
Abstract
Chromosomal instability syndromes (CIS) show autosomal recessive inheritance patterns and are identified by breakage of the chromosome and chromosomal instability which are mostly direct results of DNA repair machinery defects that lead to many phenotypic manifestations including an increased predisposition to malignancies. The most commonly known CIS are Fanconi anaemia (FA), Nijmegen syndrome (NS), Bloom’s syndrome (BS), Ataxia-telangiectasia (A-T) and Ataxia telangiectasia like disorder (ATLD). Inherited bone marrow failure syndromes constitute a group of diseases that are extremely rare and can be described by inadequate blood cell production of single or multiple hematopoietic lineages. A major proportion of IBMFs is caused by defective genes that are involved in cellular pathways like ribosome function, telomere biology, transcription regulation, DNA repair, and telomere maintenance. In these diseases, bone marrow failure is frequently linked to other somatic abnormalities. IBMFS includes disorders such as Thrombocytopenia absent radii, Congenital amegakaryocytic thrombocytopenia and Fanconi anemia. The clinical phenotypes of these diseases are often overlapping and hence proper and accurate diagnosis is a challenge. Moreover, these diseases show great clinical heterogeneity and disease penetrance which can be explained to some extent by somatic mosaicism. Mosaicism can be a result of several mutations that have proliferated to only a small cluster of adult cells during the early developmental stages of the organism and subsequently during aging. The tissues in the body of an individual are regularly subjected to several intrinsic and extrinsic mutagenic stresses which can result in the accumulation of many genetic variations ranging from single base pair to ploidy level changes. During the past decades, somatic mosaicism has emerged as one of the major contributing factors in many monogenic disorders. These disorders are often associated with a predisposition to cancer, therefore, early and proper diagnosis is extremely important for adequate management and treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Somatic mosaicism can be defined as the existence of somatically diverse clusters of somatic cells in an individual which is derived from one fertilized egg. Mosaicism can be a result of several mutations that have proliferated to only a small cluster of adult cells during the early developmental stages of the organism and subsequently during aging. Genetic differences that are present in the tissues give rise to the mosaic phenotype and these variations are not inherited in accordance with Mendelian rules. Somatic mosaicism is an important contributor to the phenotypic variations present in an individual.
Somatic mosaicism was first identified by Curt Stem in the year 1936. He demonstrated that the phenomenon of genetic recombination, which was a normal occurrence in meiosis, was also possible in the mitotic division. He discovered the presence of somatic crossing over and segregation in Drosophila melanogaster. He also identified many insertions and deletions in the chromosome and described them as somatic mosaics (Stern, 1936). Bridges, in the year 1925, described a mosaic condition in the X chromosome of a fruit fly. Patterson, in the year 1929 demonstrated the actual segregation of chromosomes in somatic cells (Patterson, 1929). Cytological evidence confirming the presence of varying chromosome constitution in diverse cells was obtained by Mc Clintock in the year 1932 by her work on variegated Zea mays. She found varying degrees of changes in the length of chromosomes in many cells which may have been a result of somatic crossing over events (McClintock, 1932). Kaufmann in 1934 reported that homologous chromosomes of somatic cells of Drosophila melanogaster showed chiasma-like arrangements (Kaufmann, 1934). Peto reported that under radiation the root tip cells of Hordeum vulgare showed the formation of chiasmata (Peto, 1935).
The tissues in the body of an individual are regularly subjected to several intrinsic and extrinsic mutagenic stresses which can result in the accumulation of many genetic variations ranging from single base pair to ploidy level changes. Examples of extrinsic factors include nicotine usage and UV exposure which can cause damage to the DNA resulting in somatic mutations. In addition, intrinsic mutagenic factors such as ploidy changes (loss or gain of entire chromosomes), transposable elements, faulty DNA replication, errors of DNA repair machinery may give rise to genetic alterations in both diseased as well as normal tissues. During the past decades, somatic mosaicism has emerged as one of the major contributing factors in many monogenic disorders (Venugopal et al., 2019). Mosaicism has also emerged as the major source of antigenic diversity, embryo twinning and is also responsible for few aspects of mitochondrial disorders.
An increase in the rate of somatic mutation and DNA lesions arising as a result of an aberrant DNA repair pathway or error-prone polymerase has been shown to be directly related to neurodegenerative phenotypes, early aging and cancer predisposition in both human and mouse.
Though the exact role of mosaicism in disease pathology is being actively investigated, it is evident that functional mosaicism has an important role in human disease. Cytogenetic and sequencing techniques have been tremendously advanced in the last decade, enabling researchers to discover pathogenic somatic mutations for many diseases and many driver mutations for cancers.
Sources of Somatic mosaicism
Single nucleotide variations and small insertions or deletions
Single nucleotide variants (SNVs) come about in six primitive varieties: T-A>G-C, T-A>A-T, C-G>G-C and C-G>A-T (transversions) and T-A>C-G, C-G>T-A (transitions). Post-zygotic mutations studies in tumours indicate that environmental stressors cause distinct signatures of mutations (Helleday et al., 2014; Pfeifer et al., 2005; Pleasance et al., 2010). It was reported that the distribution of mosaic SNVs in healthy tissues is parallel to the distribution of mutations in cancer cells (Huang et al., 2014). Therefore, the uterine environment might make an individual susceptible to certain types of mosaicism. Small (< ~ 100–200 bps) insertions and deletions (indels) is a mutational type which is of similar scale to single nucleotide variations. Plausibly, any polymerase might cause deletions, however, certain particular human polymerases are empirically shown to lead to the production of small deletions at increased rates (Hile et al., 2012). Incorporation of substitute polymerases because of stress or damage during the course of development can affect mosaic indels distribution (Fig. 1a). Thompson et al.. identified a rare association of CAMT with proximal radioulnar synostosis (RUS) which was due to a heterozygous variant of HOXA11 (RUSAT1; MIM 605,432), which is a homeobox gene (Thompson & Nguyen, 2000; Thompson et al., 2001). Niihori et al. identified heterozygous mutations MDS1 and EVI1 complex locus (MECOM) in three unrelated RUS and AT (RUSAT 2, MIM 616,738) patients (Niihori et al., 2015).

Major sources of somatic mosaicism. a Single nucleotide variation (transition or transversion mutations lead to accumulation of single nucleotide variations), b Chromosomal aneuploidy (nondisjunction during meiotic recombination can lead to the formation of gametes with abnormal chromosome numbers, which when fertilized with normal gametes can give rise to aneuploid cells), c Loss or gain of copy number (various molecular events like deletion, duplication and inversion of gene segments can lead to copy number variations) d Trinucleotide repeat expansion (expansion of trinucleotide repeats in the coding regions arise due to mitotic recombination whereas repeat expansion in the non-coding region typically occurs via meiotic recombination)
Chromosomal Aneuploidy and large-scale structural changes in the chromosome
In the constitutional state of human life only monosomy of X, and trisomy of 13, 18, 21 and X chromosomes are tolerated. However, in the mosaic state a wide array of aneuploidies are reported. These comprise of monosomy of 7, 18 and 21 and include trisomy of 7, 8, 9, 12, 14, 15, 16, 17, 20 and 22, each with variable phenotypic features, proportions of affected cells, and occurrence. With increasing maternal age, the meiotic risk increases and might result in whole chromosomal aneuploidy through nondisjunction (Fig. 1b). Meiotic nondisjunction which may be corrected by post-zygotic formation or loss of a duplicate copy of a whole chromosome can lead to mosaic aneuploidy, which in the euploid cell line can result in uniparental disomy (UPD) or otherwise due to post-zygotic nondisjunction (Conlin et al., 2010). Isochromosomes and ring chromosomes are two other significant anomalies that are seen in the mosaic state. Pallister-Killian syndrome (MIM#601,803) caused by mosaic abnormality involving isochromosomes (of 12p) is a notable one. Isochromosome 12p is only reported in the mosaic state, most probably as such an anomaly would be fatal in the constitutional state (M. Campbell, 2015). Large scale structural changes in the chromosome locations 7p14, 7q35, 14q32 and 14q11 have been reported in AT patients by Kojis et al. (Kojis et al., 1989). Iourov et al. have also that stochastic aneuploidy in the cerebrum and cerebellum of AT patients have been reported to be were two to three folds more than the unaffected controls (Iourov, Vorsanova, Liehr, et al., 2009a, 2009b). The presence of these chromosomal anomalies in AT patients could explain tissue specific clinical features of the disease.
Loss or gain of copy number
The methods that revealed the significant correlation between copy number variations and health have amplified sensitivity and implicated mosaicism as the source of many known disease-related mutations (Boone et al., 2010; Cheung et al., 2007; Conlin et al., 2010; Pham et al., 2014). The mosaicism frequency observed for disease-related copy number variation is a balance between post-zygotic mutagenesis frequency and non-severe mosaicism-related phenotypes that dodge medical diagnosis. A major portion of recurring copy number variations are a result of nonallelic homologous recombination (NAHR) between adjoining low-copy number repeats (LCRs). NAHR usually takes place during the course of meiosis (Turner et al., 2008). Lack of obvious paternal age effect on the danger of standard genomic syndromes like Smith-Magenis (MIM#182,290) and Angelman (MIM#105,830) syndromes along with insignificant prejudice in the parent of origin of frequent copy number variations reinforces the supposition that most recurrent copy number variations have a meiotic origin (Hehir-Kwa et al., 2011). Nevertheless, sporadically, copy number variations that seem to be facilitated by LCRs have been reported in the mosaic state (Messiaen et al., 2011) (Fig. 1c). Likewise, the percentage of mosaic copy number variations that truly result from post-zygotic mitotic recombination causing the absence of heterozygosity (AOH) rather than a post-zygotic new mutation has not been completely discovered. However, the occurrence of such mitotic LOH events in humans (Choate et al., 2010) and mice (Shao et al., 2001) is known.
Two key mechanistic classes: nonhomologous end joining (NHEJ) and replicative mechanisms seem to form Nonrecurrent CNVs. While particulars of the suggested replicative mechanisms vary, common characteristics emerge: stalled replication (which leads to double-stranded DNA breaks due to collapsed replication forks), long-distance template switching and template switching which is facilitated by stretches of short homology. Independent of which mechanism of mutation is involved, mosaicism can arise due to DNA replication errors. Non- homologous end joining (NHEJ) which usually takes place throughout the full duration of the cell- cycle, also contributes to the accumulation of mosaic mutations. Around 0.5 to 5 percent of patients with genetic disorders harbour non-recurrent copy number variations, highlighting the mitotic nature of these events (Ballif et al., 2006; Conlin et al., 2010; Pham et al., 2014).
Expansion of trinucleotide repeats
Many human diseases can be attributed to expansions of tri- or seldom tetra nucleotide recurrences and other simple sequence recurrences (C. T. McMurray, 2010a, 2010b). In these syndromes, wild-type length repeat alleles are first expanded to pre-mutation alleles, further increase in the length of these alleles into longer alleles leads to accumulation of harmful expansions. Trinucleotide repeats occurring in the coding regions of the genome are inclined to accumulate in the paternal germline via mitotic replication. Expansion in the non-coding regions is reported via meiotic replicative process in the oogonia (J. J. V. McMurray, 2010a, 2010b; Rifé et al., 2004). Nevertheless, it has been observed that somatic variations occur in the non-coding and coding trinucleotide repeat lengths (Fig. 1d). Early age of onset of Huntington syndrome is associated with the expansion of trinucleotide repeat in the coding region of HD gene (Swami et al., 2009). Monozygotic twins discordant for noncoding FMR1 repeat length have also been reported, suggesting that expansion can also occur mitotically (Helderman-van Den Enden et al., 1999). The degree to which somatic mosaicism due to nucleotide repeat expansion affects disease self-expression and communication is less understood.
Expansion of D4Z4, a repeat of length 3.3 kilobases on chromosome six, causes Facioscapulohumeral muscular dystrophy (FSHD) (Lemmers et al., 2010). The exact underlying process of pathogenesis is undetermined, however, repressive chromatin marks and a reduction in methylation in the vicinity of a specific haplotype of the gene DUX4 might possibly be related (Lemmers et al., 2012; Lupski, 2012). Post zygotic mutations make up about twenty to forty percent of the de novo mutations in FSHD and these are present at comparatively higher fractions of mutant cells (Lemmers et al., 2004; Van Der Maarel et al., 2000). This points to the possibility that the genome might be highly susceptible to D4Z4 contraction during early embryogenesis.
Retrotransposition of autonomous mobile elements
Despite being traditionally labelled as ‘junk’ certain elements of the human genome such as mobile, repetitive elements are emerging as important factors with major functional roles in various physiological and disease processes. For instance, LINE-1 holds on to its intrinsic transposition capability and can traverse throughout the genome. Therefore, during organism’s life span, many DNA elements are frequently copied from one genetic location and pasted to another locus on the chromosome via a replicative mechanism which is called transposition (Shapiro, 1979). This is evident in adult cortical neurons where insertion of mobile elements is responsible for somatic mosaicism (Erwin et al., 2014; Evrony et al., 2015). Events of constitutional retrotransposition can result in Mendelian syndrome because of disruption in the coding sequence, alteration in splicing sequence, and even due to subtle positional effects (Beck et al., 2011).
Methods to detect mosaicism
Cytogenetics
Though widespread somatic mosaicism has been suspected in many human diseases as well as healthy tissues for a long time, the detection of the presence of two or more genetically distinct cellular subpopulations within the same tissue has been a challenge until recently. Initial papers that have reported mosaicism relied heavily on the use of a light microscope for the detection of mosaic events in single cells. Study of metaphase chromosomes under the light microscope provided information about ploidy level changes. Banding techniques involving stains like Giemsa and other dyes allowed the detection of large structural changes such as inter- and intra- chromosomal duplications, deletions, translocations and structural rearrangements. The limitation of these banding techniques was that they could only resolve abnormalities which were larger than 3 Mb. Another cytogenetic technique used for the detection of the specific genomic region is fluoroscent in situ hybridization (FISH) which can detect deletion and duplication with a resolution of 100 Kb. These techniques can be used to detect low levels of mosaicism (Freed et al., 2014).
Comparitive genomic hybridization
In comparative genomic hybridization (CGH), DNA from test and control sample are labelled using a fluorophore and are hybridized to a reference metaphase chromosome. Deletions or duplications are then detected by measuring the ratio of emitted fluorescence. Sample DNA having the same copy number have a ratio of 1:1. Any deviations from this are indicative of variation in copy numbers. SNP microarrays (Single nucleotide polymorphism microarrays) and array CGH (aCGH) are two array-based substitutes to CGH. Both SNP microarray and aCGH can be used for the detection of copy number variation in large genomic regions. SNP microarrays can be used to detect low-level mosaic events and genotypic analysis of individuals at the probed sites (Freed et al., 2014).
Next-generation sequencing
Our understanding of human genetics was revolutionized by the advances in next-generation sequencing techniques in recent years. Bulk tissue, a group of cells or even single cells can be sequenced and may provide information about single nucleotide polymorphisms or variations, retrotransposition events, translocations, deletions and insertions. Sequencing can also provide information about copy number changes based on the frequency of reads associated with specific genomic regions. Whole-genome or whole-exome sequencing from tissues obtained from unaffected and affected parts of an individual can be used to discover somatic variants. In addition to the identification of variants, sequencing techniques can also provide information about the extent of mosaicism in tissues as well as the developmental period of origin of the mosaicism by quantifying the affected fraction of cells (Machiela & Chanock, 2014).
Single-cell sequencing
Single-cell sequencing (SCS) depends on isolation, amplification and sequencing of DNA derived from single cells which allows the examination of mosaic events at the smallest biological scale. SCS makes almost ninety percent of the genome accessible for analysis and hence can be used to discover mosaic events within tissues, cell types and even single cells. Analysis of single-cell somatic events can help in reconstructing cell lineage and provide information about body localization, affected cell types and developmental occurrence of variation (Machiela & Chanock, 2014).
Mosaicism in IBMF and CIS diseases
Chromosomal instability syndromes (CIS) show autosomal recessive inheritance patterns and are identified by breakage of the chromosome and chromosomal instability which are mostly direct results of DNA repair machinery defects that lead to many phenotypic manifestations including an increased predisposition to malignancies. The most commonly known CIS are Fanconi anaemia (FA), Nijmegen syndrome (NS), Bloom’s syndrome (BS), Ataxia telangiectasia (A-T) and Ataxia telangiectasia like disorder (ATLD). The molecular characteristics of each CIS are distinct. Fanconi anaemia is caused by loss of function mutations in any of the 22 known genes that are involved in DNA interstrand crosslink repair. Nijmegen syndrome and ataxia telangiectasia are caused by defects in the repair mechanism of double-strand DNA breaks. The genes responsible are NBN (gene encoding nibrin) and ATM (gene encoding serine-protein kinase ATM). Bloom syndrome is caused by defects in homologous recombination pathways caused by a mutation in the BLM gene which results in instability of replication fork, DNA end resection and double Holiday junction dissolution (Wu, 2016).
Inherited bone marrow failure syndromes constitute a group of diseases that are extremely rare and can be described by inadequate blood cell production of single or multiple hematopoietic lineage (Foglesong et al., 2017). A major proportion of IBMFs is caused by defective genes that are involved in cellular pathways like ribosome function, transcription regulation, DNA repair, and telomere maintenance. In these diseases, the marrow failure is frequently linked to other somatic abnormalities. IBMFS includes disorders like Thrombocytopenia absent radii, Congenital amegakaryocytic thrombocytopenia and Fanconi anemia. These disorders are often associated with a predisposition to cancer therefore, early and proper diagnosis is extremely important for adequate management and treatment (Table 1). Proper diagnosis of these diseases is extremely complicated due to the presence of great clinical heterogeneity in terms of the symptoms presented. Moreover, designating a disease as a CIS or IBMF is very difficult because of the overlapping phenotypes and clinical manifestations of these diseases. The clinical phenotypes of these diseases are often overlapping and hence proper and accurate diagnosis is a challenge. Moreover, these diseases show great clinical heterogeneity and disease penetrance which can be explained to some extent by somatic mosaicism.
Fanconi anaemia
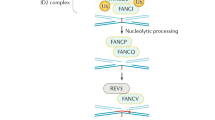
Fanconi anaemia follows an autosomal recessive inheritance pattern and is caused due to a loss of function mutation in a set of genes involved in DNA interstrand crosslink repair (ICL). Bone marrow failure, chromosomal instability, various congenital anomalies and an enhanced risk of cancers are characteristic of this disease. However, this disease shows great clinical heterogeneity as a smaller fraction of patients with FA do not display any congenital abnormality. Patients with FA have an increased susceptibility to diepoxy butane (DEB) and mitomycin C (MMC)- which are known to induce DNA crosslinks. The clinical heterogeneity displayed by FA is mirrored at the molecular level by the various complementation groups corresponding to defects in a total of 22 genes discovered till now. Out of all these complementation groups, FANCA and FANCC are the most predominant in the population. There is very little homology between the many identified FA genes.
The presence of somatic mosaicism in patients with FA was reported even before the actual identification of the FANCC gene in the year 1992 (Strathdee et al., 1992). Kwee et al., reported that around sixty to eighty percent of lymphocytes in culture isolated from FA patients were resistant to the cytotoxic concentration of alkylating agents, which was a deviation from the established action of these agents on FA patient cells (Auerbach et al., 1981; Kwee et al., 1983). Mosaicism in the somatic cells which is observed in FA may arise due to reversion or many other gain of function mutations in hematopoietic stem cell/progenitor cells (HPSCs), which, then lead to the development of blood and bone marrow cell populations with a restored capacity of repairing DNA. The presence of two distinct cell populations has been reported in the blood of FA patients with a revertant pathogenic FA gene. One of these cell populations shows susceptibility to alkylating agents that cause DNA damage such as MMC or DEB and display the known FA characteristics. The other population of blood cells has resistance to these clastogenic agents, thus leading to the mosaic state in these individuals with two cell population coexisting with distinct molecular features. Formation of a functional FA gene and protein by additional genetic events by several different molecular mechanisms such as second site mutation, back mutation, intragenic crossover and gene conversion have been reported. These molecular events may restore the mutated gene to the wildtype version (Gross et al., 2002; Kalb et al., 2007; Lo Ten Foe et al., 1997). Second site mutations typically include either a compensatory indel mutation at a specific site within the gene which is mutated which results in the formation of a functional gene and protein which cannot be distinguished from the wildtype protein.
A case study involving two monozygotic twin sisters who were diagnosed with FA very early in life but had shown hematologic stability in a twenty-eight-year follow-up study, has provided strong evidence that mosaic state can impart proliferative advantage at the hematopoietic stem cell level. Though the cultured fibroblasts from these sisters displayed the fragility characteristic of FA, their peripheral blood lymphocytes had clastogen resistance. The presence of multilineage compensatory mutations was reported by analysing the bone marrow progenitor cells. This provided support to the hypothesis that prenatal reversion events had taken place in a long-term hematopoietic stem cell which resulted in a multilineage engraftment during early developmental stages (Mankad et al., 2006; Poole et al., 1992). This case of selective growth advantage imparted by spontaneous gene reversion allowing sustained haematopoiesis provides a potential mechanism for gene therapy for hematologic correction of FA. Several studies have reported that gene-corrected FA cells have a selective advantage in IPSCs (Raya et al., 2009), human embryonic stem cells (Tulpule et al., 2010), murine models (Battaile et al., 1999; Kamimae-Lanning et al., 2013; Suzuki et al., 2017) and transplant studies of gene-corrected FA patient cells in immunocompromised mice (Río et al., 2017).
The existence of a population of revertant hematopoietic cells has frequently been considered a favourable hematologic outcome in FA patients, but long-term bone marrow failure- or AML- free survival cannot be uniformly associated with the presence of blood and marrow cells that display clastogen resistance. This is reported by Gregory et al., in a study involving a male patient who displayed hematologic stability in the initial nine to seventeen years of his life and had a stable DEB-resistant peripheral blood lymphocyte population. Multilineage revertant mutations (including erythroid, myeloid and lymphoid progenitors) were reported in the bone marrow progenitors of this individual at age fifteen. However, at fifteen and seventeen years of age, the patient displayed an 11q deletion (potentially linked to myeloid malignancy) in a non- revertant bone marrow cell population (Gregory et al., 2001).
The predictive ambiguity about the existence of revertant hematopoietic population is made additionally difficult due to the methods employed in the diagnosis of FA as they are often focused on differentiated T- lymphoid cells which may have genetically different than multilineage progenitor and stem cell population. Mosaicism in FA has been reported mostly in the genes belonging to the FA core complex and the genes involved in ubiquitination such as FANCC, FANCA and FANCD2. Reports of mosaic mutations in recently discovered FA genes and other FA genes are very limited (Asur et al., 2018; Rickman et al., 2015; Virts et al., 2015). Reports of hematologic improvement and stability are present in single lineage or often bi- and tri- lineage settings, supporting the hypothesis that mutations resulting in reversion might occur across a broad spectrum of pluripotent hematopoietic stem and progenitor cells (Nicoletti et al., 2020). A wide array of peripheral blood lymphocytes resistant to clastogens such as diepoxy butane have been reported and a few studies have reported a link between the percent of resistant peripheral blood cells and clinical manifestations. Fifteen to twenty percent of FA patients show T lymphocyte mosaicism which is often linked to the failure of hematopoietic stem cell transplant engraftment prior to the use of conditioning based on fludarabine (MacMillan et al., 2000).
Blooms syndrome
Bloom’s syndrome is an autosomal recessive disorder whose characteristics include: photosensitive skin changes, prenatal and postnatal growth deficiency, insulin resistance growth retardation, immune deficiency, susceptibility to malignancies and highly enhanced early onset of cancer risk and for the possibility of developing different types of cancers. The gene responsible for causing BS, BLM encodes for RecQ helicase in which more than 60 different types of mutations have been reported. A non-functional BLM protein results in an increase in homologous recombination, chromosome instability and a higher number of sister chromatid exchanges which are a characteristic of the disease. Around 1 percent of the population having Eastern European Jewish ancestry have the BLM Ash founder mutation (Nathan A. Ellis, Groden, et al., 1995; Ellis, Lennon, et al., 1995). Many other founder mutations are also prevalent in other populations. Nonsense, frameshift and missense variations have also been observed in this disease. BS is an ideal example of CIS and the somatic mutations that are seen as a result of that instability lead to an enhanced risk of cancer. Though at present there are no specific treatments for the causal genetic abnormality, people with Bloom’s syndrome are benefitted from aggressive treatment of infections, sun protection, monitoring for insulin resistance, and early detection of cancer (Hirschhorn, 2003).
The lymphoid cells of around 1/5th of BS patients show the presence of mosaicism. A small percentage of these mosaic cells exhibit low sister chromatid exchange. The patients with mosaicism in the frequency of sister chromatid exchange are mostly heterozygotes having 2 distinct mutations and this mosaicism resulting in low exchange of sister chromatids can be attributed to intragenic recombination (N. A. Ellis, Groden, et al., 1995; Ellis, Lennon, et al., 1995). When the homologous chromosomes are segregated after intragenic recombination event, few cells receive either both inherited mutations or only one of the mutations while few cells do not inherit even one of the mutant chromosome. The revertant cells circulating in the blood of BS patients might have some proliferative advantage over the mutated cells, which probably differs in degree based on developmental stage or contact with antigen. This might be the reason behind the variable frequencies of revertant cells seen in BS patients. Another possible phenomenon that might favour the proliferation of the revertant cell population is the characteristic immunodeficiency seen in BS. Studies focussed on the molecular pathways of BS have revealed that back mutation and somatic intragenic recombination are the genetic mechanisms responsible for the somatic mosaicism seen in BS. Back mutation was reported to be the molecular mechanism which led to phenotypic reversion in 2 patients out of the 11 having high/ low- sister chromatid exchange mosaicism (Nathan A. Ellis et al., 2001).
In 1974, the German laboratory, for the first time reported the presence of a higher than normal frequency of sister chromatid exchanges (SCEs) in patients suffering from BS (Chaganti et al., 1974; James German et al., 1974). They reported an average of 6.9 SCEs per metaphase in phytohemagglutinin-stimulated lymphocytes from normal patients, which was much lower than 89 SCEs per metaphase in cells from BS patients. German et al., reported the coexistence of two distinct population of cells in BS patients- one with an elevated number of SCEs and cells with a normal number of SCEs. An increased rate of Sister chromatid exchanges is a characteristic feature of BS and is present in all known patients, however, approximately twenty percent of BS patients have shown the presence of a smaller population of cells with normal sister chromatid exchanges in venous blood(J. German et al., 1977). High/ low- SCE mosaicism is most commonly seen in patients who are allozygous at BLM whereas autozygous BLM patients very rarely exhibit this type of mosaicism (J. German et al., 1977).
It has been indicated by studies that patients who are heterozygous the pathogenic variant of BLM have an increased predisposition to cancer despite being otherwise healthy. This might be because of a diminished DNA repair efficiency in heterozygous cells. This may also result if the normal allele is lost and a somatic clone is developed which has a highly increased rate of mutation (Gross et al., 2002).
Ataxia telangiectasia
AT (MIM 208,900) is an autosomal recessive neurodegenerative disease which manifests in early childhood. Premature ageing, gonadal atrophy, immunodeficiency, predisposition to cancer, ocular telangiectasia and cerebellar ataxia are some of the characteristics of this multi-system disorder which is currently incurable. AT is induced by a mutation in the ATM gene which is involved in the regulation of genetic recombination, DNA repair, apoptosis and cell cycle checkpoints. ATM codes for a protein kinase which is essential for processing double-stranded DNA breaks during meiotic or somatic recombination. ATM is primarily involved in the maintaining genomic integrity and DNA damage response. Gradual cerebellar degeneration is a hallmark of Ataxia telangiectasia. However, other tissues and areas of the brain are not as much affected by atrophy and progressive degeneration in AT (McKinnon, 2004).
Balanced, non-random chromosomal rearrangements in the cells of the immune system are considered to be the cellular ‘marker’ of chromosomal instability associated with AT. Common breakpoints that can be seen in the lymphocytes of AT patients are 7p14, 7q35, 14q32 and 14q11. These chromosomal locations harbour the genes that are involved in coding for T-cell receptors and immunoglobulins (Kojis et al., 1989). Kojis et al., have studied the relationship between high chromosomal breaks at these locations and the different features of the AT phenotype. They report that widespread chromosome rearrangements reported in the lymphocytes are frequently absent from fibroblasts. However, the degree of chromosome instability is relatively higher in fibroblasts than in the lymphocytes. Additionally, the variations observed in the fibroblast cells was found to be more random (Kojis et al., 1989).
Iourov et al., studied the effects of chromosomal instability in the cerebellum of AT brain. They studied ten randomly selected chromosomes 1, 7, 8, 9, 11, 14, 16, 17, 18, 19, 21, X and Y to assess the rates of aneuploidy in the cerebral cortex and cerebellum of 7 AT patients and 7 sex and age matched controls. They reported that stochastic aneuploidy in the cerebrum and the cerebellum of the AT patients were two to three folds more than the unaffected controls (Iourov, Vorsanova, Liehr, et al., 2009a, 2009b). They also analyzed the structural rearrangements of the interphase chromosomes in the neuronal cells. They identified non-random breaks of chromosome 1, 7, 14, 21 and X in the neuronal cells of the cerebellum of the AT patients. They also report an age-dependent increase in chromosome breakage and aneuploidy in these chromosomes in the cerebellum of the AT individuals (Iourov, Vorsanova, Zelenova, et al., 2009a, 2009b).
Congenital amegakaryocytic thrombocytopenia (CAMT)
CAMT commonly manifests in neonates or at infancy as simple thrombocytopenia with absent or reduced production of bone marrow megakaryocytes, and subsequently progresses to bone marrow failure in early childhood. Although patients with CAMT are not known to have any physical deformities specifically associated with the disease (Ballmaier & Germeshausen, 2011), few cases have been reported of patients with abnormal facial features, malformations of the collective system, strabismus and defect in the cardiac septa. CAMT progresses to pancytopenia at the median age of thirty-nine months. It is a rare IBMF syndrome which is caused due to a mutation in the MPL gene and follows autosomal recessive inheritance pattern (Zhao et al., 2021). Both homozygotes and compound heterozygotes positively inherit the disease (Yildirim et al., 2015). The MPL gene codes for a 635 amino acid long receptor for thrombopoietin (TPO) which comprises an intracellular domain, transmembrane domain, an extracellular cytokine receptor domain and a signal peptide. TPO plays a major role in the production and maturation of megakaryocytes and platelets. It is also an important factor in the development of early myeloid and erythroid precursor cells and pluripotent hematopoietic stem cells (12). The MPL gene has twelve exons and is positioned on chromosome 1p34 (5). Sixty percent of the forty known mutations reported in MPL gene are located in the 2nd and the 3rd exon and include intron splicing site mutations, frameshift mutations, missense mutations and exon nonsense mutations (Ballmaier & Germeshausen, 2011).
A study involving three patients belonging to two different families revealed a rare association of CAMT with proximal radioulnar synostosis (RUS) which was due to a heterozygous variant of HOXA11 (RUSAT1; MIM 605,432), which is a homeobox gene(Thompson & Nguyen, 2000; Thompson et al., 2001). Heterozygous mutations MDS1 and EVI1 complex locus (MECOM) were identified by Niihori et al. in three unrelated RUS and AT (RUSAT 2, MIM 616,738) patients (Niihori et al., 2015). Since then, many patients with IBMFs and thrombocytopenia have been identified having MECOM mutation (Kjeldsen et al., 2018). Germeshausen et al., published a study with twelve thrombocytopenia patients who had germline mutations in MECOM and identified ten novel mutations. Out of the 12 patients studied, the father of the patient P1 was suspected to have somatic mosaicism in the MECOM gene due to the presence of an only minor signal in the DNA extracted from the peripheral blood even though his son had inherited heterozygous missense mutation and had severe thrombocytopenia (Germeshausen et al., 2018).
Osumi et al., reported a case study of a female patient with possible somatic mosaicism in MECOM who had congenital bone marrow failure but did not show any radial abnormality (Osumi et al., 2018). The patient was found to be anemic and had severe thrombocytopenia with a normal leukocyte count, which was revealed by a blood test taken at birth. Skeletal abnormalities like radial dysplasia were not detected by echography and radiography, at birth. At 2 months the patient showed a severe decrease in her amount of total leukocyte and advanced to pancytopenia. A novel, nonsynonymous MECOM (NM_001105078, c.2248C > T, p.Arg750Trp) variation was identified by whole-exome sequencing. The variant to wild-type allele ratio in the foot and hand nail, umbilical cord and peripheral blood was found to be 0.933, 0.356, 0.515 and 1.028, respectively. This variation in allelic frequency in DNA obtained from different sources suggests that this variation in the MECOM was a result of somatic mosaicism (Osumi et al., 2018).
Dyskeratosis congentia
Dyskeratosis congentia (DC) is classically described as a IBMF syndrome whose symptoms include mucosal leucoplakia, nail dystrophy and abnormal skin pigmentation. In addition to these, a plethora of other abnormalities including gastrointestinal, dental, neurological, dental, skeletal, pulmonary and ophthalmic irregularities. Autosomal recessive (MIM 224,230), autosomal dominant (MIM 127,550) and X-linked recessive (MIM 305,000) modes of inheritance are recognised in this disease (Inderjeet Dokal & Vulliamy, 2003). Those diagnosed with DC have shorter chromosomes than age-matched controls (Alter et al., 2007). Around fifty percent of patients of dyskeratosis congentia have mutations in 1 of 6 genes that are involved in the synthesis of proteins that constitute the telomerase complex; TERC (MIM 602,322), NHP2 (MIM 606,470), TERT (MIM 187,270), DKC1 (MIM 300,126) and NOP10 (MIM 606,471) or the telomerase shelterin complex TINF2 (MIM 604,319). Loss of function mutations in the above-mentioned genes leads to impaired maintenance of telomere length, the effects of which are most profoundly witnessed in tissues that have rapid turnover such as nails, skin and bone marrow (Jongmans et al., 2012).
Clinical severity varies considerably in individuals with DC, a part of which can be attributed to genotype–phenotype correlations. Out of the six causal mutations responsible for DC, mutations in TINF2 and DKC1 lead to the most severe presentation in patients (Vulliamy & Dokal, 2008). Batista et al., 2011 provided evidence that mutations in DKC1 lead to more severe defects in telomere- maintenance than mutations in TERT (Batista et al., 2011).
Patients having mutations in the genes associated with DC also exhibit variation in their clinical phenotypes. Mitotic recombination is most probably initiated by shortening of telomere length in patients with DC. Skin fibroblasts obtained from DC patients have abnormal growth rate as well as morphology than age-matched controls. These fibroblasts have been reported to display abnormal chromosomal rearrangements like translocations, di- and tricentric centromeres (I Dokal et al., 1992) which underlies the propensity for chromosomal instability in DC. This instability is triggered by the shortening of the telomeres; by induction of improper joining of chromosome ends. This attachment may occur either through homologous recombination (HR) or via non-homologous end joining (NHEJ) (Verdun & Karlseder, 2007).
Jongmans et al., screened specifically for mitotic recombination events resulting in reversion to the normal phenotype (Jongmans et al., 2012). The authors have reported the presence of spontaneous reversion in 6 DC patients belonging to 4 different families with autosomal dominant inheritance pattern. In the 6 individuals studied, 7 distinct reversion events in the TERC gene were reported which could be attributed to somatic mosaicism. They further speculate that random molecular events might also lead to meiotic recombination on chromosome 3q, which may result in reversion to a normal state, thereby providing a proliferative advantage to the cells that now have two functional copies of the TERC gene (Jongmans et al., 2012).
Vulliamy et al., reported the presence of somatic mosaicism in a female carrier who was also a germline mosaic for the gene DKC1 (Vulliamy et al., 1999). The blood of the female did not show the presence of the DKC1 deletion but two of her children had inherited the affected allele. Moreover, both the affected sons had inherited the same haplotype, suggesting that the mother was a germline mosaic. This deletion was found to be also present in about five percent of the peripheral blood cells from the female suggesting that she was also a somatic mosaic for the same mutation. The percentage of cells harboring the deletion in the germline tissue was reported to be higher than the somatic tissues. This observation suggests that the mutation might have originated in a precursor cell during early developmental stages which passed on the mutation to both somatic and germline tissues (Vulliamy et al., 1999).
Discussion
Though the presence of somatic mosaicism has been well documented in FA, it is not very widely reported in the other chromosomal instability disorders and bone marrow failure syndromes. However, reports exist that point to the presence of widespread mosaicism in these disorders. Studying the mechanisms behind mosaicism in these disorders has very important implications in the clinical diagnosis and management of these diseases. The diagnosis of most of these diseases is based on the presence of disease-causing genetic mutations which are carried out on the DNA isolated from the peripheral blood of suspected patients. As is evident from the above discussion, analysis of only the peripheral blood does not provide the complete picture of all the underlying mutations present in an individual. Therefore, sequence analysis needs to be extended to DNA extracted from other sources such as skin fibroblasts for obtaining a holistic view of the mutational landscape of an individual. This will also result in a better understanding of the reasons behind variable penetrance of these diseases in individuals and might also explain some of the unexplained phenotypes observed in these disorders.
Somatic variations might provide important clues about predisposition of certain types of cells and tissues to the development of specific disease-related phenotype. For example, the chromosomal rearrangement pattern observed in AT lymphocytes is completely different from that of AT fibroblasts. Presence of LARs in AT lymphocytes which are manifestations of chromosomal changes at a molecular level can be directly correlated with deficiency of the immune system and increased rate of malignancies related to the immune system in individuals homozygous for AT. The substantial variation observed in chromosomal rearrangement frequencies in AT fibroblasts and lymphocytes advocates that the diverse characteristics of the AT phenotype can be accounted by damage at specific sites in different tissues. It has been reported that CIS and IBMF disorders have an increased predisposition to cancer as well as a high rate of chromosomal mutational events. Studying the high rates of chromosomal mosaic events in individuals with CIS and IBMF can be used as biomarkers for the risk of cancers. This might help in the early detection of cancers in patients with chromosomal instability. Thus, it is extremely important to include the study of somatic mosaic events for proper diagnosis and treatment of CIS and IBMF. Future studies that integrate clinical data, biomarkers and mosaic events can provide important insights for the development of novel screening techniques for early detection of cancer and predicting incidence rates of many other complex diseases.
Abbreviations
- BS:
-
Bloom’s syndrome
- A-T:
-
Ataxia-telangiectasia
- ATLD:
-
Ataxia telangiectasia-like disorder
- IBMFS:
-
Inherited bone marrow failure syndrome
- SNVS:
-
Single nucleotide variants
- UPD:
-
Uniparental disomy
- NAHR:
-
Nonallelic homologous recombination
- LCRs:
-
Low-copy number repeats
- NHEJ:
-
Nonhomologous end joining
- FSHD:
-
Facioscapulohumeral muscular dystrophy
- LINE:
-
Long interspersed nuclear elements
- ICL:
-
Interstrand crosslink repair
- DEB:
-
Diepoxy butane
- MMC:
-
Mitomycin C
- HPSCs:
-
Hematopoietic stem cell/progenitor cells
- IPSCs:
-
Induced pluripotent stem cells
- AML:
-
Acute myeloid leukaemia
- SCEs:
-
Sister chromatid exchanges
- CIS:
-
Chromosomal instability syndromes
- NS:
-
Nijmegen syndrome
- CAMT:
-
Congenital amegakaryocytic thrombocytopenia
- FA:
-
Fanconi anemia
- LARs:
-
Lymphocyte associated rearrangements
- FISH:
-
Fluoroscent in situ hybridization
- CGH:
-
Comparative genomic hybridization
- SNP microarrays:
-
Single nucleotide polymorphism microarrays
- aCGH:
-
Array CGH
- SCS:
-
Single-cell sequencing
References
Alter, B. P., Baerlocher, G. M., Savage, S. A., Chanock, S. J., Weksler, B. B., Willner, J. P., Peters, J. A., Giri, N., & Lansdorp, P. M. (2007). Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood, 110(5), 1439–1447. https://doi.org/10.1182/blood-2007-02-075598
Asur, R. S., Kimble, D. C., Lach, F. P., Jung, M., Donovan, F. X., Kamat, A., Noonan, R. J., Thomas, J. W., Park, M., Chines, P., Vlachos, A., Auerbach, A. D., Smogorzewska, A., & Chandrasekharappa, S. C. (2018). Somatic mosaicism of an intragenic FANCB duplication in both fibroblast and peripheral blood cells observed in a Fanconi anemia patient leads to milder phenotype. Molecular Genetics and Genomic Medicine, 6(1), 77–91. https://doi.org/10.1002/mgg3.350
Auerbach, A. D., Adler, B., & Chaganti, R. S. K. (1981). Prenatal and postnatal diagnosis and carrier detection of Fanconi anemia by a cytogenetic method. Pediatrics, 67(1), 128–135.
Ballif, B. C., Rorem, E. A., Sundin, K., Lincicum, M., Gaskin, S., Coppinger, J., Kashork, C. D., Shaffer, L. G., & Bejjani, B. A. (2006). Detection of low-level mosaicism by array CGH in routine diagnostic specimens. American Journal of Medical Genetics. Part A, 140(24), 2757–2767. https://doi.org/10.1002/ajmg.a.31539
Ballmaier, M., & Germeshausen, M. (2011). Congenital amegakaryocytic thrombocytopenia: Clinical presentation, diagnosis, and treatment. Seminars in Thrombosis and Hemostasis, 37(6), 673–681. https://doi.org/10.1055/s-0031-1291377
Batista, L. F. Z., Pech, M. F., Zhong, F. L., Nguyen, H. N., Kathleen, T., Zaug, A. J., Crary, S. M., Choi, J., Sebastiano, V., Giri, N., Wernig, M., Alter, B. P., Cech, T. R., Sharon, A., Pera, R. A. R., & Artandi, S. E. (2011). Telomere shortening and loss of self-renewal in dyskeratosis congenita iPS cells. Nature, 474(7351), 399–402. https://doi.org/10.1038/nature10084.Telomere
Battaile, B. K. P., Bateman, R. L., Mortimer, D., Mulcahy, J., Rathbun, R. K., Bagby, G., Fleming, W. H., & Grompe, M. (1999). In vivo selection of wild-type hematopoietic stem cells in a murine model of fanconi anemia. Blood, 94, 2151–2158.
Beck, C. R., Garcia-Perez, J. L., Badge, R. M., & Moran, J. V. (2011). LINE-1 elements in structural variation and disease. Annual Review of Genomics and Human Genetics, 12, 187–215. https://doi.org/10.1146/annurev-genom-082509-141802
Boone, P. M., Bacino, C. A., Shaw, C. A., Eng, P. A., Hixson, P. M., Pursley, A. N., Kang, S.-H.L., Yang, Y., Wiszniewska, J., Nowakowska, B. A., del Gaudio, D., Xia, Z., Simpson-Patel, G., Immken, L. L., Gibson, J. B., Tsai, A.C.-H., Bowers, J. A., Reimschisel, T. E., Schaaf, C. P., Stankiewicz, P., et al. (2010). Detection of clinically relevant exonic copy-number changes by array CGH. Human Mutation, 31(12), 1326–1342. https://doi.org/10.1002/humu.21360
Campbell, I. M., Shaw, C. A., Stankiewicz, P., & Lupski, J. R. (2015). Somatic mosaicism: Implications for disease and transmission genetics. Trends Genetics, 31(7), 382–392. https://doi.org/10.1016/j.tig.2015.03.013.Somatic
Chaganti, R. S. K., Schonberg, S., & German, J. (1974). A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proceedings of the National Academy of Sciences of the United States of America, 71(11), 4508–4512. https://doi.org/10.1073/pnas.71.11.4508
Cheung, S. W., Shaw, C. A., Scott, D. A., Patel, A., Sahoo, T., Bacino, C. A., Pursley, A., Li, J., Erickson, R., Gropman, A. L., Miller, D. T., Seashore, M. R., Summers, A. M., Stankiewicz, P., Chinault, A. C., Lupski, J. R., Beaudet, A. L., & Sutton, V. R. (2007). Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. American Journal of Medical Genetics Part A, 143A(15), 1679–1686. https://doi.org/10.1002/ajmg.a.31740
Choate, K. A., Lu, Y., Zhou, J., Choi, M., Elias, P. M., Farhi, A., Nelson-Williams, C., Crumrine, D., Williams, M. L., Nopper, A. J., Bree, A., Milstone, L. M., & Lifton, R. P. (2010). Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science (new York, n.y.), 330(6000), 94–97. https://doi.org/10.1126/science.1192280
Conlin, L. K., Thiel, B. D., Bonnemann, C. G., Medne, L., Ernst, L. M., Zackai, E. H., Deardorff, M. A., Krantz, I. D., Hakonarson, H., & Spinner, N. B. (2010). Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Human Molecular Genetics, 19(7), 1263–1275. https://doi.org/10.1093/hmg/ddq003
Dokal, I., Bungey, J., Williamson, P., Oscier, D., Hows, J., & Luzzatto, L. (1992). Dyskeratosis congenita fibroblasts are abnormal and have unbalanced chromosomal rearrangements. Blood, 80(12), 3090–3096. https://doi.org/10.1182/blood.v80.12.3090.3090
Dokal, I., & Vulliamy, T. (2003). Dyskeratosis congenita: Its link to telomerase and aplastic anaemia. Blood Reviews, 17(4), 217–225. https://doi.org/10.1016/S0268-960X(03)00020-1
Ellis, N. A., Lennon, D. J., Proytcheva, M., Alhadeff, B., Henderson, E. E., & German, J. (1995). Somatic intragenic recombination within the mutated locus BLM can correct the high sister-chromatid exchange phenotype of bloom syndrome cells. American Journal of Human Genetics, 57(5), 1019–1027.
Ellis, N. A., Ciocci, S., & German, J. (2001). Back mutation can produce phenotype reversion in Bloom syndrome somatic cells. Human Genetics, 108(2), 167–173. https://doi.org/10.1007/s004390000447
Ellis, N. A., Groden, J., Ye, T. Z., Straughen, J., Lennon, D. J., Ciocci, S., Proytcheva, M., & German, J. (1995). The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell, 83(4), 655–666. https://doi.org/10.1016/0092-8674(95)90105-1
Erwin, J. A., Marchetto, M. C., & Gage, F. H. (2014). Mobile DNA elements in the generation of diversity and complexity in the brain. Nature Reviews Neuroscience, 15(8), 497–506. https://doi.org/10.1038/nrn3730
Evrony, G. D., Lee, E., Mehta, B. K., Benjamini, Y., Johnson, R. M., Cai, X., Yang, L., Haseley, P., Lehmann, H. S., Park, P. J., & Walsh, C. A. (2015). Cell lineage analysis in human brain using endogenous retroelements. Neuron, 85(1), 49–59. https://doi.org/10.1016/j.neuron.2014.12.028
Foglesong, J. S., Bannon, S. A., & Dinardo, C. D. (2017). Inherited bone failure syndromes, focus on the haematological manifestations: A review. European Medical Journal, 2(3), 105–112.
Freed, D., Stevens, E. L., & Pevsner, J. (2014). Somatic mosaicism in the human genome. Genes, 5(4), 1064–1094. https://doi.org/10.3390/genes5041064
German, J., Schonberg, S., Louie, E., & Chaganti, R. S. K. (1977). Bloom’s syndrome. IV. Sister chromatid exchanges in lymphocytes. American Journal of Human Genetics, 29(3), 248–255.
German, J., Crippa, L. P., & Bloom, D. (1974). Bloom’s syndrome. III. Analysis of the chromosome aberration characteristic of this disorder. Chromosoma, 48(4), 361–366. https://doi.org/10.1007/BF00290993
Germeshausen, M., Ancliff, P., Estrada, J., Metzler, M., Ponstingl, E., Horst, R., Schwabe, D., & Scott, R. H. (2018). MECOM-associated syndrome : a heterogeneous inherited bone marrow failure syndrome with amegakaryocytic thrombocytopenia. Blood Advances, 2(6), 586–596. https://doi.org/10.1182/bloodadvances.2018016501
Gregory, J. J., Wagner, J. E., Verlander, P. C., Levran, O., Batish, S. D., Eide, C. R., Steffenhagen, A., Hirsch, B., & Auerbach, A. D. (2001). Somatic mosaicism in Fanconi anemia: Evidence of genotypic reversion in lymphohematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America, 98(5), 2532–2537. https://doi.org/10.1073/pnas.051609898
Gross, M., Hanenberg, H., Lobitz, S., Friedl, R., Herterich, S., Dietrich, R., Gruhn, B., Schindler, D., & Hoehn, H. (2002). Reverse mosaicism in Fanconi anemia: Natural gene therapy via molecular self-correction. Cytogenetic and Genome Research, 98(2–3), 126–135. https://doi.org/10.1159/000069805
Hehir-Kwa, J. Y., Rodríguez-Santiago, B., Vissers, L. E., de Leeuw, N., Pfundt, R., Buitelaar, J. K., Pérez-Jurado, L. A., & Veltman, J. A. (2011). De novo copy number variants associated with intellectual disability have a paternal origin and age bias. Journal of Medical Genetics, 48(11), 776–778. https://doi.org/10.1136/jmedgenet-2011-100147
Helderman-van Den Enden, A. T. J. M., Maaswinkel-Mooij, P. D., Hoogendoorn, E., Willemsen, R., Maat-Kievit, J. A., Losekoot, M., & Oostra, B. A. (1999). Monozygotic twin brothers with the fragile X syndrome: Different CGG repeats and different mental capacities. Journal of Medical Genetics, 36(3), 253–257. https://doi.org/10.1136/jmg.36.3.253
Helleday, T., Eshtad, S., & Nik-Zainal, S. (2014). Mechanisms underlying mutational signatures in human cancers. Nature Reviews Genetics, 15(9), 585–598. https://doi.org/10.1038/nrg3729
Hile, S. E., Wang, X., Lee, M. Y. W. T., & Eckert, K. A. (2012). Beyond translesion synthesis: Polymerase κ fidelity as a potential determinant of microsatellite stability. Nucleic Acids Research, 40(4), 1636–1647. https://doi.org/10.1093/nar/gkr889
Hirschhorn, R. (2003). In vivo reversion to normal of inherited mutations in humans. Journal of Medical Genetics, 40(10), 721–728. https://doi.org/10.1136/jmg.40.10.721
Huang, A. Y., Xu, X., Ye, A. Y., Wu, Q., Yan, L., Zhao, B., Yang, X., He, Y., Wang, S., Zhang, Z., Gu, B., Zhao, H. Q., Wang, M., Gao, H., Gao, G., Zhang, Z., Yang, X., Wu, X., Zhang, Y., & Wei, L. (2014). Postzygotic single-nucleotide mosaicisms in whole-genome sequences of clinically unremarkable individuals. Cell Research, 24(11), 1311–1327. https://doi.org/10.1038/cr.2014.131
Iourov, I. Y., Vorsanova, S. G., Liehr, T., Kolotii, A. D., & Yurov, Y. B. (2009a). Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain. Human Molecular Genetics, 18(14), 2656–2669. https://doi.org/10.1093/hmg/ddp207
Iourov, I. Y., Vorsanova, S. G., Zelenova, M. A., Korostelev, S. A., & Yurov, Y. B. (2009b). Aneuploidy in the normal, Alzheimer ’ s disease and ataxia-telangiectasia brain: Differential expression and pathological meaning. Neurobiology of Disease, 34(2), 212–220. https://doi.org/10.1016/j.nbd.2009.01.003
Jongmans, M. C. J., Verwiel, E. T. P., Heijdra, Y., Vulliamy, T., Kamping, E. J., Hehir-Kwa, J. Y., Bongers, E. M. H. F., Pfundt, R., Van Emst, L., Van Leeuwen, F. N., Van Gassen, K. L. I., Geurts Van Kessel, A., Dokal, I., Hoogerbrugge, N., Ligtenberg, M. J. L., & Kuiper, R. P. (2012). Revertant somatic mosaicism by mitotic recombination in dyskeratosis congenita. American Journal of Human Genetics, 90(3), 426–433. https://doi.org/10.1016/j.ajhg.2012.01.004
Kalb, R., Neveling, K., Hoehn, H., Schneider, H., Linka, Y., Batish, S. D., Hunt, C., Berwick, M., Callén, E., Surrallés, J., Casado, J. A., Bueren, J., Dasí, Á., Soulier, J., Gluckman, E., Zwaan, C. M., Van Spaendonk, R., Pals, G., De Winter, J. P., Schindler, D., et al. (2007). Hypomorphic mutations in the gene encoding a key Fanconi anemia protein, FANCD2, sustain a significant group of FA-D2 patients with severe phenotype. American Journal of Human Genetics, 80(5), 895–910. https://doi.org/10.1086/517616
Kamimae-Lanning, A. N., Goloviznina, N. A., & Kurre, P. (2013). Fetal origins of hematopoietic failure in a murine model of Fanconi anemia. Blood, 121(11), 2008–2012. https://doi.org/10.1182/blood-2012-06-439679
Kaufmann, B. P. (1934). Somatic mitoses of Drosophila melanogaster. Journal of Morphology, 56(1), 125–155. https://doi.org/10.1002/jmor.1050560107
Kjeldsen, E., Veigaard, C., Aggerholm, A., & Hasle, H. (2018). Congenital hypoplastic bone marrow failure associated with a de novo partial deletion of the MECOM gene at 3q26. 2. Gene, 656(February), 86–94. https://doi.org/10.1016/j.gene.2018.02.061
Kojis, T. L., Schreck, R. R., Gatti, R. A., & Sparkes, R. S. (1989). Tissue specificity of chromosomal rearrangements in ataxia-telangiectasia. Human Genetics, 83(4), 347–352. https://doi.org/10.1007/BF00291379
Kwee, M. L., Poll, E. H. A., van de Kamp, J. J. P., de Koning, H., Eriksson, A. W., & Joenje, H. (1983). Unusual response to bifunctional alkylating agents in a case of Fanconi anaemia. Human Genetics, 64, 384–387.
Lemmers, R. J. L. F., Tawil, R., Petek, L. M., Balog, J., Block, G. J., Santen, G. W. E., Amell, A. M., Van Der Vliet, P. J., Almomani, R., Straasheijm, K. R., Krom, Y. D., Klooster, R., Sun, Y., Den Dunnen, J. T., Helmer, Q., Donlin-Smith, C. M., Padberg, G. W., Van Engelen, B. G. M., De Greef, J. C., Van Der Maarel, S. M., et al. (2012). Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nature Genetics, 44(12), 1370–1374. https://doi.org/10.1038/ng.2454
Lemmers, R. J. L. F., Van Der Vliet, P. J., Klooster, R., Sacconi, S., Camaño, P., Dauwerse, J. G., Snider, L., Straasheijm, K. R., Van Ommen, G. J., Padberg, G. W., Miller, D. G., Tapscott, S. J., Tawil, R., Frants, R. R., & Van Der Maarel, S. M. (2010). A unifying genetic model for facioscapulohumeral muscular dystrophy. Science, 329(5999), 1650–1653. https://doi.org/10.1126/science.1189044
Lemmers, R. J. L. F., van der Wielen, M. J. R., Bakker, E., Padberg, G. W., Frants, R. R., & van der Maarel, S. M. (2004). Somatic mosaicism in FSHD often goes undetected. Annals of Neurology, 55(6), 845–850. https://doi.org/10.1002/ana.20106
Lo Ten Foe, J. R., Kwee, M. L., Rooimans, M. A., Oostra, A. B., Veerman, A. J. P., Van Weel, M., Pauli, R. M., Shahidi, N. T., Dokal, I., Roberts, I., Altay, C., Gluckman, E., Gibson, R. A., Mathew, C. G., Arwert, F., & Joenje, H. (1997). Somatic mosaicism in Fanconi anemia: Molecular basis and clinical significance. European Journal of Human Genetics, 5(3), 137–148. https://doi.org/10.1159/000484749
Lupski, J. R. (2012). Digenic inheritance and Mendelian disease. Nature Genetics, 44(12), 1291–1292. https://doi.org/10.1038/ng.2479
Machiela, M. J., & Chanock, S. J. (2014). Detectable clonal mosaicism in the human genome. Seminars in Hematology, 50(4), 1–21. https://doi.org/10.1053/j.seminhematol.2013.09.001
MacMillan, M. L., Auerbach, A. D., Davies, S. M., DeFor, T. E., Gillio, A., Giller, R., Harris, R., Cairo, M., Dusenbery, K., Hirsch, B., Ramsay, N. K. C., Weisdorf, D. J., & Wagner, J. E. (2000). Haematopoietic cell transplantation in patients with Fanconi anaemia using alternate donors: Results of a total body irradiation dose escalation trial. British Journal of Haematology, 109(1), 121–129. https://doi.org/10.1046/j.1365-2141.2000.01955.x
Mankad, A., Taniguchi, T., Cox, B., Akkari, Y., Rathbun, R. K., Lucas, L., Bagby, G., Olson, S., D’Andrea, A., & Grompe, M. (2006). Natural gene therapy in monozygotic twins with Fanconi anemia. Blood, 107(8), 3084–3090. https://doi.org/10.1182/blood-2005-07-2638
McClintock, B. (1932). A correlation of ring-shaped chromosomes with variegation in zea mays. Proceedings of the National Academy of Sciences, 18(12), 677–681. https://doi.org/10.1073/pnas.18.12.677
McKinnon, P. J. (2004). ATM and ataxia telangiectasia. Second in molecular medicine review series. EMBO Reports, 5(8), 772–776. https://doi.org/10.1038/sj.embor.7400210
McMurray, C. T. (2010a). Mechanisms of trinucleotide repeat instability during human development. Nature Reviews Genetics, 11(11), 786–799. https://doi.org/10.1038/nrg2828
McMurray, J. J. V. (2010b). Systolic heart failure. New England Journal of Medicine, 362(3), 228–238. https://doi.org/10.1056/nejmcp0909392
Messiaen, L., Vogt, J., Bengesser, K., Fu, C., Mikhail, F., Serra, E., Garcia-Linares, C., Cooper, D. N., Lazaro, C., & Kehrer-Sawatzki, H. (2011). Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1). Human Mutation, 32(2), 213–219. https://doi.org/10.1002/humu.21418
Nicoletti, E., Rao, G., Bueren, J. A., Río, P., Navarro, S., Surrallés, J., Choi, G., & Schwartz, J. D. (2020). Mosaicism in Fanconi anemia: Concise review and evaluation of published cases with focus on clinical course of blood count normalization. Annals of Hematology, 99(5), 913–924. https://doi.org/10.1007/s00277-020-03954-2
Niihori, T., Ouchi-Uchiyama, M., Sasahara, Y., Kaneko, T., Hashii, Y., Irie, M., Sato, A., Saito-Nanjo, Y., Funayama, R., Nagashima, T., Inoue, S. I., Nakayama, K., Ozono, K., Kure, S., Matsubara, Y., Imaizumi, M., & Aoki, Y. (2015). Mutations in MECOM, encoding oncoprotein EVI1, cause radioulnar synostosis with amegakaryocytic thrombocytopenia. American Journal of Human Genetics, 97(6), 848–854. https://doi.org/10.1016/j.ajhg.2015.10.010
Osumi, T., Tsujimoto, S. I., Nakabayashi, K., Taniguchi, M., Shirai, R., Yoshida, M., Uchiyama, T., Nagasawa, J., Goyama, S., Yoshioka, T., & Tomizawa, D. (2018). Somatic MECOM mosaicism in a patient with congenital bone marrow. Pediatric Blood & Cancer, 65(6), 1–3. https://doi.org/10.1002/pbc.26959
Patterson, J. T. (1929). The production of mutations in somatic cells of drosophila melanogaster by means of X-rays1. THB JOURNAL OF EXPEEIYENTAL ZOOLOGY, 53(3), 327–372.
Peto, F. H. (1935). Associations of somatic chromosomes induced by heat and chloral hydrate treatments. Canadian Journal of Research, 13, 301–314.
Pfeifer, G. P., You, Y. H., & Besaratinia, A. (2005). Mutations induced by ultraviolet light. Mutation Research Fundamental and Molecular Mechanisms of Mutagenesis, 571(1–2 SPEC. ISS), 19–31. https://doi.org/10.1016/j.mrfmmm.2004.06.057
Pham, J., Shaw, C., Pursley, A., Hixson, P., Sampath, S., Roney, E., Gambin, T., Kang, S.-H.L., Bi, W., Lalani, S., Bacino, C., Lupski, J. R., Stankiewicz, P., Patel, A., & Cheung, S.-W. (2014). Somatic mosaicism detected by exon-targeted, high-resolution aCGH in 10,362 consecutive cases. European Journal of Human Genetics : EJHG, 22(8), 969–978. https://doi.org/10.1038/ejhg.2013.285
Pleasance, E. D., Stephens, P. J., O’Meara, S., McBride, D. J., Meynert, A., Jones, D., Lin, M. L., Beare, D., Lau, K. W., Greenman, C., Varela, I., Nik-Zainal, S., Davies, H. R., Ordõez, G. R., Mudie, L. J., Latimer, C., Edkins, S., Stebbings, L., Chen, L., Campbell, P. J., et al. (2010). A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature, 463(7278), 184–190. https://doi.org/10.1038/nature08629
Poole, S. R., Smith, A. C. M., Hays, T., McGavran, L., & Auerbach, A. D. (1992). Monozygotic twin girls with congenital malformations resembling Fanconi anemia. American Journal of Medical Genetics, 42(6), 780–784. https://doi.org/10.1002/ajmg.1320420606
Raya, Á., Rodríguez-Piz, I., Guenechea, G., Vassena, R., Navarro, S., Barrero, M. J., Consiglio, A., Castell, M., Río, P., Sleep, E., González, F., Tiscornia, G., Garreta, E., Aasen, T., Veiga, A., Verma, I. M., Surrallés, J., Bueren, J., & Belmonte, J. C. I. (2009). Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature, 460(7251), 53–59. https://doi.org/10.1038/nature08129
Rickman, K. A., Lach, F. P., Abhyankar, A., Donovan, F. X., Sanborn, E. M., Kennedy, J. A., Sougnez, C., Gabriel, S. B., Elemento, O., Chandrasekharappa, S. C., Schindler, D., Auerbach, A. D., & Smogorzewska, A. (2015). Deficiency of UBE2T, the E2Ubiquitin ligase necessary for FANCD2 and FANCI ubiquitination, causes FA-T subtype of fanconi anemia. Cell Reports, 12(1), 35–41. https://doi.org/10.1016/j.celrep.2015.06.014
Rifé, M., Badenas, C., Quintó, L., Puigoriol, E., Tazón, B., Rodriguez-Revenga, L., Jiménez, L., Sánchez, A., & Milà, M. (2004). Analysis of CGG variation through 642 meioses in Fragile X families. MHR: Basic Science of Reproductive Medicine, 10(10), 773–776. https://doi.org/10.1093/molehr/gah102
Río, P., Navarro, S., Guenechea, G., Sánchez-Domínguez, R., Lamana, M. L., Yañez, R., Casado, J. A., Mehta, P. A., Pujol, M. R., Surrallés, J., Charrier, S., Galy, A., Segovia, J. C., Díaz De Heredia, C., Sevilla, J., & Bueren, J. A. (2017). Engraftment and in vivo proliferation advantage of gene-corrected mobilized CD34+ cells from Fanconi anemia patients. Blood, 130(13), 1535–1542. https://doi.org/10.1182/blood-2017-03-774174
Shao, C., Stambrook, P. J., & Tischfield, J. A. (2001). Mitotic recombination is suppressed by chromosomal divergence in hybrids of distantly related mouse strains. Nature Genetics, 28(2), 169–172. https://doi.org/10.1038/88897
Shapiro, J. A. (1979). Molecular model for the transposition and replication of bacteriophage Mu and other transposable elements. Proceedings of the National Academy of Sciences of the United States of America, 76(4), 1933–1937. https://doi.org/10.1073/pnas.76.4.1933
Stern, C. (1936). Somatic crossing over and segregation in drosophila melanogaster. Genetics, 21(6), 625–730. https://doi.org/10.1093/genetics/21.6.625
Strathdee, C. A., Gavish, H., & Shannon, W. R. (1992). Cloning of cDNAs for Fanconi’s anaemia by functional complementation. Nature, 356, 763–767.
Suzuki, S., Racine, R. R., Manalo, N. A., Cantor, S. B., & Raffel, G. D. (2017). Impairment of fetal hematopoietic stem cell function in the absence of Fancd2. Experimental Hematology, 48(508), 79–86. https://doi.org/10.1016/j.exphem.2016.11.006
Swami, M., Hendricks, A. E., Gillis, T., Massood, T., Mysore, J., Myers, R. H., & Wheeler, V. C. (2009). Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Human Molecular Genetics, 18(16), 3039–3047. https://doi.org/10.1093/hmg/ddp242
Thompson, A. A., & Nguyen, L. T. (2000). Amegakaryocytic thrombocytopenia and radio-ulnar synostosis are associated with HOXA11 mutation. Nature Genetics, 26(4), 397–398. https://doi.org/10.1038/82511
Thompson, A. A., Woodruff, K., Feig, S. A., Nguyen, L. T., & Carolyn Schanen, N. (2001). Congenital thrombocytopenia and radio-ulnar synostosis: A new familial syndrome. British Journal of Haematology, 113(4), 866–870. https://doi.org/10.1046/j.1365-2141.2001.02834.x
Tulpule, A., William Lensch, M., Miller, J. D., Austin, K., D’Andrea, A., Schlaeger, T. M., Shimamura, A., & Daley, G. Q. (2010). Knockdown of Fanconi anemia genes in human embryonic stem cells reveals early developmental defects in the hematopoietic lineage. Blood, 115(17), 3453–3462. https://doi.org/10.1182/blood-2009-10-246694
Turner, D. J., Miretti, M., Rajan, D., Fiegler, H., Carter, N. P., Blayney, M. L., Beck, S., & Hurles, M. E. (2008). Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nature Genetics, 40(1), 90–95. https://doi.org/10.1038/ng.2007.40
Van Der Maarel, S. M., Deidda, G., Lemmers, R. J. L. F., Van Overveld, P. G. M., Van Der Wielen, M., Hewitt, J. E., Sandkuijl, L., Bakker, B., Van Ommen, C. J. B., Padberg, G. W., & Frants, R. R. (2000). De novo facioscapulohumeral muscular dystrophy: Frequent somatic mosaicism, sex-dependent phenotype, and the role of mitotic transchromosomal repeat interaction between chromosomes 4 and 10. American Journal of Human Genetics, 66(1), 26–35. https://doi.org/10.1086/302730
Venugopal, P., Babic, M., Barnett, C., Bray, S., Brown, A., Cheah, J., D’Andrea, R., Feng, J., Fine, M., George, A., Guandalini, M., Hahn, C., Hannan, R., Lawrence, D., Moore, S., Philips, K., Poplawski, N., Saxon, B., Schreiber, A., Young, C. C., et al. (2019). Implications of revertant somatic mosaicism in bone marrow failure syndromes. Experimental Hematology, 76, S90. https://doi.org/10.1016/j.exphem.2019.06.451
Verdun, R. E., & Karlseder, J. (2007). Replication and protection of telomeres. Nature, 447(7147), 924–931. https://doi.org/10.1038/nature05976
Virts, E. L., Jankowska, A., Mackay, C., Glaas, M. F., Wiek, C., Kelich, S. L., Lottmann, N., Kennedy, F. M., Marchal, C., Lehnert, E., Scharf, R. E., Dufour, C., Lanciotti, M., Farruggia, P., Santoro, A., Savasan, S., Scheckenbach, K., Schipper, J., Wagenmann, M., Hanenberg, H., et al. (2015). AluY-mediated germline deletion, duplication and somatic stem cell reversion in UBE2T defines a new subtype of Fanconi anemia. Human Molecular Genetics, 24(18), 5093–5108. https://doi.org/10.1093/hmg/ddv227
Vulliamy, T. J., & Dokal, I. (2008). Dyskeratosis congenita: The diverse clinical presentation of mutations in the telomerase complex. Biochimie, 90(1), 122–130. https://doi.org/10.1016/j.biochi.2007.07.017
Vulliamy, T. J., Knight, S. W., Heiss, N. S., Smith, O. P., Poustka, A., Dokal, I., & Mason, P. J. (1999). Dyskeratosis congenita caused by a 3’ deletion: Germline and somatic mosaicism in a female carrier. Blood, 94(4), 1254–1260. https://doi.org/10.1182/blood.v94.4.1254
Wu, Z. H. (2016). Phenotypes and genotypes of the chromosomal instability syndromes. Translational Pediatrics, 5(2), 79–83. https://doi.org/10.21037/tp.2016.03.04
Yildirim, A. T., Güneş, B. T., Oymak, Y., Yaman, Y., Özek, G., Carti, Ö., Yeşilipek, A., & Vergin, C. (2015). Congenital amegakaryocytic thrombocytopenia: Three case reports from patients with different clinical diagnoses and somatic abnormalities. Blood Coagulation and Fibrinolysis, 26(3), 337–341. https://doi.org/10.1097/MBC.0000000000000192
Zhao, T., Zhao, T., Chen, M., & Chen, M. (2021). A case report of congenital non-megakaryocytic thrombocytopenia with inflammatory lesions of ascending colon and ileocecal area and literature review Congenital amegakaryocytic thrombocytopenia with inflammatory disease of ascending colon and ileocecum. Chinese Journal of Hematology, 41(201), 762–765. https://doi.org/10.3760/cma.j.issn.0253-2727.2020.09.010
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
PB: Writing- Original draft preparation. RSV: Supervision, Writing- Reviewing and Editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Biswas, P., Verma, R.S. Somatic mosaicism in inherited bone marrow failure and chromosomal instability syndrome. GENOME INSTAB. DIS. 2, 150–163 (2021). https://doi.org/10.1007/s42764-021-00041-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42764-021-00041-2