome by an ABCA1 inhibitor (Jaspers et al., 2015). Similar upregulation was observed in chemo-resistant ovarian cancer patients (Patch et al., 2015). Furthermore, a recent study reported that overexpression of ABCB1 induces olaparib-taxane cross-resistance in advanced prostate cancer, indicating that ABCB1 has the potential to be a target for overcoming common drug resistance (Lombard et al., 2019).
Inactivation of PARG
PARG inactivation is another newly discovered PARPi resistance mechanism, in which PARPis are overcome without HR restoration (Fig. 3e). Loss of PARG occurs frequently in mouse mammary tumors in parallel with PARPi resistance (Gogola et al., 2018). The main mechanism is that PARG and PARPi together act as barriers to the formation of the PAR chain; thus, PARG downregulation rescues PARylation upon PARPi treatment. In vitro studies have shown that treatment with high concentrations of olaparib results in a dramatic but not complete reduction in PAR and that endogenous PARG activity is necessary for effective PAR inhibition by PARPi (Gogola et al., 2018). Clinical cohorts of naive triple-negative breast cancer and ovarian cancer patients receiving PARPi treatment showed that pre-existing PARG-depleted cells could be selected by PARPi treatment (Gogola et al., 2018; Moudry et al., 2016). On the other hand, PARG deficiency confers tumors with high sensitivity to radiation and chemotherapeutics on account of replication catastrophe (Amé et al., 2009; Pillay et al., 2019). Therefore, although PARG inhibition is detrimental to PARPi efficacy, it leads to cell vulnerability, which can be utilized to target drug-resistant tumors.
Strategies to overcome PARPi resistance
Based on the increased likelihood of acquired PARPi resistance, research on PARPi combination therapy is in full swing to further optimize treatment efficacy and extend indications. Initially, the main goal of research on PARPis was to enhance the sensitivity of cancer cells to chemotherapy drugs by utilizing low-dose PARPi as a complementary therapy. Data from a phase I clinical trial showed that PARPis act as chemosensitizers with high cytotoxicity, which may be related to PARPi inhibition of other normal functions of PARP (Calabrese et al., 2004; Ferraris, 2010; Powell et al., 2010). In addition, myelosuppression was observed in patients combined with temozolomide (Plummer et al., 2008). Research on chemotherapy combinations was hampered by the above adverse effects until a phase III clinical trial showed that the planned chemotherapy dose was well tolerated by patients treated with veliparib (a PARPi), which may be attributed to the relatively low toxic effects compared with other PARPi (Coleman et al., 2019). However, whether combined with platinum, alkylating agents, or topoisomerase inhibitors, none of the combined chemotherapy and PARPi treatments are as effective as monotherapy.
Combination of PARPis and biological agents such as antiangiogenic agents is a promising therapy because these treatments do not cause myelosuppression. Antiangiogenic agents can inhibit tumor angiogenesis to form a hypoxic microenvironment, which suppresses tumor growth in parallel with downregulation of BRCA1 to form an HR-deficient phenotype that can be targeted by PARPis. Data from a phase II study showed that olaparib combined with the VEGFR inhibitor cediranib significantly improved the PFS of ovarian patients, especially patients without BRCA mutations compared with olaparib monotherapy (16.5 months versus 5.7 months) (Liu et al., 2014). At the same time, the AVANOVA2 study reported that platinum-sensitive recurrent ovarian cancer patients with wild-type BRCA benefited more from the combination of niraparib and bevacizumab (an anti-VEGFR antibody) than patients with BRCA mutations (Mirza et al., 2019). The significant efficacy of antiangiogenic agents provides the opportunity to expand the indications of PARPis, particularly for HR-proficient patients.
High levels of damaged DNA and erroneously repaired DNA are recognized by the innate immune system, which initiates an antitumor immune response, whereas tumors activate the immune checkpoint of T cells to escape from immune system attacks. Based on this mechanism, PARPi and immune checkpoint inhibitors such as anti-PD-1/PD-L1 have been used in combination for tumor treatment. PARPi increases the incidence of DNA errors and anti-PD-1/PD-L1 simultaneously encourages the antitumor response (Stewart et al., 2018). Ongoing trials of PARPi combined with immune checkpoint inhibitors show good tolerability and promising median PFS regardless of BRCA status, which suggests that patients may derive a greater benefit from the combination than one drug alone (Drew et al., 2019; Konstantinopoulos et al., 2019).
Conclusion and future perspectives
Over the past 60 years since the discovery of PARP1, additional biological functions of PARP1 have continued to be uncovered. In addition to DNA repair, chromosome remodeling, cell death, and other functions described in detail above, PARP1 has recently been found to interact with O6-methylguanine-DNA methyltransferase (MGMT) in both a non-catalytic (DNA independent) and catalytic (DNA dependent) manner to form a new DNA damage-induced PARP1-MGMT protein complex. This complex underpins pathway crosstalk in O6-methylguanine repair in Ewing sarcoma cells and provides a new potential target for cancer treatment (Cropper et al., 2022). In addition, new findings regarding the association of PARP1 with the immune system suggest that PARP1 activity can regulate the tumor microenvironment in non-small cell lung cancer (Juncheng et al., 2022). In the absence of PARP1, T cells exhibit signs of activation, such as increased expression of inducible co-stimulators and decreased expression of PD-1 in cytotoxic T lymphocytes (Juncheng et al., 2022). Beyond that, another study identified PARP1 as an effector of insulin-like growth factor binding protein 7 (IGFBP7), a biomarker used to predict different types of acute kidney injury (J. Yu et al., 2022). IGFBP7 promotes kidney injury and inflammation through a PARP1-dependent mechanism.
The rapid translation of PARPi to clinical use is attributed to the discovery of synthetic lethality, which represents a new paradigm for killing tumors bearing BRCA mutations by exploiting tumor-specific defects to create a vulnerability that causes tumor cell death. In less than a decade, four kinds of PARPis have been approved by the FDA for clinical use in the treatment and maintenance of ovarian cancer, breast cancer, prostate cancer, and pancreatic ductal adenocarcinoma. At present, the selection of a PARPi mostly depends on the assessment of BRCA mutation or HRD status, but the clinical effect is not fully in line with expectations because the anti-tumor mechanism of PARPi has not been fully clarified, and the efficacy remains to be improved. Hence, it is necessary to further explore the mechanisms of PARPis, especially the synthetic lethality with BRCA mutations, and develop biomarkers to better identify the best treatments for patients. In addition, future research should focus on overcoming the targeted drug resistance of these patients. Combined treatment may be a promising direction, as well as a way to increase the value and efficacy of such drugs through synergistic effects.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Adamowicz, M., Hailstone, R., Demin, A. A., Komulainen, E., Hanzlikova, H., Brazina, J., Gautam, A., Wells, S. E., & Caldecott, K. W. (2021). XRCC1 protects transcription from toxic PARP1 activity during DNA base excision repair. Nature Cell Biology, 23(12), 1287–1298. https://doi.org/10.1038/s41556-021-00792-w
Alano, C. C., Garnier, P., Ying, W., Higashi, Y., Kauppinen, T. M., & Swanson, R. A. (2010). NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. The Journal of Neuroscience, 30(8), 2967–2978. https://doi.org/10.1523/JNEUROSCI.5552-09.2010
Alemasova, E. E., & Lavrik, O. I. (2019). Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Research, 47(8), 3811–3827. https://doi.org/10.1093/nar/gkz120
Alvarez-Gonzalez, R., & Mendoza-Alvarez, H. (1995). Dissection of ADP-ribose polymer synthesis into individual steps of initiation, elongation, and branching. Biochimie, 77(6), 403–407. https://doi.org/10.1016/0300-9084(96)88153-3
Amé, J.-C., Fouquerel, E., Gauthier, L. R., Biard, D., Boussin, F. D., Dantzer, F., de Murcia, G., & Schreiber, V. (2009). Radiation-induced mitotic catastrophe in PARG-deficient cells. Journal of Cell Science, 122(Pt 12), 1990–2002. https://doi.org/10.1242/jcs.039115
Amé, J.-C., Rolli, V., Schreiber, V., Niedergang, C., Apiou, F., Decker, P., Muller, S., Höger, T., Murcia, J. M., & de Murcia, G. (1999). PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase*. Journal of Biological Chemistry, 274(25), 17860–17868. https://doi.org/10.1074/jbc.274.25.17860
Anderson, P., & Kedersha, N. (2008). Stress granules: The Tao of RNA triage. Trends in Biochemical Sciences, 33(3), 141–150. https://doi.org/10.1016/j.tibs.2007.12.003
Andrei, L., Kasas, S., Ochoa Garrido, I., Stanković, T., Suárez Korsnes, M., Vaclavikova, R., Assaraf, Y. G., & Pešić, M. (2020). Advanced technological tools to study multidrug resistance in cancer. Drug Resistance Updates, 48, 100658. https://doi.org/10.1016/j.drup.2019.100658
Barazas, M., Annunziato, S., Pettitt, S. J., de Krijger, I., Ghezraoui, H., Roobol, S. J., Lutz, C., Frankum, J., Song, F. F., Brough, R., Evers, B., Gogola, E., Bhin, J., van de Ven, M., van Gent, D. C., Jacobs, J. J. L., Chapman, R., Lord, C. J., Jonkers, J., & Rottenberg, S. (2018). The CST complex mediates end protection at double-strand breaks and promotes PARP inhibitor sensitivity in BRCA1-deficient cells. Cell Reports, 23(7), 2107–2118. https://doi.org/10.1016/j.celrep.2018.04.046
Barber, L. J., Sandhu, S., Chen, L., Campbell, J., Kozarewa, I., Fenwick, K., Assiotis, I., Rodrigues, D. N., Reis Filho, J. S., Moreno, V., Mateo, J., Molife, L. R., De Bono, J., Kaye, S., Lord, C. J., & Ashworth, A. (2013). Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. The Journal of Pathology, 229(3), 422–429. https://doi.org/10.1002/path.4140
Beck, C., Boehler, C., Guirouilh Barbat, J., Bonnet, M.-E., Illuzzi, G., Ronde, P., Gauthier, L. R., Magroun, N., Rajendran, A., Lopez, B. S., Scully, R., Boussin, F. D., Schreiber, V., & Dantzer, F. (2014). PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Research, 42(9), 5616–5632. https://doi.org/10.1093/nar/gku174
Bilokapic, S., Suskiewicz, M. J., Ahel, I., & Halic, M. (2020). Bridging of DNA breaks activates PARP2–HPF1 to modify chromatin. Nature, 585(7826), 609–613. https://doi.org/10.1038/s41586-020-2725-7
Boamah, E. K., Kotova, E., Garabedian, M., Jarnik, M., & Tulin, A. V. (2012). Poly(ADP-Ribose) POLYMERASE 1 (PARP-1) regulates ribosomal biogenesis in drosophila nucleoli. PLoS Genetics, 8(1), e1002442. https://doi.org/10.1371/journal.pgen.1002442
Bock, F. J., Todorova, T. T., & Chang, P. (2015). RNA Regulation by poly(ADP-Ribose) polymerases. Molecular Cell, 58(6), 959–969. https://doi.org/10.1016/j.molcel.2015.01.037
Boehler, C., Gauthier, L. R., Mortusewicz, O., Biard, D. S., Saliou, J.-M., Bresson, A., Sanglier-Cianferani, S., Smith, S., Schreiber, V., Boussin, F., & Dantzer, F. (2011). Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proceedings of the National Academy of Sciences, 108(7), 2783–2788. https://doi.org/10.1073/pnas.1016574108
Brochu, G., Duchaine, C., Thibeault, L., Lagueux, J., Shah, G. M., & Poirier, G. G. (1994). Mode of action of poly(ADP-ribose) glycohydrolase. Biochimica Et Biophysica Acta, 1219(2), 342–350. https://doi.org/10.1016/0167-4781(94)90058-2
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., Kyle, S., Meuth, M., Curtin, N. J., & Helleday, T. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature, 434(7035), 913–917. https://doi.org/10.1038/nature03443
Bunting, S. F., Callén, E., Wong, N., Chen, H.-T., Polato, F., Gunn, A., Bothmer, A., Feldhahn, N., Fernandez-Capetillo, O., Cao, L., Xu, X., Deng, C.-X., Finkel, T., Nussenzweig, M., Stark, J. M., & Nussenzweig, A. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection OF DNA breaks. Cell, 141(2), 243–254. https://doi.org/10.1016/j.cell.2010.03.012
Calabrese, C. R., Almassy, R., Barton, S., Batey, M. A., Calvert, A. H., Canan-Koch, S., Durkacz, B. W., Hostomsky, Z., Kumpf, R. A., Kyle, S., Li, J., Maegley, K., Newell, D. R., Notarianni, E., Stratford, I. J., Skalitzky, D., Thomas, H. D., Wang, L.-Z., Webber, S. E., & Curtin, N. J. (2004). Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. Journal of the National Cancer Institute, 96(1), 56–67. https://doi.org/10.1093/jnci/djh005
Caldecott, K. W., Aoufouchi, S., Johnson, P., & Shall, S. (1996). XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular “nick-sensor” in vitro. Nucleic Acids Research, 24(22), 4387–4394. https://doi.org/10.1093/nar/24.22.4387
Caldecott, K. W., McKeown, C. K., Tucker, J. D., Ljungquist, S., & Thompson, L. H. (1994). An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Molecular and Cellular Biology, 14(1), 68–76. https://doi.org/10.1128/mcb.14.1.68-76.1994
Ceccaldi, R., Liu, J. C., Amunugama, R., Hajdu, I., Primack, B., Petalcorin, M. I. R., O’Connor, K. W., Konstantinopoulos, P. A., Elledge, S. J., Boulton, S. J., Yusufzai, T., & D’Andrea, A. D. (2015). Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature, 518(7538), 258–262. https://doi.org/10.1038/nature14184
Ceccaldi, R., Rondinelli, B., & D’Andrea, A. D. (2016). Repair pathway choices and consequences at the double-strand break. Trends in Cell Biology, 26(1), 52–64. https://doi.org/10.1016/j.tcb.2015.07.009
Chambon, P., Weill, J. D., & Mandel, P. (1963). Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochemical and Biophysical Research Communications, 11, 39–43. https://doi.org/10.1016/0006-291x(63)90024-x
Chen, C.-C., Feng, W., Lim, P. X., Kass, E. M., & Jasin, M. (2018a). Homology-directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Annual Review of Cancer Biology, 2, 313–336. https://doi.org/10.1146/annurev-cancerbio-030617-050502
Chen, Q., Kassab, M. A., Dantzer, F., & Yu, X. (2018b). PARP2 mediates branched poly ADP-ribosylation in response to DNA damage. Nature Communications. https://doi.org/10.1038/s41467-018-05588-5
Chiu, L.-Y., Ho, F.-M., Shiah, S.-G., Chang, Y., & Lin, W.-W. (2011). Oxidative stress initiates DNA damager MNNG-induced poly(ADP-ribose)polymerase-1-dependent parthanatos cell death. Biochemical Pharmacology, 81(3), 459–470. https://doi.org/10.1016/j.bcp.2010.10.016
Clarke, N., Wiechno, P., Alekseev, B., Sala, N., Jones, R., Kocak, I., Chiuri, V. E., Jassem, J., Fléchon, A., Redfern, C., Goessl, C., Burgents, J., Kozarski, R., Hodgson, D., Learoyd, M., & Saad, F. (2018). Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: A randomised, double-blind, placebo-controlled, phase 2 trial. The Lancet Oncology, 19(7), 975–986. https://doi.org/10.1016/S1470-2045(18)30365-6
Coleman, R. L., Fleming, G. F., Brady, M. F., Swisher, E. M., Steffensen, K. D., Friedlander, M., Okamoto, A., Moore, K. N., Efrat Ben-Baruch, N., Werner, T. L., Cloven, N. G., Oaknin, A., DiSilvestro, P. A., Morgan, M. A., Nam, J.-H., Leath, C. A., Nicum, S., Hagemann, A. R., Littell, R. D., & Bookman, M. A. (2019). Veliparib with first-line chemotherapy and as Maintenance therapy in ovarian cancer. New England Journal of Medicine, 381(25), 2403–2415. https://doi.org/10.1056/NEJMoa1909707
Coleman, R. L., Oza, A. M., Lorusso, D., Aghajanian, C., Oaknin, A., Dean, A., Colombo, N., Weberpals, J. I., Clamp, A., Scambia, G., Leary, A., Holloway, R. W., Gancedo, M. A., Fong, P. C., Goh, J. C., O’Malley, D. M., Armstrong, D. K., Garcia-Donas, J., Swisher, E. M., ARIEL3 investigators. (2017). Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (london, England), 390(10106), 1949–1961. https://doi.org/10.1016/S0140-6736(17)32440-6
Cropper, J. D., Alimbetov, D. S., Brown, K. T. G., Likhotvorik, R. I., Robles, A. J., Guerra, J. T., He, B., Chen, Y., Kwon, Y., & Kurmasheva, R. T. (2022). PARP1-MGMT complex underpins pathway crosstalk in O6-methylguanine repair. Journal of Hematology & Oncology, 15(1), 146. https://doi.org/10.1186/s13045-022-01367-4
DaRosa, P. A., Wang, Z., Jiang, X., Pruneda, J. N., Cong, F., Klevit, R. E., & Xu, W. (2015). Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature, 517(7533), 223–226. https://doi.org/10.1038/nature13826
Daugas, E., Susin, S. A., Zamzami, N., Ferri, K. F., Irinopoulou, T., Larochette, N., Prévost, M. C., Leber, B., Andrews, D., Penninger, J., & Kroemer, G. (2000). Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB Journal, 14(5), 729–739.
Day, T. A., Layer, J. V., Cleary, J. P., Guha, S., Stevenson, K. E., Tivey, T., Kim, S., Schinzel, A. C., Izzo, F., Doench, J., Root, D. E., Hahn, W. C., Price, B. D., & Weinstock, D. M. (2017). PARP3 is a promoter of chromosomal rearrangements and limits G4 DNA. Nature Communications. https://doi.org/10.1038/ncomms15110
Del Campo, J. M., Matulonis, U. A., Malander, S., Provencher, D., Mahner, S., Follana, P., Waters, J., Berek, J. S., Woie, K., Oza, A. M., Canzler, U., Gil-Martin, M., Lesoin, A., Monk, B. J., Lund, B., Gilbert, L., Wenham, R. M., Benigno, B., Arora, S., & Mirza, M. R. (2019). Niraparib maintenance therapy in patients with recurrent ovarian cancer after a partial response to the last platinum-based chemotherapy in the ENGOT-OV16/NOVA trial. Journal of Clinical Oncology, 37(32), 2968–2973. https://doi.org/10.1200/JCO.18.02238
Demple, B., & Harrison, L. (1994). Repair of oxidative damage to DNA: Enzymology and biology. Annual Review of Biochemistry, 63, 915–948. https://doi.org/10.1146/annurev.bi.63.070194.004411
Dias, M. P., Moser, S. C., Ganesan, S., & Jonkers, J. (2021). Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nature Reviews Clinical Oncology, 18(12), 773–791. https://doi.org/10.1038/s41571-021-00532-x
Drew, Y., Kaufman, B., Banerjee, S., Lortholary, A., Hong, S. H., Park, Y. H., Zimmermann, S., Roxburgh, P., Ferguson, M., Alvarez, R. H., Domchek, S., Gresty, C., Angell, H. K., Ros, V. R., Meyer, K., Lanasa, M., Herbolsheimer, P., & de Jonge, M. (2019). 1190PD - phase II study of olaparib + durvalumab (MEDIOLA): Updated results in germline BRCA-mutated platinum-sensitive relapsed (PSR) ovarian cancer (OC). Annals of Oncology, 30, v485–v486. https://doi.org/10.1093/annonc/mdz253.016
Drew, Y., Ledermann, J., Hall, G., Rea, D., Glasspool, R., Highley, M., Jayson, G., Sludden, J., Murray, J., Jamieson, D., Halford, S., Acton, G., Backholer, Z., Mangano, R., Boddy, A., Curtin, N., & Plummer, R. (2016). Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. British Journal of Cancer, 114(12), e21. https://doi.org/10.1038/bjc.2016.133
Dungrawala, H., Bhat, K. P., Le Meur, R., Chazin, W. J., Ding, X., Sharan, S. K., Wessel, S. R., Sathe, A. A., Zhao, R., & Cortez, D. (2017). RADX promotes genome stability and modulates Chemosensitivity by regulating RAD51 at replication forks. Molecular Cell, 67(3), 374-386.e5. https://doi.org/10.1016/j.molcel.2017.06.023
Eustermann, S., Videler, H., Yang, J.-C., Cole, P. T., Gruszka, D., Veprintsev, D., & Neuhaus, D. (2011). The DNA-binding domain of human PARP-1 interacts with DNA single-strand breaks as a monomer through its second zinc finger. Journal of Molecular Biology, 407(1), 149–170. https://doi.org/10.1016/j.jmb.2011.01.034
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N. J., Johnson, D. A., Richardson, T. B., Santarosa, M., Dillon, K. J., Hickson, I., Knights, C., Martin, N. M. B., Jackson, S. P., Smith, G. C. M., & Ashworth, A. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature, 434(7035), 917–921. https://doi.org/10.1038/nature03445
Ferraris, D. V. (2010). Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. Journal of Medicinal Chemistry, 53(12), 4561–4584. https://doi.org/10.1021/jm100012m
Gagné, J.-P., Hunter, J. M., Labrecque, B., Chabot, B., & Poirier, G. G. (2003). A proteomic approach to the identification of heterogeneous nuclear ribonucleoproteins as a new family of poly(ADP-ribose)-binding proteins. The Biochemical Journal, 371(Pt 2), 331–340. https://doi.org/10.1042/BJ20021675
Gagné, J.-P., Isabelle, M., Lo, K. S., Bourassa, S., Hendzel, M. J., Dawson, V. L., Dawson, T. M., & Poirier, G. G. (2008). Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Research, 36(22), 6959–6976. https://doi.org/10.1093/nar/gkn771
Ganesan, S. (2018). Tumor suppressor tolerance: Reversion mutations in BRCA1 and BRCA2 and resistance to PARP inhibitors and platinum. JCO Precision Oncology, 2, 1–4. https://doi.org/10.1200/PO.18.00001
Gelmon, K. A., Tischkowitz, M., Mackay, H., Swenerton, K., Robidoux, A., Tonkin, K., Hirte, H., Huntsman, D., Clemons, M., Gilks, B., Yerushalmi, R., Macpherson, E., Carmichael, J., & Oza, A. (2011). Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. The Lancet Oncology, 12(9), 852–861. https://doi.org/10.1016/S1470-2045(11)70214-5
Gibbs-Seymour, I., Fontana, P., Rack, J. G. M., & Ahel, I. (2016). HPF1/C4orf27 Is a PARP-1-interacting protein that regulates PARP-1 ADP-ribosylation activity. Molecular Cell, 62(3), 432–442. https://doi.org/10.1016/j.molcel.2016.03.008
Gibson, B. A., & Kraus, W. L. (2012). New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nature Reviews Molecular Cell Biology, 13(7), 411–424. https://doi.org/10.1038/nrm3376
Gogola, E., Duarte, A. A., de Ruiter, J. R., Wiegant, W. W., Schmid, J. A., de Bruijn, R., James, D. I., Guerrero Llobet, S., Vis, D. J., Annunziato, S., van den Broek, B., Barazas, M., Kersbergen, A., van de Ven, M., Tarsounas, M., Ogilvie, D. J., van Vugt, M., Wessels, L. F. A., Bartkova, J., & Rottenberg, S. (2018). Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell, 33(6), 1078-1093.e12. https://doi.org/10.1016/j.ccell.2018.05.008
Golan, T., Hammel, P., Reni, M., Van Cutsem, E., Macarulla, T., Hall, M. J., Park, J.-O., Hochhauser, D., Arnold, D., Oh, D.-Y., Reinacher-Schick, A., Tortora, G., Algül, H., O’Reilly, E. M., McGuinness, D., Cui, K. Y., Schlienger, K., Locker, G. Y., & Kindler, H. L. (2019). Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. The New England Journal of Medicine, 381(4), 317–327. https://doi.org/10.1056/NEJMoa1903387
González-Martín, A., Pothuri, B., Vergote, I., DePont Christensen, R., Graybill, W., Mirza, M. R., McCormick, C., Lorusso, D., Hoskins, P., Freyer, G., Baumann, K., Jardon, K., Redondo, A., Moore, R. G., Vulsteke, C., O’Cearbhaill, R. E., Lund, B., Backes, F., & Barretina-Ginesta, P. (2019). Niraparib in patients with newly diagnosed advanced ovarian cancer. The New England Journal of Medicine, 381(25), 2391–2402. https://doi.org/10.1056/NEJMoa1910962
Goodall, J., Mateo, J., Yuan, W., Mossop, H., Porta, N., Miranda, S., Perez-Lopez, R., Dolling, D., Robinson, D. R., Sandhu, S., Fowler, G., Ebbs, B., Flohr, P., Seed, G., Rodrigues, D. N., Boysen, G., Bertan, C., Atkin, M., Clarke, M., TOPARP-A investigators. (2017). Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discovery, 7(9), 1006–1017. https://doi.org/10.1158/2159-8290.CD-17-0261
Gorodetska, I., Kozeretska, I., & Dubrovska, A. (2019). BRCA genes: The role in genome stability, cancer stemness and therapy resistance. Journal of Cancer, 10(9), 2109–2127. https://doi.org/10.7150/jca.30410
Guillemette, S., Serra, R. W., Peng, M., Hayes, J. A., Konstantinopoulos, P. A., Green, M. R., & Cantor, S. B. (2015). Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes & Development, 29(5), 489–494. https://doi.org/10.1101/gad.256214.114
Haince, J.-F., McDonald, D., Rodrigue, A., Déry, U., Masson, J.-Y., Hendzel, M. J., & Poirier, G. G. (2008). PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. The Journal of Biological Chemistry, 283(2), 1197–1208. https://doi.org/10.1074/jbc.M706734200
Hanzlikova, H., Gittens, W., Krejcikova, K., Zeng, Z., & Caldecott, K. W. (2017). Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Research, 45(5), 2546–2557. https://doi.org/10.1093/nar/gkw1246
Hochegger, H., Dejsuphong, D., Fukushima, T., Morrison, C., Sonoda, E., Schreiber, V., Zhao, G. Y., Saberi, A., Masutani, M., Adachi, N., Koyama, H., de Murcia, G., & Takeda, S. (2006). Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. The EMBO Journal, 25(6), 1305–1314. https://doi.org/10.1038/sj.emboj.7601015
Hu, Q., Botuyan, M. V., Zhao, D., Cui, G., Mer, E., & Mer, G. (2021). Mechanisms of BRCA1–BARD1 nucleosome recognition and ubiquitylation. Nature. https://doi.org/10.1038/s41586-021-03716-8
Huang, D., & Kraus, W. L. (2022). The expanding universe of PARP1-mediated molecular and therapeutic mechanisms. Molecular Cell, 82(12), 2315–2334. https://doi.org/10.1016/j.molcel.2022.02.021
Isabelle, M., Gagné, J.-P., Gallouzi, I.-E., & Poirier, G. G. (2012). Quantitative proteomics and dynamic imaging reveal that G3BP-mediated stress granule assembly is poly(ADP-ribose)-dependent following exposure to MNNG-induced DNA alkylation. Journal of Cell Science, 125(Pt 19), 4555–4566. https://doi.org/10.1242/jcs.106963
Jankevicius, G., Hassler, M., Golia, B., Rybin, V., Zacharias, M., Timinszky, G., & Ladurner, A. G. (2013). A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nature Structural & Molecular Biology, 20(4), 508–514. https://doi.org/10.1038/nsmb.2523
Jaspers, J. E., Sol, W., Kersbergen, A., Schlicker, A., Guyader, C., Xu, G., Wessels, L., Borst, P., Jonkers, J., & Rottenberg, S. (2015). BRCA2-deficient sarcomatoid mammary tumors exhibit multidrug resistance. Cancer Research, 75(4), 732–741. https://doi.org/10.1158/0008-5472.CAN-14-0839
Ji, Y., & Tulin, A. V. (2013). Post-transcriptional regulation by poly(ADP-ribosyl)ation of the RNA-binding proteins. International Journal of Molecular Sciences, 14(8), 16168–16183. https://doi.org/10.3390/ijms140816168
Juncheng, P., Joseph, A., Lafarge, A., Martins, I., Obrist, F., Pol, J., Saavedra, E., Li, S., Sauvat, A., Cerrato, G., Lévesque, S., Leduc, M., Kepp, O., Durand, S., Aprahamian, F., Nirmalathansan, N., Michels, J., Kroemer, G., & Castedo, M. (2022). Cancer cell-autonomous overactivation of PARP1 compromises immunosurveillance in non-small cell lung cancer. Journal for Immunotherapy of Cancer, 10(6), e004280. https://doi.org/10.1136/jitc-2021-004280
Kang, H. C., Lee, Y.-I., Shin, J.-H., Andrabi, S. A., Chi, Z., Gagné, J.-P., Lee, Y., Ko, H. S., Lee, B. D., Poirier, G. G., Dawson, V. L., & Dawson, T. M. (2011). Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proceedings of the National Academy of Sciences of the United States of America, 108(34), 14103–14108. https://doi.org/10.1073/pnas.1108799108
Kashima, L., Idogawa, M., Mita, H., Shitashige, M., Yamada, T., Ogi, K., Suzuki, H., Toyota, M., Ariga, H., Sasaki, Y., & Tokino, T. (2012). CHFR protein regulates mitotic checkpoint by targeting PARP-1 protein for ubiquitination and degradation. The Journal of Biological Chemistry, 287(16), 12975–12984. https://doi.org/10.1074/jbc.M111.321828
Kassner, I., Barandun, M., Fey, M., Rosenthal, F., & Hottiger, M. O. (2013). Crosstalk between SET7/9-dependent methylation and ARTD1-mediated ADP-ribosylation of histone H1.4. Epigenetics & Chromatin, 6(1), 1. https://doi.org/10.1186/1756-8935-6-1
Kondrashova, O., Nguyen, M., Shield-Artin, K., Tinker, A. V., Teng, N. N. H., Harrell, M. I., Kuiper, M. J., Ho, G.-Y., Barker, H., Jasin, M., Prakash, R., Kass, E. M., Sullivan, M. R., Brunette, G. J., Bernstein, K. A., Coleman, R. L., Floquet, A., Friedlander, M., Kichenadasse, G., AOCS Study Group. (2017). Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discovery, 7(9), 984–998. https://doi.org/10.1158/2159-8290.CD-17-0419
Konstantinopoulos, P. A., Waggoner, S., Vidal, G. A., Mita, M., Moroney, J. W., Holloway, R., Van Le, L., Sachdev, J. C., Chapman-Davis, E., Colon-Otero, G., Penson, R. T., Matulonis, U. A., Kim, Y. B., Moore, K. N., Swisher, E. M., Färkkilä, A., D’Andrea, A., Stringer-Reasor, E., Wang, J., & Munster, P. (2019). Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncology, 5(8), 1141–1149. https://doi.org/10.1001/jamaoncol.2019.1048
Krishnakumar, R., Gamble, M. J., Frizzell, K. M., Berrocal, J. G., Kininis, M., & Kraus, W. L. (2008). Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science, 319(5864), 819–821. https://doi.org/10.1126/science.1149250
Kristeleit, R., Shapiro, G. I., Burris, H. A., Oza, A. M., LoRusso, P., Patel, M. R., Domchek, S. M., Balmaña, J., Drew, Y., Chen, L.-M., Safra, T., Montes, A., Giordano, H., Maloney, L., Goble, S., Isaacson, J., Xiao, J., Borrow, J., Rolfe, L., & Shapira-Frommer, R. (2017). A phase I-II study of the oral PARP inhibitor rucaparib in patients with germline BRCA1/2-mutated ovarian carcinoma or other solid tumors. Clinical Cancer Research, 23(15), 4095–4106. https://doi.org/10.1158/1078-0432.CCR-16-2796
Langelier, M.-F., Planck, J. L., Roy, S., & Pascal, J. M. (2011). Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: Structural and functional insights into DNA-dependent PARP-1 activity. The Journal of Biological Chemistry, 286(12), 10690–10701. https://doi.org/10.1074/jbc.M110.202507
Langelier, M.-F., Planck, J. L., Roy, S., & Pascal, J. M. (2012). Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science, 336(6082), 728–732. https://doi.org/10.1126/science.1216338
Langelier, M.-F., Ruhl, D. D., Planck, J. L., Kraus, W. L., & Pascal, J. M. (2010). The Zn3 domain of human poly(ADP-ribose) polymerase-1 (PARP-1) functions in both DNA-dependent poly(ADP-ribose) synthesis activity and chromatin compaction. The Journal of Biological Chemistry, 285(24), 18877–18887. https://doi.org/10.1074/jbc.M110.105668
Ledermann, J., Harter, P., Gourley, C., Friedlander, M., Vergote, I., Rustin, G., Scott, C. L., Meier, W., Shapira-Frommer, R., Safra, T., Matei, D., Fielding, A., Spencer, S., Dougherty, B., Orr, M., Hodgson, D., Barrett, J. C., & Matulonis, U. (2014). Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. The Lancet Oncology, 15(8), 852–861. https://doi.org/10.1016/S1470-2045(14)70228-1
Lepeltier, E., Rijo, P., Rizzolio, F., Popovtzer, R., Petrikaite, V., Assaraf, Y. G., & Passirani, C. (2020). Nanomedicine to target multidrug resistant tumors. Drug Resistance Updates, 52, 100704. https://doi.org/10.1016/j.drup.2020.100704
Leung, A. K. L., Vyas, S., Rood, J. E., Bhutkar, A., Sharp, P. A., & Chang, P. (2011). Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Molecular Cell, 42(4), 489–499. https://doi.org/10.1016/j.molcel.2011.04.015
Li, N., Zhang, Y., Han, X., Liang, K., Wang, J., Feng, L., Wang, W., Songyang, Z., Lin, C., Yang, L., Yu, Y., & Chen, J. (2015). Poly-ADP ribosylation of PTEN by tankyrases promotes PTEN degradation and tumor growth. Genes & Development, 29(2), 157–170. https://doi.org/10.1101/gad.251785.114
Liptay, M., Barbosa, J. S., & Rottenberg, S. (2020). Replication fork remodeling and therapy escape in DNA damage response-deficient cancers. Frontiers in Oncology, 10, 670. https://doi.org/10.3389/fonc.2020.00670
Litton, J. K., Rugo, H. S., Ettl, J., Hurvitz, S. A., Gonçalves, A., Lee, K.-H., Fehrenbacher, L., Yerushalmi, R., Mina, L. A., Martin, M., Roché, H., Im, Y.-H., Quek, R. G. W., Markova, D., Tudor, I. C., Hannah, A. L., Eiermann, W., & Blum, J. L. (2018). Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. The New England Journal of Medicine, 379(8), 753–763. https://doi.org/10.1056/NEJMoa1802905
Liu, J. F., Barry, W. T., Birrer, M., Lee, J.-M., Buckanovich, R. J., Fleming, G. F., Rimel, B., Buss, M. K., Nattam, S., Hurteau, J., Luo, W., Quy, P., Whalen, C., Obermayer, L., Lee, H., Winer, E. P., Kohn, E. C., Ivy, S. P., & Matulonis, U. A. (2014). Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. The Lancet Oncology, 15(11), 1207–1214. https://doi.org/10.1016/S1470-2045(14)70391-2
Loehr, A., Hussain, A., Patnaik, A., Bryce, A. H., Castellano, D., Font, A., Shapiro, J., Zhang, J., Sautois, B., Vogelzang, N. J., Chatta, G., Courtney, K., Harzstark, A., Ricci, F., Despain, D., Watkins, S., King, C., Nguyen, M., Simmons, A. D., & Abida, W. (2022). Emergence of BRCA reversion mutations in patients with metastatic castration-resistant prostate cancer after treatment with rucaparib. European Urology, S0302–2838(22), 02639–02642. https://doi.org/10.1016/j.eururo.2022.09.010
Lombard, A. P., Liu, C., Armstrong, C. M., D’Abronzo, L. S., Lou, W., Chen, H., Dall’Era, M., Ghosh, P. M., Evans, C. P., & Gao, A. C. (2019). Overexpressed ABCB1 induces olaparib-taxane cross-resistance in advanced prostate cancer. Translational Oncology, 12(7), 871–878. https://doi.org/10.1016/j.tranon.2019.04.007
Lord, C. J., & Ashworth, A. (2017). PARP inhibitors: Synthetic lethality in the clinic. Science, 355(6330), 1152–1158. https://doi.org/10.1126/science.aam7344
Mallery, D. L., Vandenberg, C. J., & Hiom, K. (2002). Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. The EMBO Journal, 21(24), 6755–6762. https://doi.org/10.1093/emboj/cdf691
Manco, G., Lacerra, G., Porzio, E., & Catara, G. (2022). ADP-ribosylation post-translational modification: An overview with a focus on RNA biology and new pharmacological perspectives. Biomolecules, 12(3), 443. https://doi.org/10.3390/biom12030443
Manke, I. A., Lowery, D. M., Nguyen, A., & Yaffe, M. B. (2003). BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science, 302(5645), 636–639. https://doi.org/10.1126/science.1088877
Maruta, H., Okita, N., Takasawa, R., Uchiumi, F., Hatano, T., & Tanuma, S. (2007). The involvement of ATP produced via (ADP-Ribose)n in the maintenance of DNA replication apparatus during DNA repair. Biological & Pharmaceutical Bulletin, 30(3), 447–450. https://doi.org/10.1248/bpb.30.447
Mashimo, M., Onishi, M., Uno, A., Tanimichi, A., Nobeyama, A., Mori, M., Yamada, S., Negi, S., Bu, X., Kato, J., Moss, J., Sanada, N., Kizu, R., & Fujii, T. (2021). The 89-kDa PARP1 cleavage fragment serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis. Journal of Biological Chemistry. https://doi.org/10.1074/jbc.RA120.014479
Matveeva, E., Maiorano, J., Zhang, Q., Eteleeb, A. M., Convertini, P., Chen, J., Infantino, V., Stamm, S., Wang, J., Rouchka, E. C., & Fondufe-Mittendorf, Y. N. (2016). Involvement of PARP1 in the regulation of alternative splicing. Cell Discovery, 2, 15046. https://doi.org/10.1038/celldisc.2015.46
Messner, S., Altmeyer, M., Zhao, H., Pozivil, A., Roschitzki, B., Gehrig, P., Rutishauser, D., Huang, D., Caflisch, A., & Hottiger, M. O. (2010). PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Research, 38(19), 6350–6362. https://doi.org/10.1093/nar/gkq463
Mirza, M. R., Åvall Lundqvist, E., Birrer, M. J., dePont Christensen, R., Nyvang, G.-B., Malander, S., Anttila, M., Werner, T. L., Lund, B., Lindahl, G., Hietanen, S., Peen, U., Dimoula, M., Roed, H., Ør Knudsen, A., Staff, S., Krog Vistisen, A., Bjørge, L., Mäenpää, J. U., AVANOVA investigators. (2019). Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): A randomised, phase 2, superiority trial. The Lancet. Oncology, 20(10), 1409–1419. https://doi.org/10.1016/S1470-2045(19)30515-7
Mirza, M. R., Monk, B. J., Herrstedt, J., Oza, A. M., Mahner, S., Redondo, A., Fabbro, M., Ledermann, J. A., Lorusso, D., Vergote, I., Ben-Baruch, N. E., Marth, C., Mądry, R., Christensen, R. D., Berek, J. S., Dørum, A., Tinker, A. V., du Bois, A., González-Martín, A., ENGOT-OV16/NOVA Investigators. (2016). Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. The New England Journal of Medicine, 375(22), 2154–2164. https://doi.org/10.1056/NEJMoa1611310
Moore, K. N., Secord, A. A., Geller, M. A., Miller, D. S., Cloven, N., Fleming, G. F., Wahner Hendrickson, A. E., Azodi, M., DiSilvestro, P., Oza, A. M., Cristea, M., Berek, J. S., Chan, J. K., Rimel, B. J., Matei, D. E., Li, Y., Sun, K., Luptakova, K., Matulonis, U. A., & Monk, B. J. (2019). Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. The Lancet Oncology, 20(5), 636–648. https://doi.org/10.1016/S1470-2045(19)30029-4
Moudry, P., Watanabe, K., Wolanin, K. M., Bartkova, J., Wassing, I. E., Watanabe, S., Strauss, R., Troelsgaard Pedersen, R., Oestergaard, V. H., Lisby, M., Andújar-Sánchez, M., Maya-Mendoza, A., Esashi, F., Lukas, J., & Bartek, J. (2016). TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. The Journal of Cell Biology, 212(3), 281–288. https://doi.org/10.1083/jcb.201507042
Mukhopadhyay, A., Elattar, A., Cerbinskaite, A., Wilkinson, S. J., Drew, Y., Kyle, S., Los, G., Hostomsky, Z., Edmondson, R. J., & Curtin, N. J. (2010). Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clinical Cancer Research, 16(8), 2344–2351. https://doi.org/10.1158/1078-0432.CCR-09-2758
Murai, J., Huang, S.-Y.N., Renaud, A., Zhang, Y., Ji, J., Takeda, S., Morris, J., Teicher, B., Doroshow, J. H., & Pommier, Y. (2014). Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Molecular Cancer Therapeutics, 13(2), 433–443. https://doi.org/10.1158/1535-7163.MCT-13-0803
Niere, M., Mashimo, M., Agledal, L., Dölle, C., Kasamatsu, A., Kato, J., Moss, J., & Ziegler, M. (2012). ADP-ribosylhydrolase 3 (ARH3), not poly(ADP-ribose) glycohydrolase (PARG) isoforms, Is responsible for degradation of mitochondrial matrix-associated poly(ADP-ribose)*. Journal of Biological Chemistry, 287(20), 16088–16102. https://doi.org/10.1074/jbc.M112.349183
Norquist, B., Wurz, K. A., Pennil, C. C., Garcia, R., Gross, J., Sakai, W., Karlan, B. Y., Taniguchi, T., & Swisher, E. M. (2011). Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. Journal of Clinical Oncology, 29(22), 3008–3015. https://doi.org/10.1200/JCO.2010.34.2980
Oka, S., Kato, J., & Moss, J. (2006). Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. The Journal of Biological Chemistry, 281(2), 705–713. https://doi.org/10.1074/jbc.M510290200
Palazzo, L., Leidecker, O., Prokhorova, E., Dauben, H., Matic, I., & Ahel, I. (2018). Serine is the major residue for ADP-ribosylation upon DNA damage. eLife, 7, e34334. https://doi.org/10.7554/eLife.34334
Park, H., Kam, T.-I., Dawson, T. M., & Dawson, V. L. (2020). Poly (ADP-ribose) (PAR)-dependent cell death in neurodegenerative diseases. International Review of Cell and Molecular Biology, 353, 1–29. https://doi.org/10.1016/bs.ircmb.2019.12.009
Patch, A.-M., Christie, E. L., Etemadmoghadam, D., Garsed, D. W., George, J., Fereday, S., Nones, K., Cowin, P., Alsop, K., Bailey, P. J., Kassahn, K. S., Newell, F., Quinn, M. C. J., Kazakoff, S., Quek, K., Wilhelm-Benartzi, C., Curry, E., Leong, H. S., Australian Ovarian Cancer Study Group, & Bowtell, D. D. L. (2015). Whole-genome characterization of chemoresistant ovarian cancer. Nature, 521(7553), 489–494. https://doi.org/10.1038/nature14410
Patel, A. G., Sarkaria, J. N., & Kaufmann, S. H. (2011). Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proceedings of the National Academy of Sciences of the United States of America, 108(8), 3406–3411. https://doi.org/10.1073/pnas.1013715108
Peterson, F. C., Chen, D., Lytle, B. L., Rossi, M. N., Ahel, I., Denu, J. M., & Volkman, B. F. (2011). Orphan macrodomain protein (human C6orf130) is an O-acyl-ADP-ribose deacylase: Solution structure and catalytic properties. The Journal of Biological Chemistry, 286(41), 35955–35965. https://doi.org/10.1074/jbc.M111.276238
Pillay, N., Tighe, A., Nelson, L., Littler, S., Coulson-Gilmer, C., Bah, N., Golder, A., Bakker, B., Spierings, D. C. J., James, D. I., Smith, K. M., Jordan, A. M., Morgan, R. D., Ogilvie, D. J., Foijer, F., Jackson, D. A., & Taylor, S. S. (2019). DNA replication vulnerabilities render ovarian cancer cells sensitive to poly(ADP-Ribose) glycohydrolase inhibitors. Cancer Cell, 35(3), 519-533.e8. https://doi.org/10.1016/j.ccell.2019.02.004
Plummer, R., Jones, C., Middleton, M., Wilson, R., Evans, J., Olsen, A., Curtin, N., Boddy, A., McHugh, P., Newell, D., Harris, A., Johnson, P., Steinfeldt, H., Dewji, R., Wang, D., Robson, L., & Calvert, H. (2008). Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clinical Cancer Research, 14(23), 7917–7923. https://doi.org/10.1158/1078-0432.CCR-08-1223
Poveda, A., Floquet, A., Ledermann, J. A., Asher, R., Penson, R. T., Oza, A. M., Korach, J., Huzarski, T., Pignata, S., Friedlander, M., Baldoni, A., Park-Simon, T.-W., Tamura, K., Sonke, G. S., Lisyanskaya, A., Kim, J.-H., Filho, E. A., Milenkova, T., Lowe, E. S., SOLO2/ENGOT-Ov21 investigators. (2021). Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. The Lancet Oncology, 22(5), 620–631. https://doi.org/10.1016/S1470-2045(21)00073-5
Powell, C., Mikropoulos, C., Kaye, S. B., Nutting, C. M., Bhide, S. A., Newbold, K., & Harrington, K. J. (2010). Pre-clinical and clinical evaluation of PARP inhibitors as tumour-specific radiosensitisers. Cancer Treatment Reviews, 36(7), 566–575. https://doi.org/10.1016/j.ctrv.2010.03.003
Prokhorova, E., Agnew, T., Wondisford, A. R., Tellier, M., Kaminski, N., Beijer, D., Holder, J., Groslambert, J., Suskiewicz, M. J., Zhu, K., Reber, J. M., Krassnig, S. C., Palazzo, L., Murphy, S., Nielsen, M. L., Mangerich, A., Ahel, D., Baets, J., O’Sullivan, R. J., & Ahel, I. (2021). Unrestrained poly-ADP-ribosylation provides insights into chromatin regulation and human disease. Molecular Cell, 81(12), 2640-2655.e8. https://doi.org/10.1016/j.molcel.2021.04.028
Rack, J. G. M., Perina, D., & Ahel, I. (2016). Macrodomains: structure, function, evolution, and catalytic activities. Annual Review of Biochemistry, 85, 431–454. https://doi.org/10.1146/annurev-biochem-060815-014935
Rafiei, S., Fitzpatrick, K., Liu, D., Cai, M.-Y., Elmarakeby, H. A., Park, J., Ricker, C., Kochupurakkal, B. S., Choudhury, A. D., Hahn, W. C., Balk, S. P., Hwang, J. H., Van Allen, E. M., & Mouw, K. W. (2020). ATM loss confers greater sensitivity to ATR inhibition than PARP inhibition in prostate cancer. Cancer Research, 80(11), 2094–2100. https://doi.org/10.1158/0008-5472.CAN-19-3126
Raphael, B. J., Hruban, R. H., Aguirre, A. J., Moffitt, R. A., Yeh, J. J., Stewart, C., Robertson, A. G., Cherniack, A. D., Gupta, M., Getz, G., Gabriel, S. B., Meyerson, M., Cibulskis, C., Fei, S. S., Hinoue, T., Shen, H., Laird, P. W., Ling, S., Lu, Y., & Zenklusen, J. C. (2017). Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell, 32(2), 185-203.e13. https://doi.org/10.1016/j.ccell.2017.07.007
Ray Chaudhuri, A., Callen, E., Ding, X., Gogola, E., Duarte, A. A., Lee, J.-E., Wong, N., Lafarga, V., Calvo, J. A., Panzarino, N. J., John, S., Day, A., Crespo, A. V., Shen, B., Starnes, L. M., de Ruiter, J. R., Daniel, J. A., Konstantinopoulos, P. A., Cortez, D., & Nussenzweig, A. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature, 535(7612), 382–387. https://doi.org/10.1038/nature18325
Ray Chaudhuri, A., & Nussenzweig, A. (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nature Reviews Molecular Cell Biology, 18(10), 610–621. https://doi.org/10.1038/nrm.2017.53
Ray-Coquard, I., Pautier, P., Pignata, S., Pérol, D., González-Martín, A., Berger, R., Fujiwara, K., Vergote, I., Colombo, N., Mäenpää, J., Selle, F., Sehouli, J., Lorusso, D., Guerra Alía, E. M., Reinthaller, A., Nagao, S., Lefeuvre-Plesse, C., Canzler, U., Scambia, G., PAOLA-1 Investigators. (2019). Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. The New England Journal of Medicine, 381(25), 2416–2428. https://doi.org/10.1056/NEJMoa1911361
Rebbeck, T. R., Mitra, N., Wan, F., Sinilnikova, O. M., Healey, S., McGuffog, L., Mazoyer, S., Chenevix-Trench, G., Easton, D. F., Antoniou, A. C., Nathanson, K. L., CIMBA Consortium, Laitman, Y., Kushnir, A., Paluch-Shimon, S., Berger, R., Zidan, J., Friedman, E., Ehrencrona, H., & Andrulis, I. (2015). Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA, 313(13), 1347–1361. https://doi.org/10.1001/jama.2014.5985
Robinson, D., Van Allen, E. M., Wu, Y.-M., Schultz, N., Lonigro, R. J., Mosquera, J.-M., Montgomery, B., Taplin, M.-E., Pritchard, C. C., Attard, G., Beltran, H., Abida, W., Bradley, R. K., Vinson, J., Cao, X., Vats, P., Kunju, L. P., Hussain, M., Feng, F. Y., & Chinnaiyan, A. M. (2015). Integrative clinical genomics of advanced prostate cancer. Cell, 161(5), 1215–1228. https://doi.org/10.1016/j.cell.2015.05.001
Robson, M., Im, S.-A., Senkus, E., Xu, B., Domchek, S. M., Masuda, N., Delaloge, S., Li, W., Tung, N., Armstrong, A., Wu, W., Goessl, C., Runswick, S., & Conte, P. (2017). Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. The New England Journal of Medicine, 377(6), 523–533. https://doi.org/10.1056/NEJMoa1706450
Rolli, V., O’Farrell, M., Ménissier-de Murcia, J., & de Murcia, G. (1997). Random mutagenesis of the poly(ADP-ribose) polymerase catalytic domain reveals amino acids involved in polymer branching. Biochemistry, 36(40), 12147–12154. https://doi.org/10.1021/bi971055p
Rondinelli, B., Gogola, E., Yücel, H., Duarte, A. A., van de Ven, M., van der Sluijs, R., Konstantinopoulos, P. A., Jonkers, J., Ceccaldi, R., Rottenberg, S., & D’Andrea, A. D. (2017). EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nature Cell Biology, 19(11), 1371–1378. https://doi.org/10.1038/ncb3626
Rosenthal, F., Feijs, K. L. H., Frugier, E., Bonalli, M., Forst, A. H., Imhof, R., Winkler, H. C., Fischer, D., Caflisch, A., Hassa, P. O., Lüscher, B., & Hottiger, M. O. (2013). Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nature Structural & Molecular Biology, 20(4), 502–507. https://doi.org/10.1038/nsmb.2521
Ruf, A., Mennissier de Murcia, J., de Murcia, G., & Schulz, G. E. (1996). Structure of the catalytic fragment of poly(AD-ribose) polymerase from chicken. Proceedings of the National Academy of Sciences of the United States of America, 93(15), 7481–7485. https://doi.org/10.1073/pnas.93.15.7481
Ruf, A., Rolli, V., de Murcia, G., & Schulz, G. E. (1998). The mechanism of the elongation and branching reaction of poly(ADP-ribose) polymerase as derived from crystal structures and mutagenesis. Journal of Molecular Biology, 278(1), 57–65. https://doi.org/10.1006/jmbi.1998.1673
Schlacher, K., Christ, N., Siaud, N., Egashira, A., Wu, H., & Jasin, M. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell, 145(4), 529–542. https://doi.org/10.1016/j.cell.2011.03.041
Sharifi, R., Morra, R., Appel, C. D., Tallis, M., Chioza, B., Jankevicius, G., Simpson, M. A., Matic, I., Ozkan, E., Golia, B., Schellenberg, M. J., Weston, R., Williams, J. G., Rossi, M. N., Galehdari, H., Krahn, J., Wan, A., Trembath, R. C., Crosby, A. H., & Ahel, I. (2013). Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. The EMBO Journal, 32(9), 1225–1237. https://doi.org/10.1038/emboj.2013.51
Slade, D., Dunstan, M. S., Barkauskaite, E., Weston, R., Lafite, P., Dixon, N., Ahel, M., Leys, D., & Ahel, I. (2011). The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature, 477(7366), 616–620. https://doi.org/10.1038/nature10404
Smith, R., Lebeaupin, T., Juhász, S., Chapuis, C., D’Augustin, O., Dutertre, S., Burkovics, P., Biertümpfel, C., Timinszky, G., & Huet, S. (2019). Poly(ADP-ribose)-dependent chromatin unfolding facilitates the association of DNA-binding proteins with DNA at sites of damage. Nucleic Acids Research, 47(21), 11250–11267. https://doi.org/10.1093/nar/gkz820
Solmaz, A. E., Onay, H., Yeniay, L., Gökmen, E., Özdemir, N., Alanyalı, S., Oktay, A., Özsaran, Z., Kapkaç, M., & Özkınay, F. (2020). BRCA1-BRCA2 mutation analysis results in 910 individuals: Mutation distribution and 8 novel mutations. Cancer Genetics, 241, 20–24. https://doi.org/10.1016/j.cancergen.2019.12.008
Stewart, R. A., Pilié, P. G., & Yap, T. A. (2018). Development of PARP and Immune-checkpoint inhibitor combinations. Cancer Research, 78(24), 6717–6725. https://doi.org/10.1158/0008-5472.CAN-18-2652
Vyas, S., Chesarone-Cataldo, M., Todorova, T., Huang, Y.-H., & Chang, P. (2013). A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nature Communications, 4, 2240. https://doi.org/10.1038/ncomms3240
Vyas, S., Matic, I., Uchima, L., Rood, J., Zaja, R., Hay, R. T., Ahel, I., & Chang, P. (2014). Family-wide analysis of poly(ADP-ribose) polymerase activity. Nature Communications, 5, 4426. https://doi.org/10.1038/ncomms5426
Waks, A. G., Cohen, O., Kochupurakkal, B., Kim, D., Dunn, C. E., Buendia Buendia, J., Wander, S., Helvie, K., Lloyd, M. R., Marini, L., Hughes, M. E., Freeman, S. S., Ivy, S. P., Geradts, J., Isakoff, S., LoRusso, P., Adalsteinsson, V. A., Tolaney, S. M., Matulonis, U., & Wagle, N. (2020). Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Annals of Oncology, 31(5), 590–598. https://doi.org/10.1016/j.annonc.2020.02.008
Whitehouse, C. J., Taylor, R. M., Thistlethwaite, A., Zhang, H., Karimi-Busheri, F., Lasko, D. D., Weinfeld, M., & Caldecott, K. W. (2001). XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell, 104(1), 107–117. https://doi.org/10.1016/s0092-8674(01)00195-7
Xu, Y., Huang, S., Liu, Z.-G., & Han, J. (2006). Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. The Journal of Biological Chemistry, 281(13), 8788–8795. https://doi.org/10.1074/jbc.M508135200
Yang, L., Zhang, Y., Shan, W., Hu, Z., Yuan, J., Pi, J., Wang, Y., Fan, L., Tang, Z., Li, C., Hu, X., Tanyi, J. L., Fan, Y., Huang, Q., Montone, K., Dang, C. V., & Zhang, L. (2017). Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Science Translational Medicine, 9(400), eaal1645. https://doi.org/10.1126/scitranslmed.aal1645
Yu, J., Hu, X., Yang, Q., Shan, R., Zhang, Y., Dong, Z., Li, H., Wang, J., Li, C., Xie, S., Dong, Y., Ni, W., Jiang, L., Liu, X., Wei, B., Wen, J., Liu, M., Chen, Q., Yang, Y., & Meng, X. (2022). Insulin-like growth factor binding protein 7 promotes acute kidney injury by alleviating poly ADP ribose polymerase 1 degradation. Kidney International, 102(4), 828–844. https://doi.org/10.1016/j.kint.2022.05.026
Yu, S.-W., Andrabi, S. A., Wang, H., Kim, N. S., Poirier, G. G., Dawson, T. M., & Dawson, V. L. (2006). Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proceedings of the National Academy of Sciences of the United States of America, 103(48), 18314–18319. https://doi.org/10.1073/pnas.0606528103
Yu, S.-W., Wang, H., Poitras, M. F., Coombs, C., Bowers, W. J., Federoff, H. J., Poirier, G. G., Dawson, T. M., & Dawson, V. L. (2002). Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science, 297(5579), 259–263. https://doi.org/10.1126/science.1072221
Zhang, Y., Liu, S., Mickanin, C., Feng, Y., Charlat, O., Michaud, G. A., Schirle, M., Shi, X., Hild, M., Bauer, A., Myer, V. E., Finan, P. M., Porter, J. A., Huang, S.-M.A., & Cong, F. (2011). RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nature Cell Biology, 13(5), 623–629. https://doi.org/10.1038/ncb2222
Zhou, B., Yan, J., Guo, L., Zhang, B., Liu, S., Yu, M., Chen, Z., Zhang, K., Zhang, W., Li, X., Xu, Y., Xiao, Y., Zhou, J., Fan, J., Hung, M.-C., Li, H., & Ye, Q. (2020). Hepatoma cell-intrinsic TLR9 activation induces immune escape through PD-L1 upregulation in hepatocellular carcinoma. Theranostics, 10(14), 6530–6543. https://doi.org/10.7150/thno.44417
Acknowledgements
This work was supported by National Natural Science Foundation of China (T2225006) and Beijing Municipal Natural Science Foundation (Z220011) to ML.
Author information
Authors and Affiliations
Contributions
ML designed and wrote the manuscript. YG wrote the manuscript. BF revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Ethical approval
Not applicable.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guo, Y., Fan, B. & Li, M. PARP molecular functions and applications of PARP inhibitors in cancer treatment. GENOME INSTAB. DIS. 4, 137–153 (2023). https://doi.org/10.1007/s42764-023-00100-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42764-023-00100-w