
Abstract
Rhizome rot is the leading debilitating disease affecting ginger, prevalent in various regions of India where the crop is cultivated. This term refers to all diseases impacting the rhizome, regardless of the pathogens involved, as they ultimately result in partial or total rhizome loss. The disease arises from the interaction of multiple pathogens, creating a complex scenario that complicates management strategies. The specific pathogens involved influence both the nature of the damage and the symptoms exhibited. The present study carried out to understand the complex nature of the disease and pathogens involved in the disease development. Molecular characterization identified the causative organisms at the species level, including Pythium aphanidermatum, Fusarium oxysporum, Fusarium solani, Sclerotium rolfsii, Ralstonia solanacearum, Meloidogyne hapla, and Meloidogyne enterolobii.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ginger, one of the earliest known oriental spices, has been cultivated in India for both fresh vegetables and dried spices since time immemorial. The crop is susceptible to a variety of diseases, including soft rot or rhizome rot, leaf spot, and bacterial wilt. Among these, rhizome rot is particularly significant due to the severe crop losses it can cause, occurring in several parts of India where ginger is grown. The term rhizome rot is broadly used to describe all diseases affecting the rhizome, regardless of the pathogens involved, as they ultimately result in partial or total rhizome loss. The specific pathogens involved determine the nature of the damage and symptom expression (Aravind et al. 2024). High incidence of rhizome rot typically occurs during warm and humid conditions, particularly in regions with poor drainage and heavy rainfall. In India, the disease is prevalent in major ginger-growing states like Kerala, Karnataka, and Assam, with frequency of attack peaking during the monsoon season. The economic impact is severe, often resulting in up to 50% crop loss, which drastically affects the livelihoods of farmers dependent on ginger cultivation. Studies show that while India experiences significant losses, other ginger-producing countries such as China, Nepal, and Thailand also report high incidences, albeit with varying causal agents and climatic influences. Statistical comparisons indicate that India faces one of the highest economic impacts due to the disease, underscoring the need for improved disease management practices and resistant ginger varieties to mitigate these losses (Archana et al. 2023; Chauhan and Patel 1990). Rhizome rot is a major constraint and, under severe conditions, can lead to 100% yield loss. The term rhizome rot is generally used for all diseases affecting the rhizome, despite the various pathogens involved. The number and nature of pathogens involved in disease development determine the intensity of damage and yield loss. Common pathogens involved in this complex disease include Fusarium spp., Pythium spp., Sclerotium rolfsii, Rhizoctonia solani, Ralstonia solanacearum, and Meloidogyne spp. (Islam et al. 2019; Chebte 2015). Each pathogen produces distinct symptoms: Pythium mainly causes soft rot symptoms, Fusarium spp. causes yellowing and wilting, and Ralstonia solanacearum causes bacterial wilt and green wilt symptoms (Elliott 2003; Bhardwaj and Mukherjee 2011; Archana et al. 2023). Sclerotium rolfsii typically infects during the later stages of plant growth, causing basal rotting. The interaction or combination of these pathogens with plant-parasitic nematodes leads to synergistic effects, increasing disease severity and leading to complete yield loss (Subedi et al. 2024; Kaushal et al. 2023). Understanding the pathogens involved in this disease complex and identifying them at the species level is crucial for developing effective management strategies. Molecular characterization of pathogens helps to understand the variability and diversity among them, which is vital for effective management under field conditions. The present study aims to understand the complex nature of pathogens involved in disease development and identify them at the species level, which is essential for effective management strategies in the field.
Materials and methods
Isolation, purification, and maintenance of culture
Infected samples were collected from major ginger-growing areas of Karnataka (Latitude: Approximately 11.6° N to 18.45° N and Longitude: Approximately 74.5° E to 78.5° E) and Kerala (Latitude: Approximately 8.0° N to 12.5° N and Longitude: Approximately 74.5° E to 77.5° E). Rhizomes exhibiting typical symptoms were utilized for pathogen isolation. Cornmeal agar (CMA) supplemented with Pimaricin was employed for isolating Pythium spp., while potato dextrose agar (PDA) media supplemented with pentachloronitrobenzene (PCNB) was used for isolating Fusarium spp. For isolating virulent colonies of Ralstonia solanacearum, specific TZC agar medium (Tyrosine, Zinc, and Calcium agar medium) was used. Nematodes were extracted from infected soil using the sieving method and from plant roots using the Baermann funnel method. Pure cultures were obtained through the hyphal tip technique and single spore isolation methods. The pathogenicity tests for all isolated samples, including Pythium, Fusarium, Sclerotium, Ralstonia, and Meloidogyne, were conducted using Koch’s postulates under in vitro conditions. The popular ginger variety Humnabad Local was used for these tests to verify the pathogenicity of the isolated organisms.
Molecular characterization of pathogens involved in complex disease
Extraction of fungal DNA by modified CTAB method
All fungal DNA isolation was carried out using 5-day-old mycelium mats with the modified CTAB method. Mycelial mats were transferred to sterilized pestles and mortars and ground with liquid nitrogen. To the crushed mycelial powder, 5 ml of 2X CTAB solution (composed of 100 mM Tris pH 8, 20 mM EDTA pH 8, and 1.4 M NaCl), 50 mg of PVP, and 50 µl of 1% β-mercaptoethanol were added immediately before incubation. The mixture was incubated in a water bath at 65 °C for 1 h with gentle mixing.
After incubation, the mixture was cooled to room temperature. An equal volume of a chloroform: isoamyl alcohol mixture (24:1) was added and the solution was centrifuged. The supernatant was collected, and 1/10th volume of 10% CTAB buffer (10% CTAB and 0.7 M NaCl) was gently mixed in. Another equal volume of the chloroform: isoamyl alcohol mixture (24:1) was added, and the mixture was centrifuged at 5000 rpm for 5 min at 4 °C. The supernatant was then collected, and 0.6 volumes of ice-cold isopropanol were added. This mixture was mixed well and stored overnight at 4 °C.
The next day, the mixture was centrifuged at 11,000 rpm for 20 min at 4 °C. The pellet was air-dried and dissolved in 500 µl of nuclease-free water. An equal volume of phenol-chloroform: isoamyl alcohol was added and centrifuged at 11,000 rpm for 10 min at 4 °C. The supernatant was collected, and an equal volume of the chloroform: isoamyl alcohol mixture (24:1) was added again and centrifuged at 11,000 rpm for 20 min at 4 °C. The supernatant was collected once more, and 1/10th volume of 3 M sodium acetate and 2.5 volumes of ethanol were added, then stored overnight at -20 °C.
The next day, the solution was centrifuged at 11,000 rpm for 20 min at 4 °C. The pellet was collected, the supernatant was discarded, and the pellet was air-dried. Finally, the pellet was dissolved in 50 µl of nuclease-free water and stored overnight at -4 °C for further use.
Bacterial DNA extraction
Ralstonia solanacearum was cultured in 5 ml of nutrient broth at 25 °C with shaking. A 1.5 ml aliquot of the culture was pipetted into a 1.5 ml tube and centrifuged for 2 min at 14,000 rpm. The supernatant was discarded, and the pellet was washed with distilled water. To the pellet, 550 µl of TE buffer with lysozyme was added, and the suspension was resuspended by pipetting. The mixture was incubated for 30 min at 37 °C. Following this, 76 µl of 10% SDS and proteinase K were added, mixed by flipping the tube, and incubated for another 15 min at 65 °C. Then, 100 µl of 5 M NaCl was added and mixed well by flipping the tube. Subsequently, 80 µl of CTAB-NaCl was added, mixed by flipping the tube, and incubated for 10 min at 65 °C.Next, 700 µl of chloroform: isoamyl alcohol was added, and the contents were mixed by flipping the tube for at least 15 s. The tubes were then centrifuged at 14,000 rpm for 5 min, and the aqueous layer was carefully transferred to a new 1.5 ml tube, avoiding the white middle layer. This step was repeated as necessary to ensure proteins were not carried over. An equal volume of isopropanol was added to the aqueous layer and inverted several times to mix, followed by centrifugation for 15 min at 4 °C. The supernatant was gently drained, and 1 ml of ice-cold 70% ethanol was added. The mixture was centrifuged at 14,000 rpm, and the ethanol was removed. The remaining ethanol was evaporated in a laminar flow cabinet. The DNA pellet was dissolved in 50–100 µl of 10 mM Tris (pH 8.0) by incubating overnight at 4 °C. To remove contaminating RNA, 6 µl of RNase was added to the DNA solution and incubated for 30 min at 37 °C. The extracted DNA was stored at -20 °C.
Purification of DNA
RNase A (10 µg/ml) was added to the water before dissolving the DNA to remove any RNA in the preparation (10 µl RNase A in 10 ml H2O). After resuspension, the DNA was incubated at 65 °C for 20 min to destroy any DNases that might be present. The pellet was then dissolved in 50 µl TE buffer or DNase-free sterile distilled water and stored at 4 °C.
DNA quantification
DNA was quantified using a Nanodrop spectrophotometer, which provided the concentration and purity of the DNA. Based on the quantification at 260 nm and 280 nm, samples with a 260/280 ratio of 1.8 were considered pure and of high quality. The stock DNA was then diluted in nuclease-free water to a concentration of 50 ng/100 µl for use in PCR.
Polymerase chain reaction (PCR)
Species-specific oligonucleotides were used as primers for the molecular detection of the isolated pathogens, following the protocol described by Wang and Chang (2003) with slight modifications. A set of species-specific oligonucleotide primers was selected to identify the species isolated from infected plants. The genomic DNA of the isolate was diluted to 50 ng/µl and used as a template for amplification. PCR reactions were performed in 0.5 ml PCR tubes. Each 20 µl PCR reaction mixture consisted of 2.0 µl of 50 ng/µl DNA template, 10 µl of 2X PCR master mix, 1 µl of primer, and 7 µl of nuclease-free water. The reaction mixture was mixed thoroughly by vertexing and briefly centrifuged to ensure that all components were well combined and to prevent reagents from sticking to the tube walls. PCR thermal conditions are optimized to ensure the denaturation of DNA strands, annealing of primers to the target sequences, and extension of new DNA strands by DNA polymerase. Denaturation carried at 94 °C, to separate the double-stranded DNA into single strands. Annealing temperatures were 55 °C, allowing the primers to hybridize specifically to the target sequences. Extension carried out at a temperature of approximately 72 °C, where DNA polymerase synthesizes new DNA strands complementary to the target sequences. These thermal cycling conditions are repeated for 30 cycles, to exponentially amplify the target DNA.
Nematode DNA extraction and sequencing
Genomic DNA from nematode egg masses and females was extracted using the Macherey-Nagel NucleoSpin DNA isolation kit. The extracted DNA was sequenced by Barcode Bioscience, Bangalore, for species-level identification. The Basic Local Alignment Search Tool (BLAST) was employed to identify regions of local similarity between the obtained Meloidogyne nucleotide sequences and those in WarmBase, an open-access resource providing nematode genome sequences. A phylogenetic tree was then constructed using the FASTA format sequences and the Mega 5.2 software. The analysis was carried out by Maximum Likelihood (ML) method with a bootstrap value of 1000.
Results
Pythium aphanidermatum produced hyaline, coenocytic, and sometimes lustrous, occasionally slightly brownish due to abundant oospores or hyphal swellings. Sporangium was consisting of terminal complexes of swollen hyphal branches of varying lengths. Oogonia were terminally formed and antheridia were mostly intercalary. Oospores size varied from 18.50 to 20.20 μm.
Fusarium oxysporum, Fusarium solani produced septate and hyaline mycelium as well observed production of both macro and microconidia. Macro conidia were stout, thick walled and curved at the end with 2–5 septate. Micro conidia were one to two celled. Chlamydospores formation was seen both terminally and intercalary.
Sclerotium rolfsii on PDA produced silky, fluffy, white profuse mycelial mat. Aerial hyphae was not uniformly distributed. Initiation of sclerotial bodies were obtained from the sixth day after inoculation. In the beginning, the sclerotial bodies were white but gradually turned to buff-brown color and then to chocolate brown at maturity. The fully matured sclerotial bodies were spherical to ellipsoidal and measured 1.5 mm in diameter.
Bacteria grown on TZC medium produced two types of colonies, some colonies isolated with reddish pink colored covered with white mucilage are virulent ones and other type colony observed was dark red avirulent colonies without white mucilage. Often colonies were observed in clusters. Electron microscopic observations revealed that bacterium is rod shaped with polar tuff flagellum. Based on these characters, the bacterium was identified as Ralstonia solanacearum.
To identify the Meloidogyne species, adult females were picked from galled roots and used for perineal pattern preparation and identified as Meloidogyne spp. The pure culture of each pathogen isolated is depicted in Fig. 1.
Isolation, purification, and maintenance of culture

Pure culture of pathogens involved in the disease complex. A) Fusarium spp, B) Sclerotium rolfsii, C) Pythium spp, D) Ralstonia solanacearum
Molecular characterization of pathogens involved in the complex
To identify Pythium at the species level, species-specific oligonucleotides were used as primers in the present experiment. The specificity of the designed primers was verified through PCR and gel documentation. Species-specific primer sets, Pm1F/Pm1R, Pu1F/Pu1R, and Pa1/ITS2, were used to detect P. myriotylum, P. ultimum, and P. aphanidermatum, respectively. Among these, the P. aphanidermatum-specific primer (5’ TCCACGTGAACCGTTGAAATC 3’ / 5’ GCTGCGTTCTTCATCGATGC 3’) amplified DNA at 210 bp for all 26 isolates collected from major ginger-growing areas (Fig. 2). These results confirmed that the isolated causal agent is P. aphanidermatum.

PCR amplification of pathogens involved in the disease complex
Species-level identification of Fusarium spp. was conducted using species-specific primer sets MB2F/MB2R and AY212027F/AY212027R to detect F. oxysporum and F. solani, respectively. Among the 26 isolates, 10 collected from Bagalkote, Bidar, Uttar Kannada, Chikkamagaluru, and Haveri were amplified with F. solani-specific primers AY212027F/AY212027R (5’ GAGCTGTGCGCGAGTCTGTG 3’ / 5’ ACTGCTCCTTCGAGTCGTCA 3’), with DNA amplified at 371 bp (Plate 8b). Isolates from Hassan, Kodagu, Mysuru, Shimoga, Calicut, and Wayanad were amplified with the MB2F/MB2R primer set at 250 bp, confirming them as F. oxysporum (Fig. 2).
Molecular characterization of Sclerotium rolfsii isolates was carried out by using ITS1 and ITS 4 primers. All the isolates DNA was amplified at 600 bp.
Similarly, detection and characterization of Ralstonia solanacearum from 26 isolates were carried out by using specific primer viz., RsolfliC-F/ RsolfliC-R (5’GAACGCCAACGGTGCGAACT3’/5’ GGCGGCCTTCAGGGAGGTC 3’) against the DNA template. All the isolates irrespective of the place confirmed the presence of Ralstonia solanacearum. All the isolates were amplified at 400 bp, which confirmed the presence of Ralstonia solanacearum in major ginger growing areas of Kerala and Karnataka (Fig. 2). Molecular characterization and species level identification of nematodes were carried out by genomic DNA isolation from nematodes followed by PCR with 18 s primers. The species level identification was carried out by sequencing.
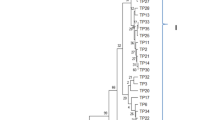
The nematode isolated showed remarkable similarity with M. hala and M. enterolobii when subjected to phylogenetic analysis by all the methods such as neighbour joining tree, maximum likelihood, maximum parsimony method. The phylogenic tree presented in the Fig. 3 represents the evolutionary history among other lineages of Meloidogyne. The nematode isolated clustered with M. hala and M. enterolobii and confirmed the species level identity and association causing rhizome rot of ginger.

Phylogenetic tree showing relationship among the related Meloidogyne spp
Discussion
Species specific primers, 5’TCCACGTGAACCGTTGAAATC3’/5’ GCTGCG TTCTTCATCGATGC 3’ was used to detect the species of Pythium and got amplified at 210 bp, thus it was confirmed as Pythium aphanidermatum. This result of molecular level confirmation of Pythium aphanidermatum are in agreement with Wang and Chang (2003), they differentiated the different species of Pythium from the soil through PCR technique for early detection of the pathogen by using species specific oligonucleotides. Because of its rapidness and ease, PCR amplification with the specific primers could be used to distinguish Pythium species and confirm their identification.
Similarly the isolated Fusarium spp. were subjected for molecular identification using Fusarium solani and F. oxysporum species specific primers (5’GAGCTGTGC GCGAGTCTGTG3’/5’ACTGCTCCTTCGAGTCGTCA3’) and 5’TGCTGTGTATGG ATGGATGG3’/ 5’CATGGTCGATAGCTTGTCTCAG3’. Fusarium solani got amplified at 371 bp and F. oxysporum got amplified at 250 bp. Thus it was confirmed that the pathogen was Fusarium solani and F. oxysporum were associated with rhizome rot of ginger. These results were supported earlier by Bahar and Shahab (2011) who reported that Fusarium solani amplified between 168 and 474 bp and F. oxysporum amplified between at 237–275 bp. Similar study were also carried out by Sharma et al. 2024: Sui et al. 2024. Their fundings also identified and characterized species level Fusarium spp causing rhizome rot in ginger.
Confirmation of Ralstonia solanacearum was done using specific primres viz., RsolfliC-F/ RsolfliC-R (5’GAACGCCAACGGTGCGAACT3’/5’ GGCGGCCTTCAG GGAGGTC 3’) against the single template. The template DNA gets amplified at 400 bp that confirmed the presence of Ralstonia solanacearum. Similar results recorded by Schonfeld et al. (2003), they reported the species level identification of Ralstonia solanacearum isolated from soil. Similar characterization of Ralstonia solanacearium causing rhizome rot were carried out by Ariute et al. 2023; Subedi et al. 2024.
Molecular characterization of Sclerotium rolfsii was carried out by using genomic DNA. Internal transcribed region of ribosomal DNA was amplified using ITS1-ITS4 universal primers. Based on cultural, morphological and molecular characteristics, the fungal pathogen was identified as Sclerotium rolfsii (Kannahi et al. 2016; Kifelew et al. 2015; Silué et al. 2023). The PCR product after gel run resulted in a product size of 550 bp. Similar results were obtained by Mahadevakumar et al. (2016), they characterized different Sclerotium rolfsii isolates from infected cucumber by morphological and molecular way.
Moleular characterization and species level identification of nematode was carried out by genomic DNA isolation from nematodes and amplification with 18 s primers. The species level identification was carried out by sequencing. The nematode isolated showed remarkable similarity with Meloidogyne hala and Meloidogye enterolobii. The results obtained from the current study confirmed the association of Meloidogyne hala as well as Meloidogyne enterolobii causing rhizome rot in ginger (Schonfeld et al. 2003; Jan and Khan 2002; Kaushal et al. 2023).
Conclusion
The molecular characterization of the isolated pathogens confirmed their identities with high specificity. Pythium aphanidermatum was identified by a distinct band at 210 bp. Fusarium solani produced bands at 371 bp, while F. oxysporum specifically exhibited a band at 250 bp. Sclerotium rolfsii was confirmed with a band size of 600 bp. The bacterial DNA was amplified at 400 bp, confirming the presence of Ralstonia solanacearum. Nematode species were identified through genomic DNA isolation and sequencing, revealing the association of M. hapla and M. enterolobii with rhizome rot of ginger. These results provide crucial insights into the specific pathogens affecting ginger crops, facilitating targeted disease management strategies.
References
Aravind S, Chaithanya KC, Sivaranjani R, Kandiannan K, Srinivasan V, Sankar SM, Babu KN (2024) Induction of in vitro micro rhizomes and assessment of yield, quality, and clonal fidelity in ex vitro established plants of ginger (Zingiber officinale Rosc). Plant Cell Tissue Organ Cult (PCTOC) 157(2):1–13
Archana TS, Mesta RK, Basavarajappa MP, Kumar KCK (2023) Promoting resilience in ginger: Elicitor-driven strategies to combat the rhizome rot disease. J Phytopathol 171(11–12):627–641
Ariute JC, Felice AG, Soares S, da Gama MAS, de Souza EB, Azevedo V, Benko-Iseppon AM (2023) Characterization and association of Rips repertoire to host range of novel Ralstonia solanacearum strains by in silico approaches. Microorganisms 11(4):954
Bahar M, Shahab H (2011) Analysis of Iranian isolates of Fusarium solani using morphological, pathogenicity and microsatellite DNA marker characterization. Afr J Biotechnol 11(2):474–482
Bhardwaj RL, Mukherjee S (2011) Effects of fruit juice blending ratios on kinnow juice preservation at ambient storage condition. Afr J Food Sci 5(5):281–286
Chauhan HL, Patel MH (1990) Etiology of complex rhizome rot of ginger (Zingiber officinale) in Gujarat and in vitro screening of fungicides against its causal agents. Indian J Agric Sci 60(1):80–81
Chebte A (2015) Genetic diversity of Fusarium isolates from onion basal rot and their inhibition by antagonistic Bacillus isolates under laboratory and glasshouse conditions. M.Sc Thesis, Addis Ababa University
Elliott SM (2003) Rhizome rot disease of ginger. Crop and Plant Protection Unit, Ministry of Agriculture, Bodies Research Station, sent. Catherina, Jamaica, West Indies
Islam MM, Khatun F, Faruk MI, Rahman MM, Hossain MA (2019) Incidence of rhizome rot of ginger in some selected areas of Bangladesh and the causal pathogens associated with the disease. Bangladesh J Agricultural Res 44(3):569–576
Jan S, Khan TA (2002) Interactive effect of Fusarium solani, Rotylenchulus reniformis and Meloidogyne javanica on tomato. Indian J Nematology 32(2):135–138
Kannahi M, Dhivya U, Kumar RS (2016) Biocontrol efficacy of Trichoderma spp. against rhizome rot disease of Zingiber officinale Rosc. Int J Pharm Sci Rev Res 36(1):63–66
Kaushal N, Sharma N, Sharma P (2023) An innovative approach for biocontrol of Meloidogyne incognita in ginger using potential bacteria isolated from Indian Himalayas. Curr Microbiol 80(12):381
Kifelew H, Kassa B, Sadessa K, Hunduma T (2015) Prevalence of bacterial wilt of ginger (Zingiber officinale) caused by Ralstonia solanacearum (Smith) in Ethiopia. Int J Res Stud Agricultural Sci 1:14–22
Mahadevakumar, S., Yadav, V., Tejaswini, G.S. & Janardhana, G.R. (2016). Morphological and molecular characterization of Sclerotium rolfsii associated with fruit rot of Cucurbita maxima. European Journal of Plant Pathology, 145(1), 215-219.
Schonfeld, J., Heuer, H., Elsas, J.D.V. & Smalla, K. (2003). Specific and sensitive detection of Ralstonia solanacearum in soil on the basis of PCR amplification of fliC fragments. Applied and Environmental Microbiology, 69(12), 7248–7256.
Sharma V, Thakur M, Chauhan A (2024) Isolation, identification, pathogenicity test and molecular characterization of Fusarium oxysporum f. sp. zingiberi causing Fusarium yellows disease from infected rhizomes of ginger (Zingiber officinale Rosc). J Plant Pathol, 1–6
Silué NS, Kouamé AEP, Séka K, Cissé N (2023) Assessment of the pathogenicity of fungi associated with ginger (Zingiber officinale) cultivated in two production areas in Côte d’Ivoire. Journal of Phytopathology
Subedi N, Cowell T, Cope-Arguello M, Paul P, Cellier G, Bkayrat H, Miller SA (2024) Characterization of Ralstonia pseudosolanacearum diversity and screening tomato, pepper, and eggplant resistance to manage bacterial wilt in South Asia. PhytoFrontiers™, pp PHYTOFR–10
Sui Y, Huang K, Sun X, Li Y, Xu P, Li N, Pang M (2024) The potential pathogenic fungus of ginger (Zingiber officinale) wilt disease: Fusarium spp. Journal of Plant Pathology
Wang, P.H. & Chang, C.W. (2003). Detection of the low-germination-rate resting oospores of Pythium myriotylum from soil by PCR. Letters in Applied Microbiology, 36, 157–161.
Funding
This research received no external funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest involved in this manuscript.
Ethics approval and consent to participate
No ethical rules are violated while conducting this research work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Archana, T.S., Mesta, R.K., Basavarajappa, M.P. et al. Elucidating the molecular profile of pathogens inducing rhizome rot in ginger (Zingiber officinale Rosc.). Vegetos (2024). https://doi.org/10.1007/s42535-024-00944-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42535-024-00944-x