
Abstract
To study the correlation between root microbiome and its community structure and the health, survival, and growth of its host which is the key to solve the problem of the diversity relationship between wild plants and root microorganism. In this study, we used high-throughput techniques of next generation sequencing (NGS) which was applied to study the endophytic and rhizosphere bacterial and fungal community in hulless barley (Hordeum vulgare) plants, by assessing its PCR amplicon of 16S rDNA sequences and ITS region. The results of the principal component analysis (PCA) showed that bacterial phyla Proteobacteria, Actinobacteria, and Acidobacteria dominate the bacterial community and that the phyla of Ascomycota and Basidiomycota dominate the mycobiota community in the root-soil interface of hulless barley. In both 16S and ITS data, the alpha diversity in bulk soil samples was significantly higher than that of rhizosphere and root samples, and root sample was least diverse, suggesting the microbial selection from the plant host. Beta diversity analysis indicated a clear separation from samples with different sample types (bulk soil, rhizosphere, and root samples). Lastly, the overall microbiota profile and differentially presented taxa were studied to assess the function. It can be concluded that the microbial diversity of wild hulless barley in different soil samples was significantly different and related to host genotypes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
As major staple crops, barley appeared early in the history of domestication of human agriculture, but surprisingly, it is one of the least utilized cereal for human food consumption worldwide (Achatz et al. 2010). During the period of intensive crop improvement in the twentieth century, barley was largely neglected by plant breeders in Europe due to multiple reasons. In recent decades, it gained great attention back worldwide as an economic effective food which is also good for health (Bhatty 1999a). Barley grains are known to possess healthy related minerals and proteins and have high β-glucan content, which could inhibit cholesterol synthesis (Bhatty 1999b; Schreiber et al. 2014). Barley is normally considered as the third option after wheat and rice by the local farmers, unless it is specifically used for cultural and religious purposes. As one of the main regions of domestication and diversity of cultivated barley, the Qinghai-Tibet Plateau in western China has abundant hulless barley resources (Narwal et al. 2017). Hulless barley is the main ingredient for traditional Tibetan food and it is also an indispensable element for the local religion with great demands.
Low production yield and high disease rate compared with other cereal products are the main disadvantages of hulless barley cultivation. In the past, decades of efforts were taken to tackle the problem. Pesticides and fertilizers were applied since the modern agriculture era began but often led to the rise of other problems such as pesticide residues and soil compaction. Besides, people continuously cultivate wild hulless barley populations to develop cultivars with increased grain yield (Yang et al. 2008; Zeng 2015). Prominent cultivars include ZQ2000, ZQ320, and ZQ690, which are also the research objectives in this study. The emergence of these new hulless barley cultivars greatly contribute to the overall crop yield in the Tibetan Plateau area wherein the total crop production reached one million tons in 2015. Especially for ZQ2000, its growing area has accounted for more than half of the cultivated area of barley, which exceeded 100 million acres in 2016. Alternatively, the availability of draft genome sequences and transcriptome study of the hulless barley offered a fundamental basis for solving the yield problem with genetic approaches (Chen et al. 2014; Zeng et al. 2015). Many other crop species have been shown successfully with the application of those genetic modification approaches to prevent them from diseases and hence to improve the yield. Recently, utilizing specific microbes to optimize the crop productivity became an available approach, especially for the crops in the area with unfavorable weather or stress-related conditions (van der Heijden et al. 2008; Latch 1993; Lugtenberg and Kamilova 2009).
Root endophyte is a group of endosymbiotic microorganisms living in intercellular and intracellular spaces of a compartment inside of plant root (Miliute et al. 2015; Murphy et al. 2014). Cells from root constantly secrete a plethora of photosynthesis-derived organic compounds into its surrounding rhizosphere (soil attached with the root) during the process of rhizodeposition, which helps the surrounding attached soil to sustain with a stable composition of microbiota (Jones et al. 2004). Microbiota that exists in both rhizosphere and endophytic compartment normally does not cause plant disease or significant morphological changes in plants. Increasing evidence has even demonstrated that those microbes acted an important role in plant development as mutualists and commensals, contributing many essential functionalities, such as to enhance host growth and nutrient acquisition and to improve the plant’s ability to tolerate abiotic stresses (Tkacz and Poole 2015). For instances, some Proteobacteria species in endophytic and rhizospheric Rhizobiales transformed N2 from the atmosphere into plant-available ammonia for promoting crop growth. Soil bacteria and fungi are involved in crops’ carbon cycling or soil formation, which could further boost the crops’ nutrient acquisition (Kogel et al. 2006). In addition, soil fungi promote the absorption of phosphorus in crops. Ninety percent of phosphorus acquisition is accomplished by fungi and phosphorus-solubilizing bacteria in many plants. The same goes the plant nitrogen acquisition in which soil uptake from the microbes contributed to about 80% of the total amount (van der Heijden et al. 2008). In the aspect of disease resistance, the endophytic and rhizosphere microbes also directly or indirectly contribute to the generation of various disease resistance–related chemical compounds and fungitoxic metabolites (van der Heijden et al. 2008). Collaborating with each other, soil-related microbes and crops could regulate crops’ productivity via plant symbionts especially in the condition of poor nutrition. Thus, close inspection and characterization of the molecular composition and the research of potential mechanisms underlying plant and root microbe community associations are necessary and beneficial for agriculture-related purposes.
With the advancement of next generation sequencing (NGS) technology, new aspects of the microbial diversity have emerged with the application of amplicon and metagenomic sequencing methods in the studies of microbial endophytes (Gottel et al. 2011; Kaul et al. 2016). By analyzing the association of microbes and environmental metadata, it is known that endophytic microbial community structure is linked with factors such as plant genotype, abiotic, and biotic factors including environmental conditions, microbe–microbe interactions and plant–microbe interactions in many other plants (Hardoim et al. 2015). In this study, high-throughput techniques of NGS were applied to the study of endophytic bacterial and fungal community in hulless barley plant by Illumina 16S and ITS sequencing. We identified abundant Proteobacteria, Actinobacteria, and Bacteroidetes species in all 16S samples, and Ascomycota and Basidiomycota are constantly presented in all ITS samples. We also confirmed that the diversity was progressively decreased from bulk soil sample to rhizosphere samples and then lastly root samples, which is in accordance with previous studies of other plants (Hauben et al. 1999). Lastly, the function of the Operational Taxonomic Units (OTUs) was predicted and projected to the physical characterization of cultivars of hulless barley to assess its potential association.
2 Materials and Methods
2.1 Study Site and Sample Collection
Surface-sterilized seeds of hulless barley (Hordeum vulgare L.) cultivars ZQ320, ZQ690, and ZQ2000 were sown onto pots filled with natural field soil collected from the Agricultural and Animal Husbandry College of Tibet University in 2015. The sample collection was repeated twice to collect the root and rhizosphere soil material for 16S and ITS sequencing. For each variety, three biological replicates were prepared. Soil and root samples collected were carried out on the same day simultaneously when the plants were ranging from 110 to 125 days old. A combination of washing and ultrasound treatments was employed to simultaneously separate the rhizosphere fraction from the roots to enrich root endophytes. In parallel, bulk soil controls (pots filled with the same soil and exposed to the same environmental conditions as the plant-containing pots) were prepared. DNA was extracted and prepared for sequencing libraries from samples following the previously described method (Fadrosh et al. 2014).
2.2 OTU Generation and Annotation
16S and ITS amplicon sequencing were performed on the Illumina MiSeq platform. The quality of raw reads was analyzed by FastQC v0.11.5 software. Regions with low complexity and low quality scores were trimmed using SeqTK v1.2 software. After trimming, clean sequences were subjected to the QIIME2 (2017.10 version) pipeline (Caporaso et al. 2010). Briefly, reads were demultiplexed using the QIIME2 demux plugin according to their barcode sequence. Demultiplexed sequences were further quality filtered and clustered using QIIME2 DADA2 plugin to generate the OTU table. The representative sequences for each OTU were aligned using QIIME2 alignment plugin then QIIME2 phylogeny plugin was applied to construct the rooted phylogenetic tree by employing the FastTree program. The taxonomic analysis was carried out with the QIIME2 feature-classifier plugin, using the Greengenes database at the similarity threshold of 99% (for 16S data) and UNITE database developer version (for ITS data) (DeSantis et al. 2006; Kõljalg et al. 2005). The chloroplast- and mitochondria-related OTUs were excluded from the 16S downstream analysis, and the non-fungal OTUs were excluded from the ITS downstream analysis. The generated BIOM file and phylogenetic trees were further imported into Phyloseq for comparison and visualization (McMurdie and Holmes 2013).
2.3 Statistical Analysis of Microbiome Community Profiles
A variety of alpha and beta diversity indexes (observed OTUs, Shannon, PD whole tree, evenness, Jaccard distance, Bray-Curtis distance, unweighted UniFrac distance, and weighted UniFrac distance) were assessed using QIIME2 diversity core-metrics-phylogenetic plugin, and their differences among groups of interest were calculated by pairwise Kruskal-Wallis analysis and pairwise permutational multivariate analysis of variance (PERMANOVA) using alpha-group-significance and beta-group-significance commands respectively (Anderson and Walsh 2013). Also, to compare and visualize those above alpha diversity metrics at multiple sample depth, QIIME2 diversity alpha-rarefaction plugin was utilized. Besides the overall diversity comparison among samples, the individual OTU was examined to see if any are represented differently among groups of interest: OTUs were collapsed into different classification levels and then the QIIME2 composition plugin was used to recruit analysis of composition of microbiomes (ANCOM) to calculate the significance (Mandal et al. 2015).
2.4 Measurements for Nutrition Ingredient
Beta-glucan was measured following GB 5009.2 protocol. Protein content was measured following the Kjeldahl method. Water content was measured following the GB 5009.3 protocol. Ash content was measured following the GB 5009.4 protocol. Fat content was measured following the GB 5009.6 protocol. Starch content was measured following the GB/T 5009.7 protocol. To note, all GB protocols are recorded in the National Food Safety Standard Determination of materials in foods (China).
2.5 Functional Prediction Analysis
Metagenomic inference and functional analysis of 16S data were performed using PICRUSt (Langille et al. 2013). PICRUSt utilizes a computational approach to predict the potential metagenome using the 16S data. Output data from PICRUSt were filtered and re-formatted using the microbiome helper package. Differences were calculated between groups for each metabolic pathway to estimate the functional differences comparing different sample types in the STAMP software.
3 Results
3.1 Composition of Hulless Barley Bacteria Community
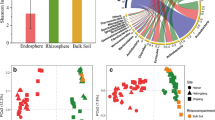
The 16S amplicon sequencing of the root and rhizosphere samples from three hulless barley cultivars, along with the bulk soil samples, yielded 1,170,139 high-quality, nonchimeric sequences across all samples, with a median of 56,500 (range 38,276–65,915) sequence frequency per sample (Fig. 1 and Supplementary Table 1). Using QIIME2 DADA2 denoise-single plugin, 9017 OTUs were identified. The majority of the 16S findings are bacteria, but a small portion of species detected were assigned to the kingdom of Archaea (0.79%). We assessed the taxonomic distributions of identified OTUs at different levels. With respect to the level of phylum, all samples contained abundant Proteobacteria, Actinobacteria, and Bacteroidetes species, which accounted for 49.8%, 10.8%, and 10.8% of the entire community. Among them, the Proteobacteria is the most abundant phylum, and it was also significant to discriminate the bulk soil samples from plant rhizosphere and root samples using ANCOM (q value = 0.032). As described in the heat map (Supplementary Fig. 1), Proteobacteria, along with Actinobacteria and Bacteroidetes, formed a cluster that abundantly appears in all samples. The second cluster consisted of phyla Chloroflexi, Acidobacteria, Planctomycetes, Gemmatimonadetes, and Verrucomicrobia, which exhibited a greater abundance when comparing root samples to bulk soil and rhizosphere samples.

The stacked bar chart for bacteria distribution at the phylum level. The Y axis explains the proportion of phylum. The X axis represents different samples
We confirmed that the endophyte microbiome in hulless barley followed the typical patterns that were observed in other similar root microbial studies. The taxa distribution of soil microbiome was rich and phylogenetically more diverse than the endophytic microbiome, as indicated by the alpha diversity of 8.8 for the root samples and 9.3 for the bulk soil and rhizosphere samples measured by the average Shannon’s diversity index (Fig. 2). The pairwise Kruskal-Wallis analysis also indicated that Shannon’s diversity of root microbiome was significantly different from the rhizosphere (q value = 0.001046) and the bulk soil (q value = 0.018832). Also, for all three hulless barley cultivars, they exhibited similar but a higher diversity in the rhizosphere comparing with its endophytic microbiome, which was in accordance with previous research as well (Hauben et al. 1999). Using principal coordinates analysis (PCoA) on weighted UniFrac distances, we quantified the major components driving differences between samples and found a clear separation along axis 1 (explaining 59.53% of the overall variation) and confirmed the general pattern that soil and roots harbor distinct microbiome (Fig. 3). Axis 2 explained 9.273% of the variation overall and partially separated the bulk soil samples from the root rhizosphere samples, although we did not notice an obvious separation as one of the bulk soil samples mingle with the cluster rhizosphere samples, suggesting negligible effects of the plant host on beta diversity. Pairwise PERMANOVA results indicated that the beta diversity among soil types was significantly different in all three pairwise comparisons (q valueroot vs. rhizosphere = 0.003, q valuebulksoil vs. rhizosphere = 0.0075, q valuebulksoil vs. root = 0.012). Possible effects due to variety difference were generally not obvious as indicated by both alpha diversity and beta diversity analysis. In the following, we break down the dissimilarities between soil and root samples to compositional patterns evident in the taxonomic profiles of the samples.

The box plot of bacterial Shannon’s diversity index distribution of different sample types. The X axis represents the samples types and the number of samples in the specific sample type. b stands for bulk soil sample, r stands for root sample, and s stands for rhizosphere sample. The Y axis represents the Shannon’s diversity index

The PCoA plot for the bacterial weighted unifraction distance comparing different sample types. The color of the dot represents the sample type
For detailed characterization of the hulless barley root microbiome, the OTUs were collapsed into different taxonomy levels. Using ANCOM, four were identified as significantly different. Namely, Pseudomonas veronii at the species level and Rubrivivax and Stenotrophomonas at the genus level were detected most enriched in the rhizosphere and endophyte microbial community of all three cultivars but few sequences were detected in the bulk soil. Comamonas at the genus level was detected prevalent in the bulk soil but not in the rhizosphere and endophytic samples. Pedosphaera at the genus level, Gemmataceae and Pedosphaeraceae at the family level, and BD7–11 at the class level were detected most abundant in the bulk soil sample, then less in the rhizosphere and almost vanished in root samples. In contrast, Pseudomonas veronii at the species, Rubrivivax and Stenotrophomonas at the genus level, and TM7–3 at the level of class were detected to be depleted in bulk soil samples, then the abundance gradually increased from rhizosphere samples to the root samples.
3.2 Composition of Hulless Barley Fungal Community
We have also examined the fungal community by running the ITS sequencing with a similar experimental design used for the 16S analysis above. The MiSeq run yielded 517,990 high-quality, nonchimeric sequences across all samples, with a median of 20,789 (range 1675–54,947) sequence frequency per sample (Supplementary Table 2). We identified 2305 OTUs in total across all 21 samples. The taxonomy of ITS amplicons shows that 11 phylum mycobiota were founded, including five main phyla: Ascomycota, Basidiomycota, Mortierellomycota, Mucoromycota, and Rozellomycota. Of these, Ascomycota was the predominant mycobiota which makes up 62.5% of the relative abundance when the unclassified OTU were excluded.
To gain insights into the richness of the microbiota, we compared the total number of observed OTUs, Chao1, and the Shannon diversity indices of the communities retrieved from bulk soil and plant-associated microhabitats. All the indices revealed a significant reduction of the fungus richness and diversity in the root samples (Kruskal-Wallis, q value = 0.002), while the rhizosphere’s displayed an intermediate composition between soil and root samples. The rarefaction curve of observed OTU index showed that most microbes could be captured when the ITS rDNA sequences more than 2000 for each sample. The measures of within sample diversity using Shannon and Faith PD index indicated that the diversity decreased from rhizosphere to root for hulless barley, and the bulk soil had the highest alpha diversity (Supplementary Fig. 1). This result was consistent with previous observations in rice and barley (Bulgarelli et al. 2015; Murphy et al. 2014; Sengupta et al. 2017). Principal coordinates analysis plotted to visualize the differences among groups of samples was performed based on the weighted unifraction distances. The rhizosphere compartments were separated across the first principal coordinate, indicating the largest source of variation in root-associated mycobiota communities. All root samples were grouped into one tight cluster, in which the axis 1 exhibits an obvious separation from bulk soil and rhizosphere samples. The axis 2 drives the separation of bulk soil samples and rhizosphere sample though one of the rhizosphere samples is mingled with the bulk soil cluster. The root samples from three cultivars were grouped together and separated with rhizosphere soil samples. While the bulk soil samples and rhizosphere soil samples from three cultivars grouped together show high similarity (Supplementary Fig. 2).
By studying the differently represented OTU from different levels using ANCOM, the only identified OTU was Ceratobasidiaceae (family level) that it was only found in ZQ320 samples but depleted in the other two cultivars nor the bulk soil samples.
3.3 Endophyte Bacterial and Fungal May Influence the Growth of Hulless Barley
The plant from these three cultivars (ZQ2000, ZQ690, and ZQ320) exhibited similar phenotype, but ZQ320 was known with better cold tolerance and ZQ690 was known with better drought tolerance. Comparing with ZQ2000 and ZQ690, ZQ320 plants seemed to have a higher number of solid grains per ear (57.5), and the average weight was 50 g per thousand grains. The yield of ZQ690 was the most in three cultivars (100.05 kg/mu), and the most yield per plant (1.45 kg). The nutrition profile of three cultivars seeds was also obtained and measured using biochemistry approaches (Table 1).
In order to obtain an insight of the relationship between the root microbiome with the yield and the food quality of the plant, we analyzed and predicted the potential functionality of OTUs. Firstly, we explored specifically the predicted functional capacity of the microbiota involved using PICRUSt, which predicts the functional capacity of a community based on 16S rRNA data. The results predicted a number of KEGG gene orthologs (KOs; Kyoto Encyclopedia of Genes and Genomes, release 67.1) associated with known KEGG pathways to be enriched or depleted within the bacterial community (Fig. 4). Predicted functions indicated that the majority of predicted functions were enriched for metabolism including pathways involved in amino acid, carbohydrate, energy, vitamins, and lipid metabolism as well as those involved in xenobiotics biodegradation metabolism. In the second level of the KEGG pathway, the most abundant function was membrane transport, indicating that both entophytic and rhizosphere microbiome played an important role in nutrition transportation for the plant. Compared with bacteria, fungus was known to be more active in improving the growth of hulless barley. Several studies have demonstrated that mycorrhizal fungi could improve crop water and phosphorus acquisition through the exchange for carbon. Compared with similar reactions known through bacteria, this mechanism performs more efficiently for phosphate acquisition. Ascomycota and Basidiomycota were the two most abundant phyla in our study. In barley, endophytic Ascomycota promoted resistance to pathogen and tolerance to abiotic stresses. Besides improving disease resistance, endophytic Basidiomycota provided increased biomass and seed yield for barley. Though the correlation between the relative abundance of the above two phyla with the yield and nutrition contents was not detected significantly, we still believe they are indispensable for the growth of hulless barley.

The predicted function (level 1) distribution from PICRUSt. The Y axis explains the relative abundance. The X axis explains the sample names
4 Discussion
Using metagenomics approaches, people have obtained a better understanding of the diversity of microbes in various habitats in the recent decade (van der Heijden et al. 2008). It includes microbes associated with plants, which thrive under ground in the rhizosphere and microbes inside of plant tissues as endophytes. The interaction of microbes between root and rhizosphere plays a unique role in plant development. The microbiota inhabiting this niche may provide the plant with physiologically accessible nutrients and phytohormones that improve plant growth, may suppress phytopathogens, or may help plants withstand heat, salt, and drought; meanwhile, they can dramatically undermine plant health by introducing several pathogen-related diseases. With further understanding of the function of endophytic and rhizosphere microbiota of plants, how to effectively shift this balance of microbiota composition in the root and rhizosphere niche became an important agronomic interest in supporting plant growth and improved crop yield. In this study, we characterized the rhizosphere and endophytic microbial community composition of three popular hulless barley plant cultivars, along with comparable bulk soil samples as control. Our design permitted us to test the influence of hulless barley host genotype on its endophytic and rhizosphere microbial community across the field environments. The alpha and beta diversity were closely looked and compared among different compartment and cultivars. Together, these results further support the hypothesis that the barley rhizosphere and root are two microhabitats colonized by communities with taxonomically distinct profiles, which emerge from the soil biota through progressive differentiation. The study also allowed us to assess the degree to how these bacterial species interact with fungal species, and hence result in a possible function.
Regarding the 16S analysis, Proteobacteria, Bacteroidetes, Actinobacteria, Verrucomicrobia, and Acidobacteria were the most dominant five phyla in the rhizosphere and root communities, representing 83.2% of the total sequences at the level of phylum. Of note, other members of the soil biota, such as Firmicutes and Chloroflexi, were almost excluded from the plant-associated assemblages but took a higher proportion in the bulk soil samples. In the meantime, a small portion of sequences were identified from the domain of archaea, this was because the Greengenes database we used in taxonomic annotation includes both. The enrichment of members of the phylum Proteobacteria significantly discriminated rhizosphere and root samples from bulk soil samples irrespective of the cultivars tested. We suspected this fact was associated with Nitrogen uptake for the hulless barley. Nitrogen is an essential nutrient required by plants for their growth and metabolism. Although Nitrogen is abundant in the atmosphere in the form of diatomic (N2) molecule, its own molecular structure makes itself inert and difficult to be absorbed by plants. As described in previous study, Proteobacteria is one of the main functional diazotrophs microbes that fix the atmospheric N2 in the form of ammonia through its metabolic process (Bahulikar et al. 2014). In addition, Pseudomonas veronii at the species level and Rubrivivax and Stenotrophomonas at the genus level designated a conserved barley microbiota whose enrichment differentiated the rhizosphere and endophytic communities from that of the bulk soil community. Pseudomonas veronii is known to degrade a variety of simple aromatic organic compounds (Anzai et al. 2000). Rubrivivax gelatinosus is a purple nonsulfur photosynthetic bacterium capable of producing their own food via photosynthesis (Nagashima et al. 2012). Stenotrophomonas is a genus of Gram-negative bacteria with species ranging from common soil organisms to opportunistic human pathogen (Hauben et al. 1999). The implication of these differentially represented OTUs is still unclear to us, as not much knowledge is known. Surprisingly, even rhizosphere and endosphere bacterial community were enriched with the other Proteobacteria genera, the bulk soil was enriched with a unique Proteobacteria genus Comamonas, implying a strong exclusion for it from the plant. The trend of selective exclusion was also observed for Pedosphaera at the genus level, Gemmataceae and Pedosphaeraceae at the family level, and BD7–11 at the class level. It is either specifically targeted by the plant immune system or it could not co-exist with the microbiome of the rhizosphere and endosphere.
Our results have shown that the dominant fungi were Ascomycota and Basidiomycota at phylum level. It is known that Ascomycota promoted resistance to pathogen and tolerance to abiotic stresses. Basidiomycota not only provides increased biomass and seed yield for barley, but also improves the plant’s disease resistance. The only significantly presented fungal OTU Ceratobasidiaceae (family level) in ZQ320 is known to contain many species of opportunistic parasites for plants, causing a variety of economically important diseases. A better understanding of its significance requires future colonization experiment. Though the UNITE database is the most comprehensive one for fungal sequences, it is initially designed for ectomycorrhizal fungi in Northern Europe and more ITS sequences were collected worldwide through the years, but it will not be surprising that the environmental soil fungi from the Qinghai-Tibet Plateau cannot be fully deciphered. The association between fungal species and bacterial species was also investigated by the Spearman correlation analysis (data is not shown) but failed with identifying a significant association. It is also questionable for the illegality of identifying true correlation based on two separate high-throughput sequencing runs. With our preliminary characterization of endophytic and rhizosphere microbe from amplicon sequencing result, we believe a follow-up metagenomics analysis for the same experiment subjects could provide a more comprehensive and accurate interpretation of the microbe community abundance and function.
By comparing Shannon’s diversity of root-associated bacterial community, we found that bacterial communities within bulk soil and rhizosphere samples were equally diverse but those associated with the endosphere were significantly less rich, which is both true in the bacterial and fungal community in our study. Interestingly, when we use phylogenetic diversity index, PD whole tree’s diversity, we found that endosphere was still the least, rhizosphere presented a median level of diversity, and bulk soil samples possesses the highest diversity, displaying a progressive increase of diversity. These findings were in accordance with previous result which showed that endophytic bacterial communities are often simple, encompassing up to hundreds of OTUs, in contrast with the soil or rhizosphere microbiome, and confirmed the effect of plant root in selecting the rhizosphere microbiome and emphasized the intricate interactions between plant and soil environment (Lundberg et al. 2012). Our data confirms that plants can influence the structure of soil microorganisms as a filter, by selecting a less diverse fraction of those as the competent endophytes. We would speculate that diversity should decrease as the association with the plant is intensified, which result in a stronger impact. However, the different genotype of host cultivars did not influence the endosphere and rhizosphere microbiome as indicated by the beta diversity PCoA plot. Taken together, these results highlighted a shift in community composition at the barley root-soil interface, which progressively differentiated the rhizosphere and root bacterial assemblages from the bulk soil biota.
Deep-sequencing efforts that allow multiplexing of many samples simultaneously, such as the ones used in this study, present an opportunity to scale up these types of analyses and to potentially unravel the links between the hulless barley root microbiome with crop yield. Here, we investigated relationships of bacterial and fungal communities inside and outside hulless barley root of the three cultivars of hulless barley. Our study will certainly aid in developing better cultivation method. The key OTUs identified could potentially be used as growth-promoting endophytes for colonization and inoculation in order to improve the crop yields. The growth-promoting and bio-controlling mechanisms of the endophytes will be investigated in the future, so as to lay a foundation for the theory and practice of development and utilization of endophyte resource of hulless barley.
5 Conclusion
This paper reviewed the general situation and different groups of the overall microflora of three wild hulless barley species, evaluated their functions, and drew the conclusion that the main structure relationships of the rhizosphere and endogenous microflora of the species were Proteobacteria, actinomycetes, acidomycetes, ascomycetes, and basidiomycetes. The results demonstrated that the different types of samples (soil, rhizosphere, and root system samples) had a significant separation relationship and supports hypotheses regarding rhizosphere and root system classification. According to this study, it can provide a basis for studying the growth promotion and biological control mechanism of endogenous plants. It was used for reference by the related research on the richness of rhizosphere microbial population. It could contribute to the improvement of the yield of wild hulless barley after artificial domestication.
References
Achatz B, von Rüden S, Andrade D, Neumann E, Pons-Kühnemann J, Kogel K-H, Franken P, Waller F (2010) Root colonization by Piriformospora Indica enhances grain yield in barley under diverse nutrient regimes by accelerating plant development. Plant Soil 333(1–2):59–70
Anderson MJ, Walsh DCI (2013) PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: what null hypothesis are you testing? Ecol Monogr 83(4):557–574
Anzai Y, Kim H, Park JY, Wakabayashi H, Oyaizu H (2000) Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int J Syst Evol Microbiol 50(Pt 4):1563–1589
Bahulikar RA, Torres-Jerez I, Worley E, Craven K, Udvardi MK (2014) Diversity of nitrogen-fixing bacteria associated with switchgrass in the native tallgrass prairie of Northern Oklahoma, ed. J. E. Kostka. Appl Environ Microbiol 80(18):5636–5643
Bhatty RS (1999a) The potential of hull-less barley. Cereal Chem 76(5):589–599
Bhatty RS (1999b) β-Glucan and flour yield of hull-less barley. Cereal Chem 76(2):314–315
Bulgarelli D, Garrido-Oter R, Münch PC, Weiman A, Dröge J, Pan Y, McHardy AC, Schulze-Lefert P (2015) Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17(3):392–403
Caporaso JG et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336
Chen X, Long H, Gao P, Deng G, Pan Z, Liang J, Tang Y, Tashi N, Yu M (2014) Transcriptome assembly and analysis of Tibetan hulless barley (Hordeum Vulgare L. Var. Nudum) developing grains, with emphasis on quality properties. PLoS One 9(5):e98144
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7):5069–5072
Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, Ravel J (2014) An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2(1):6
Gottel NR, Castro HF, Kerley M, Yang Z, Pelletier DA, Podar M, Karpinets T, Uberbacher E, Tuskan GA, Vilgalys R, Doktycz MJ, Schadt CW (2011) Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Appl Environ Microbiol 77(17):5934–5944
Hardoim PR, van Overbeek LS, Berg G, Pirttilä AM, Compant S, Campisano A, Döring M, Sessitsch A (2015) The hidden world within plants: ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol Mol Biol Rev 79(3):293–320
Hauben L, Vauterin L, Moore ERB, Hoste B, Swings J (1999) Genomic diversity of the genus Stenotrophomonas. Int J Syst Bacteriol 49(Pt 4):1749–1760
Jones DL, Hodge A, Kuzyakov Y (2004) Plant and mycorrhizal regulation of rhizodeposition. New Phytol 163(3):459–480
Kaul S, Sharma T, K. Dhar M (2016) ‘Omics’ tools for better understanding the plant–endophyte interactions. Front Plant Sci 7
Kogel K-H, Franken P, Hückelhoven R (2006) Endophyte or parasite--what decides? Curr Opin Plant Biol 9(4):358–363
Kõljalg U, Larsson K-H, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, Sen R, Taylor AFS, Tedersoo L, Vrålstad T (2005) UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol 166(3):1063–1068
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31(9):814–821
Latch GCM (1993) Physiological interactions of endophytic fungi and their hosts. Biotic stress tolerance imparted to grasses by endophytes. Agric Ecosyst Environ 44(1–4):143–156
Lugtenberg B, Kamilova F (2009) Plant-growth-promoting rhizobacteria. Annu Rev Microbiol 63:541–556
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, Rio TG d, Edgar RC, Eickhorst T, Ley RE, Hugenholtz P, Tringe SG, Dangl JL (2012) Defining the core Arabidopsis Thaliana root microbiome. Nature 488(7409):86–90
Mandal S et al (2015) Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis 26:27663
McMurdie PJ, Holmes S (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data, ed. Michael Watson. PLoS One 8(4):e61217
Miliute I, Buzaite O, Baniulis D, Stanys V (2015) Bacterial endophytes in agricultural crops and their role in stress tolerance: a review. Zemdirbyste-Agriculture 102(4):465–478
Murphy BR, Doohan FM, Hodkinson TR (2014) Fungal endophytes of barley roots. J Agric Sci 152(4):602–615
Nagashima S, Kamimura A, Shimizu T, Nakamura-Isaki S, Aono E, Sakamoto K, Ichikawa N, Nakazawa H, Sekine M, Yamazaki S, Fujita N, Shimada K, Hanada S, Nagashima KVP (2012) Complete genome sequence of phototrophic betaproteobacterium Rubrivivax Gelatinosus IL144. J Bacteriol 194(13):3541–3542
Narwal S, Kumar D, Sheoran S, Verma RPS, Gupta RK (2017) Hulless Barley as a promising source to improve the nutritional quality of wheat products. J Food Sci Technol 54(9):2638–2644
Schreiber M et al (2014) The barley genome sequence assembly reveals three additional members of the CslF (1,3;1,4)-β-glucan synthase gene family, ed. Samuel P. Hazen. PLoS One 9(3):e90888
Sengupta S, Ganguli S, Singh PK (2017) Metagenome analysis of the root endophytic microbial community of Indian rice ( O. Sativa L.). Genomics Data 12:41–43
Tkacz A, Poole P (2015) Role of root microbiota in plant productivity. J Exp Bot 66(8):2167–2175
van der Heijden MGA, Bardgett RD, van Straalen NM (2008) The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett 11(3):296–310
Yang P et al (2008) Genetic diversity analysis of the developed qingke (hulless barley) cultivars from the plateau regions of Sichuan province in China revealed by SRAP markers. Yi Chuan 30(1):115–122
Zeng XQ (2015) Genetic variability in agronomic traits of a germplasm collection of hulless barley. Genet Mol Res 14(4):18356–18369
Zeng X, Long H, Wang Z, Zhao S, Tang Y, Huang Z, Wang Y, Xu Q, Mao L, Deng G, Yao X, Li X, Bai L, Yuan H, Pan Z, Liu R, Chen X, WangMu QM, Chen M, Yu L, Liang J, DunZhu DW, Zheng Y, Yu S, LuoBu ZX, Guang X, Li J, Deng C, Hu W, Chen C, TaBa XN, Gao L, Lv X, Abu YB, Fang X, Nevo E, Yu M, Wang J, Tashi N (2015) The draft genome of Tibetan hulless barley reveals adaptive patterns to the high stressful Tibetan Plateau. Proc Natl Acad Sci U S A 112(4):1095–1100
Funding
This study is funded by the program of Junior Teacher Innovation Project of Tibet Autonomous Region (QCZ2016-45) and the Natural Science Foundation of Tibet Autonomous Region (2015212-14-23).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Figure 1
(TIFF 1.11 mb)
Supplementary Table 1
(CSV 4.11 kb)
Supplementary Table 2
(CSV 1.54 mb)
Rights and permissions
About this article

Cite this article
Liu, Z.D., Li, L., Zhuo, G. et al. Characterizing Structure and Potential Function of Bacterial and Fungal Root Microbiota in Hulless Barley Cultivars. J Soil Sci Plant Nutr 19, 420–429 (2019). https://doi.org/10.1007/s42729-019-00045-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42729-019-00045-8