
Abstract
The cloning of the laccase gene encoding industrially relevant laccase enzyme was carried out from Coriolopsis caperata into heterologous E.coli. Laccase enzymes play a role in the oxidation reactions of one electron of a wide range of phenols, aromatic amines and heterocyclic compounds such as lignin. They are multi-copper oxidoreductases that catalyze the one-electron (e-) oxidation coupled with the transfer of four electrons to the catalytic copper atoms, which leads to the reduction of two water molecules from reducing oxygen. In the present study, C. caperata was utilized for cloning the laccase gene and the protocol for cloning is outlined in the present study. The full-length gene (~ 1622bp) was amplified from Coriolopsis caperata by extracting RNA and synthesizing cDNA and PCR using gene-specific primers. By including the restriction site sequence of NcoI and XhoI enzymes into the synthetic primers, the directed cloning of the laccase gene fragment in the pET28b vector was performed.The construct was transformed into the E.coli BL-21 DE3 strain for expression. The transformed E.coli strain was screened on the kanamycin marker plate and the recombinant colonies were selected for plasmid isolation. Laccase gene cloning was confirmed by DNA sequencing of extracted plasmid. The cells were further processed for inclusion bodies isolation for enzyme recovery. The protein obtained was of size ~ 57kDa and confirmed on SDS PAGE which was same as expected size. The enzyme activity was found to be 73.1U/mL using ABTS as substrate. The laccase gene was successfully cloned from the fungus C. caperata and because laccase enzyme has widespread applications in the pharmaceutical, food, and environmental industries the E.coli recombinant host expressing laccase gene could serve as a warehouse for higher laccase enzyme production in these industries.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Laccases are multicopper oxidases ubiquitous in fungi and also present in higher plants, bacteria, and insects (Shekher et al. 2011). In plants, laccases participate in the lignification process, wound healing in response to pathogen defense, and iron metabolism (Piscitelli et al. 2010). Laccases are involved in a variety of physiological processes in fungi, including stress defense, pathogenesis, pigmentation, lignin degradation and morphogenesis. Laccases participate in the one-electron oxidation of a wide range of substrate molecules, including phenolic compounds, aromatic amines and heterocyclic compounds. They are environment friendly enzymes that require just molecular oxygen as a co-substrate for catalysis and produce only water molecules as a by-product. Due to their substrate promiscuity and eco-friendly nature, laccases possess enormous potential in the food, pharmaceutical, industrial, and environmental industries (Mayolo-Deloisa et al. 2020). Moreover, fungal laccases have been recovered from the residual compost of industrial production of edible mushrooms including Agaricus bisporus and Pleurotus ostreatus, due to their extracellular production. The ability to degrade lignocellulosic material renders laccase enzyme suitable for application in biorefinery processes for the production of clean biofuels.
Laccases play a role in bioremediation, detoxification of wastes, textile finishing (decolorization, detoxification), dye discoloration of paper pulp and transformation of xenobiotics (antibiotics, steroids) (Singh and Kumar 2019). Despite their valuable properties, the biotechnological applications of laccases are hampered by high production costs due to low enzyme activity and low yield, owing to the inability of traditional fermentation procedures to replicate the natural conditions required for cultivating many organisms. The native sources were shown to be insufficient to supply the enormous market demand for these enzymes. As a result, research is focused on scaling up laccase production while both increasing activity and lowering costs (Chenthamarakshan et al. 2017).
To address these bottle necks, recombinant protein expression in amenable hosts offers great solutions to improve productivity in shorter time and lowering down production costs. Another method is to screen for the hypersecretory strains. Enzyme engineering, enzyme evolution and immobilization are also viable options for improving the stability and activity of laccase enzymes. Basidiomycetes (white rot fungi) are the prolific producers of laccases, while other phyla such as Ascomycetes and Deuteromycetes also produce this enzyme (Yang et al. 2017; Wadgaonkar et al. 2021). In addition to a few examples of homologous expression (Alves et al. 2004), heterologous expression of laccases has been successfully performed in bacteria (Cheng et al. 2021), yeasts such as Yarrowia lipolytica, and Pichia pastoris (Iimura et al. 2018; Liu et al. 2020), filamentous fungi for e.g., Aspergillus niger, Aspergillus oryzae, and Trichoderma atroviride (Larrondo et al. 2003) and plants such as Zea mays and Arabidopsis thaliana (Li and Steffens 2002). E.coli serve as the excellent heterologous host for expression of laccases (Suzuki et al. 2003; Fernandes et al. 2007; Tan et al. 2017). Recently, a thermostable and organic solvent-tolerant laccase from Bacillus pumilus ARA has been expressed in E.coli which showed potential decolorization activities against various industrial synthetic dyes (Jiang et al. 2021). Remarkably, the application of laccase enzymes for decontaminating environmental pollutants holds great value (Yang et al. 2017). In another example, a white-rot fungus, Obba rivulosa derived recombinant laccase, has shown potential in mediator promoted oxidation of biorefinery lignin at acidic pH (Kontro et al. 2020).
Noteworthy, the recombinant enzymes tend to form aggregates and pose challenges in the purification, in case of heterologous expression in the E.coli host. On contrary, the expression of the laccase enzyme in other bacterial hosts such as Streptomyces lividans has resulted in significant amount of laccase enzyme with superior purity (Dubé et al. 2008). Previously, various studies have shown the higher level production of laccases by the white rot-fungus Coriolopsis caperata using media and fermentation optimization approaches (Nandal et al. 2013; Wadgaonkar et al. 2021).
Following the present literature, the present study attempted to clone and express the full length 1622-base-pair gene, lac-2 gene (laccase gene) from Coriolopsis caperata into the E.coli system.
Materials and methods
Strain, plasmids and reagents
The Coriolopsis caperata culture was isolated and cultured on the Potato Dextrose Agar (PDA) medium at 25°C. The heterologous expression strain, Escherichia coli BL21 DE3 strain was procured from Thermo Fisher Scientific. Coriolopsis caperata was cultivated in the liquid Potato dextrose broth medium for 72h, followed by centrifugation of suspension culture at 4000rpm for 10min to pellet the cells for RNA isolation. The RNA extraction was performed using the Trizol (Thermo Fisher) reagent as per the manufacturer’s protocol. The restriction enzymes NcoI and XhoI were purchased from New England Biolabs, Inc, which were utilized for directional cloning of laccase gene fragment into the pET28b vector (Merck). The pET28b vector linearization was done using the calf intestinal alkaline phosphatase. The Promega DNA ligation kit containing T4 DNA Ligase, Ligase 10X Buffer was utilized for ligating the digested laccase gene fragment and pET28b vector.
RNA extraction and cDNA synthesis
The RNA extraction from the 72h grown culture of Coriolopsis caperata was performed using Trizol reagent (Life Technologies, USA) and chloroform as per the manufacturer’s protocol. The integrity and quality of extracted RNA were checked on the 1% agarose gel and the RNA estimation was done on the Qubit fluorometer (Thermo Fisher Scientific). Next, the cDNA synthesis was carried out using 5 mM oligo dT18 primer, 200 U of Moloney murine leukemia virus reverse transcriptase (MuMLV-RT Invitrogen, USA), 1 mMdNTP solution and 3 mM Mg2+ in a volume of 20µl.
PCR amplification of laccase (Lac2) gene
The gene sequence of the lac-2 gene (GenBank, accession number: AGE13770) was retrieved from the NCBI GenBank database from the Coriolopsis caperata cDNA database. Next, the gene-specific PCR primers were designed to amplify the entire gene fragment for cloning using the Primer 3 tool available online at https://primer3.ut.ee/. The primer sequences and details were as per Table 1. Additionally, to introduce the restriction sites for cloning into the pET28b vector, the site-specific sequences and adapter nucleotide sequences were added to the primers.
The laccase enzyme encoding gene (Lac2 gene) was amplified using standard PCR reaction with gene-specific forward and reverse primers with an annealing temperature of 57°C. The PCR amplified product was resolved on the 2% agarose gel containing 0.5µg/ml of EtBr, prepared in 0.5X TBE buffer. 5.0µl of PCR product was mixed with 6X gel tracking dye for loading along with 5µl of molecular marker 1Kb size (APS Lifetech, Pune) DNA ladder.The bands were visualized under a UV Trans illuminator, and the gel images were recorded using BIO-RAD GelDoc- XR gel documentation system. A single band of PCR amplified product of laccase gene (size 1622bp) was expected.
Cloning of laccase gene (lac-2)
For cloning of the laccase gene into the pET28b vector, the amplified PCR was extracted from the gel using a gel extraction kit (Thermo Fisher Scientific), followed by ligation of the double digested PCR product and pET28b vector with XhoI and NcoI restriction enzymes (New England Biolabs, Inc). Prior to ligation the vector DNA was dephosphorylated. The ligation of the purified lac-2 gene into the cloning vector pET28b (Merck) was performed using theT4 DNA ligase as per the manufacturer’s instructions (Promega Ligation kit). The ligated construct (5µl) was transformed into E. coli BL21 DE3 strain and the transformed cells were plated onto the Luria-Bertani (LB) media plates containing kanamycin (30µg/mL). After overnight incubation at 37°C, the transformants were allowed to grow on the selection media and few colonies were selected and grown in 5ml LB media with kanamycin overnight (12–16h) at 37°C for screening for positive recombinants through plasmid isolation followed by DNA sequencing. Sanger DNA sequencing was performed for confirmation of presence gene of interest at geneOmbio Technologies Pvt Ltd., Pune with primer specific to vector pET28b T7 promoter and also the laccase gene specific primers used for PCR. The positive clone harboring the Laccase 2 gene in correct orientation according to the sequencing results was termed as pET28b-Lac-2MW.
Inclusion bodies isolation and SDS PAGE
The overnight grown culture of recombinant E. coli was inoculated in 1 lit of LB media with kanamycin. The flask was incubated at 37°C overnight. The 1mM IPTG and 0.25 mMCuSO4 were added as inducers in the culture and further incubated at 16°C for 5h. The cells from the culture were harvested after 5h and the cell pellet processed for inclusion bodies isolation. Inclusion bodies were isolated by sonicating the cell pellet on ice for 3 cycles of 20min. The lysed cell pellet was treated with with Urea, Sodium chloride, DTT and Triton X solution. On centrifugation the pellet was treated with 25mM and 50mM Trizma one by one. The centrifugation steps were performed at 9000g for 10min each at low temperature. The pellet obtained from the procedure was resuspended in 2M Urea in 100mM Tris buffer. The dissolved IBs were loaded on SDS PAGE for the confirmation of molecular weight of protein. The protein bands were stained with Bromophenol blue. The sample was run along with molecular marker of range 3.5kDa – 205kDa manufactured by Genei (cat No. 3,110,475,001,730).
The protein was initially denatured at 950C for 10min along with the 2X reducing dye and then loaded on gel. The 12% gel was used for gel electrophoresis.

Shows the image of RNA extracted from the Coriolopsis caperata in 72 hr liquid culture. 5µl of RNA sample was loaded and the RNA bands were resolved on to the 1% agarose gel prepared in 0.5X TBE buffer
Enzyme activity
The enzyme activity was measured at 420nm under UV-Vis spectrophotometer by using substrate 0.5 mM of 2,2’- azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS, Himedia) (extinction coeffar 420, 36,000M cm-1) at room temperature in 1mM sodium acetate buffer (pH5.5). The absorbance was used to calculate enzyme activity (Baltierra-Trejo et al. 2015).

Shows the PCR amplification of laccase gene (lac-2) using gene specific primers. Left to right: Lane M -1Kb molecular marker, Lane NTC-Negative test control, and Lane Laccase PCR-lac-2gene of Coriolopsiscaperata
Results
The aim of the present study was cloning and expression of laccase gene (lac-2) from the Coriolopsis caperata into the heterologous host, E.coli. The C.caperata culture was grown in the liquid medium for 72h for isolation of RNA to synthesize cDNA using oligo dT primers. Figure 1 shows the gel image of isolated RNA, the integrity of the RNA was confirmed by the presence of 3 distinct bands on the gel. Next, the cDNA was utilized as the template for the amplification of laccase gene using gene-specific primers (Fig. 2). Figure 2 shows the PCR amplification of the lac-2 gene using the cDNA template and gene-specific primers. A single band of size 1622 band was expected for the lac-2 gene. Following PCR, the amplicon was resolved on gel and extracted for cloning into the pET28b vector by direction cloning. The ligated lac-2 gene fragment into the vector was transformed into the expression BL-21 DE3 E.coli strain and the recombinant strains were screened on the kanamycin antibiotic plate. The positive clones were screened by isolating plasmids and confirmed through DNA sequencing. Figure 3 shows the lac-2 constructs in the pET28b vector. The identity of the recombinant plasmid was further checked with DNA sequencing, and the positive clone was named pET28b-Lac-2MW.
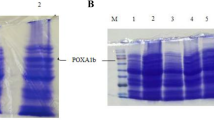
Figure 4 shows the laccase band which was expected to be present at ~ 57kDa on SDS PAGE as per ex-situ size evaluation of the protein. Molecular weight of native laccase from Coriolopsis caperata and recombinant enzyme was found to be similar respectively. The enzyme activity was found to be 73.1U/mL using ABTS as substrate.

Shows the gel image of plasmid DNA isolated from recombinant strains of BL-21 DE3 strains of E.coli. Lanes represents the plasmids isolated from two different colonies for screening; 5µl of plasmid DNA was loaded in each well. The bands were resolved on 1% agarose gel prepared in 0.5X TBE buffer. Five microliter of Coriolopsis caperata plasmid DNA was loaded on gel.

Protein loaded on 12% SDS PAGE gel Well no. 1: Molecular marker; Well no. 2: Laccase from native organism Coriolopsis caperata; Well no. 2: Recombinant Laccase from E. coli; Well no. 3 Parial purified recombinant IBs from E. coli
Discussion
In this study, the cloning and expression of a laccase gene (lac-2) was performed from C.caperata into the heterologous host E.coli using directional cloning. Successful cloning and expression was achieved as detailed in the present study. Expression of the laccase enzyme was confirmed by SDS PAGE, and further the activity of the laccase enzyme also evaluated. Next, a comparative study of native and recombinant enzyme for their stability and production cost is to be done for its industrial application. Purification and characterization of enzyme could help in exploring the other parameters of recombinant laccase production.
References
Alves AM, Record E, Lomascolo A, Scholtmeijer K, Asther M, Wessels JG, Wösten HA (2004) Highlyefficient production of laccase by the basidiomycete Pycnoporus cinnabarinus. Appl Environ Microbiol 70(11):6379–6384
Baltierra-Trejo E, Márquez-Benavides L, Sánchez-Yáñez JM (2015) Inconsistencies and ambiguities in calculating enzyme activity: The case of laccase. J Microbiol Methods 119:126–131
Cheng C-M, Patel AK, Singhania RR, Tsai C-H, Chen S-Y, Chen C-W, Di Dong C (2021) “Heterologous expression of bacterial CotA-laccase, characterization and its application for biodegradation of malachite green. " Bioresource Technology 340:125708
Chenthamarakshan A, Parambayil N, Miziriya N, Soumya P, Lakshmi M, Ramgopal A, Dileep A, Nambisan P (2017) Optimization of laccase production from Marasmiellus palmivorus LA1 by Taguchi method of design of experiments. BMC Biotechnol 17(1):1–10
Dubé E, Shareck F, Hurtubise Y, Daneault C, Beauregard M (2008) Homologous cloning, expression, and characterisation of a laccase from Streptomyces coelicolor and enzymatic decolourisation of an indigo dye. Appl Microbiol Biotechnol 79(4):597–603
Fernandes AT, Soares CM, Pereira MM, Huber R, Grass G, Martins LO (2007) “A robust metallo-oxidase from the hyperthermophilic bacterium Aquifex aeolicus”. FEBS J 274(11):2683–2694
Iimura Y, Sonoki T, Habe H (2018) Heterologous expression of Trametes versicolor laccase in Saccharomyces cerevisiae. Protein expression and purification 141:39–43
Jiang Y-p, Cai J-l, Pei J-j, Li Q, Zhao L-g (2021) “Cloning, overexpression, and characterization of a thermostable, organic solvent-tolerant laccase from Bacillus pumilus ARA and its application to dye decolorization.“ ACS omega. 6:9741–974914
Kontro J, Maltari R, Mikkilä J, Kähkönen M, Mäkelä MR, Hildén K, Nousiainen P, Sipilä J (2020) “Applicability of recombinant laccases from the white-rot fungus Obba rivulosa for mediator-promoted oxidation of biorefinery lignin at low pH.“Frontiers in bioengineering and biotechnology:1387
Larrondo LF, Avila M, Salas L, Cullen D, Vicuna R (2003) “Heterologous expression of laccase cDNA from Ceriporiopsis subvermispora yields copper-activated apoprotein and complex isoform patterns.“ Microbiology 149(5): 1177–1182
Li L, Steffens JC (2002) “Overexpression of polyphenol oxidase in transgenic tomato plants results in enhanced bacterial disease resistance.“ Planta. 215:239–2472
Liu C, Zhang W, Qu M, Pan K, Zhao X (2020) Heterologous expression of laccase from Lentinula edodes in Pichia pastoris and its application in degrading rape straw. Front in microbiology 11:1086
Mayolo-Deloisa K, González-González M, Rito-Palomares M (2020) Laccases in food industry: Bioprocessing, potential industrial and biotechnological applications. Front Bioeng Biotechnol 8:222
Nandal P, Ravella SR, Kuhad RC (2013) “Laccase production by Coriolopsis caperata RCK2011: optimization under solid state fermentation by Taguchi DOE methodology.“ Scientific reports. 3:1–71
Piscitelli A, Pezzella C, Giardina P, Faraco V, Sannia G (2010) “Heterologous laccase production and its role in industrial applications.“ Bioengineered bugs. 1:254–2644
Shekher R, Sehgal S, Kamthania M, Kumar A (2011) “Laccase: microbial sources, production, purification, and potential biotechnological applications.“ Enzyme research 2011
Singh P, Kumar S (2019) Microbial enzyme in food biotechnology. Enzymes in food biotechnology. Elsevier, pp 19–28
Suzuki T, Endo K, Ito M, Tsujibo H, Miyamoto K, Inamori Y (2003) “A thermostable laccase from Streptomyces lavendulae REN-7: purification, characterization, nucleotide sequence, and expression.“ Bioscience, biotechnology, and biochemistry. 67:2167–217510
Tan S-I, Ng I-S, Yu Y-J (2017) Heterologous expression of an acidophilic multicopper oxidase in Escherichia coli and its applications in biorecovery of gold. Bioresources and Bioprocessing 4(1):1–10
Wadgaonkar M, Tale V, Chavan Y (2021) “Media Optimization and Comparative Study of White-Rot Fungi Species for Their Capacity to Produce Laccase Enzyme.“ Annals of the Romanian Society for Cell Biology: 12487–12499
Yang J, Li W, Ng TB, Deng X, Lin J, Ye X (2017) Laccases: production, expression regulation, and applications in pharmaceutical biodegradation. Front Microbiol 8:832
Acknowledgements
I would like to thank geneOmbio Technologies Pvt Ltd, Pune for the providing the laboratory facility required for the lab work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wadgaonkar, M., Tale, V. & Chavan, Y. Cloning of laccase gene from Coriolopsis caperata into heterologous E.coli host. Vegetos 36, 195–200 (2023). https://doi.org/10.1007/s42535-022-00480-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42535-022-00480-6