
Abstract
To develop advanced and elaborate nanotechnologies, the behavior of materials must be understood at the nanoscale. Since direct observation is not generally possible experimentally, molecular dynamics simulations have been used to estimate nanoscale behavior, although simulations still have spatio-temporal limitations. Thus, coarse-grained molecular dynamics (CGMD) simulations have been suggested to study the physical properties and molecular behavior of mesoscale systems. A ‘bead’ composed of several atoms or molecules can represent the physical properties of a materials. In this study, we performed CGMD simulations of water and ethylene glycol, represented by Lennard–Jones parameters with various numbers of molecules within a single bead, to determine interaction parameters by comparing our results against empirically determined physical properties. Our results show the possible range of the number of molecules per bead satisfying a particular physical property such as density and self-diffusion coefficient. These data yielded the most suitable number of molecules to be included in a bead for CGMD simulations containing water and ethylene glycol. Moreover, we identified and discussed the effects of time scale factor, of which the empirically applicable range of 4–10, on self-diffusivity coefficients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
To design advanced and elaborate nanoscale materials, it is essential to observe the behavior of materials at the nanoscale. Experimentally, nanoscale microscopy techniques, such as transmission electron microscopy and scanning electron microscopy, have allowed for morphological characterization and composition determination [1]. However, these technologies require solid-phase samples, or samples that are not affected by sample preparation. To overcome such experimental limitations, several researchers have examined the nanoscale behavior via quantum and molecular dynamics (MD) simulations, which allow for the visualization of small particles such as atoms and molecules. Currently, simulations are used primarily to estimate physical properties depending on atomic substitution [2], molecular composition [3], or structural modification [4]. Using density functional theory, which is a quantum mechanics modelling method, Choi et al. estimated the improved thermal properties of a particular sludge composition to be applied in the casting of advanced alloys for use in automotive engines [5]. Cha et al. described the behavior of nanoparticles in nanofluids and quantitatively evaluated particle dispersion via MD simulations. This study overcame experimental limitations, resulting in significant improvements in the thermal properties of the nanofluid [6].
In modern computing systems, quantum and MD simulations typically cover systems containing hundreds of thousands of atoms, and can handle up to millions of atoms [7]. Depending on the simulation conditions, the total simulation time can reach up to tens of nanoseconds [8]. However, a large number of atoms in the simulation system results in increased calculation times. To overcome spatio-temporal limitations, many studies have reported MD simulations at the mesoscale using a single, arbitrary particle—‘bead’, which is composed of several atoms or molecules [9, 10]. These coarse-grained molecular dynamics (CGMD) studies have mainly been performed to simulate the behavior of macromolecules, such as proteins, and can accurately predict the physical properties and experimental phenomena of these systems [11, 12], as well as molecular behavior and/or inter-molecular interactions [13, 14]. Dissipative particle dynamics (DPD) and CGMD simulations are representative methods used to describe molecular behavior at the mesoscale. DPD, in which interparticle repulsion dominates phase dynamics, is generally used to describe phase separation or the self-assembly of beads. Son et al. [15] used a DPD method to investigate the phase diagrams of mixtures of water, ethanol, and vanillin. CGMD simulations take into account both repulsive and attractive interactions between beads to describe molecular behavior. These systems generally employ a Lennard–Jones (LJ) potential, such as a MARTINI force field [16, 17]. Interaction parameters of CG models are determined by comparing the results of all-atom MD or experiment.
For CGMD simulations, the most important factors are selecting appropriate type of potential function and parameterization methods. In the past decades, many researchers have attempted to develop a proper force field that ensures the reliability of the CGMD simulation which reproduces realistic macroscopic properties of interest, such as density, diffusivity, and microstructure. However, matching all the macroscopic properties simultaneously is hardly available because different molecular-scale mechanisms determine different physical properties such as density and diffusivity, so called mutually exclusive relation [18, 19]. Izvekov and Voth reported that diffusivity and radial distribution functions cannot be matched simultaneously [20], and Tóth found that total energy and structure could not be fitted at the same time [21]. To develop appropriate coarse-grained method, the desired physical properties should be well chosen. In this study, we performed CGMD simulations and their parameters are tuned to reproduce the density and self-diffusion coefficient of water and ethylene glycol, respectively. Based on the simulation, we optimized the parameters according to the number of atoms in a single-bead. The parameterization of a CG model is generally divided into two steps: bead mapping and establishing bead interactions. In the bead mapping step, the number of atoms or molecules within a single bead are determined. Interaction parameters in CGMD directly affect the predicted physical properties [22]. Therefore, the parameters are iteratively determined by comparing the results against data obtained using other MD or experimental methods. These data may include density, mean square displacement, radial distribution function, and free energy [23].
In this study, we performed CGMD simulations of water and ethylene glycol to determine the interaction parameters of single beads composed of various numbers of molecules. The predicted physical properties of the material were compared against experimental results. Our simulations yielded the possible range of the number of molecules per bead that satisfies the observed physical properties, i.e., the density and self-diffusion coefficient (SDC). We believe that our results can be incorporated into a database for suitable bead mapping, to facilitate and design CGMD simulation systems of materials that include water or ethylene glycol.
Simulation Details
CGMD simulation systems are composed of CG beads and can yield predicted characteristics, such as physical properties and molecular behavior, identical to those calculated in MD simulations. The number of atoms or molecules that are included within a bead and the bead mass needs to be considered in a coarse-graining process. Time scale factors, which affect calculations of physical properties, are adjusted according to the bead mapping scale. In previous studies, they examined mixtures of water and ethylene glycol, which are used in automotive coolants [24, 25]. The number of each water and ethylene glycol molecules in a single bead were selected to have a similar mass of beads for each material in given mixture. Note that the masses and volumes of water (H2O) and ethylene glycol ((CH2OH)2) molecules are 18.015 and 62.068 amu, and 29.93 and 103.10 Å3, respectively. The numbers of molecules within a single bead of water and ethylene glycol were 4, 7, 10, 17, 24, and 34 and 1, 2, 3, 5, 7, and 10, respectively. Here, we decided the minimum number of water and ethylene glycol molecules in a bead to 4 and 1, respectively, so that each bead contains 12 and 10 atoms with similar weight. The maximum number of molecules in a bead is empirically determined by considering allowable range of time scale factor. Figure 1 shows snapshots of CGMD simulations composed of beads containing four molecules of water and one molecule of ethylene glycol. The box size was controlled to fix the density of the simulation system, which was 0.997 g/cm3 and 1.11 g/cm3 for water and ethylene glycol, respectively. The length of the cubic cell was varied from 100 to 208 Å. Table 1 shows masses and system volumes according to the number of molecules per bead. Details regarding the system densities are discussed in Sect. 3.1.

Snapshots of the coarse-grained molecular dynamics (CGMD) simulation system. a Four molecules of water comprised a single bead and the total number of beads was 8,332 in a 100 × 100 × 100 Å3 box. b One molecule of ethylene glycol was included in a single bead and the total number of beads was 10,770 in a 100 × 100 × 100 Å3 box. Red, gray and white spheres represent oxygen, carbon and hydrogen atoms, respectively
LJ potentials (V) were used to describe the interactions between a pair of van der Waals potentials composed of both repulsive and attractive terms (r12 and r6, respectively). In particular, the intermolecular force depends strongly on the intermolecular distance (r). The expression for calculating the LJ potential is shown below:
where ε is the depth of the potential well as an energy parameter and σ is the finite distance parameter when the potential between a pair of molecules is zero. Interaction parameters (i.e., ε and σ) were examined with various numbers of molecules in a single CG bead.
The Mesocite module of the Materials Studio software package (ver. 8.0; BIOVIA Software Inc., San Diego, CA, USA) was used for CGMD simulations. All simulations were performed over 8 ns using NPT ensemble, where N is the number of atoms, V is the volume, and T is the temperature, at 298 K with a Berendsen thermostat and barostat [26]. The time step was 10 fs and 20 independent simulations were run for each system.
Results and Discussions
Parameters for Density of the System
When composing the CGMD simulation system, we firstly considered the density as a factor of similarity comparing to experimental results. Figure 2 shows LJ parametric maps, in which parametric surfaces are composed of pairs of interaction parameters, for the densities of water and ethylene glycol as a function of the number of molecules per bead. The density of each system was calculated according to the pairs of interaction parameters, i.e., epsilon and sigma. Interaction parameters, which cross the density surface, represent the experimental densities of water (0.997 g/cm3) and ethylene glycol (1.11 g/cm3), as shown in Fig. 2a, b. Note that black dots represent the actual simulation data for constructing LJ parametric maps, and the colors of the density surface intend to divide into higher and lower area by the color from red to blue over the range employed in this study. Since the parameters and the number of molecules contained in a bead are inter-dependent, the number of molecules contained in the bead was limited in range. For example, a bead containing 10 water molecules only can be fitted to the water density by the epsilon ranged from 0.8 to 1.1 kcal/mol corresponding to the sigma ranged from 6.35 to 6.56 Å. The interaction parameters shown in Fig. 2c, d depended on the number of molecules per bead. Sigma increased with the number of molecules per bead due to bead enlargement. In contrast, the values of epsilon greater than 1.2 kcal/mol resulted in crystallization of both water and ethylene glycol CG beads. It means that CG system is structurally stabilized with crystallization when the value of epsilon decreased below a certain threshold value. Epsilon values ranged from 0.8 to 1.1 kcal/mol, while sigma values ranged from 4.0 to 11.0 Å for both water and ethylene glycol. To estimate the molecular behaviors of water and ethylene glycol, we investigated the mean-square displacement (MSD) of the system and compared it with the experimental results [2]. MSD indicates the spatial extent of random particle motion throughout the system. It is measured by the deviation of particle position from the initial position as a function of time, as shown below:
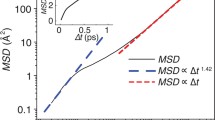
where x, x0 and T indicate the present and original positions of the particle and the average time, respectively. Figure 3 shows the MSD of a water bead, which included four water molecules, depending on the interaction parameters. We found that the slope of an MSD curve changed despite pairs of the parameters exhibiting the same density, i.e., 0.997 g/cm3.

Lennard–Jones (LJ) parametric maps for the densities of a water and b ethylene glycol as a function of the number of molecules per bead, indicated in parentheses. Black dots represent the actual simulation data for constructing LJ parametric maps, and the colors of the density surface intend to divide into higher and lower area by the color from red to blue over the range employed in this study. Epsilon and sigma indicated densities similar to those obtained experimentally for c water and d ethylene glycol. Note that interaction parameters were obtained by intersected lines between the density surfaces and parametric surfaces

Mean-square displacement (MSD) of water beads containing four water molecules each as a function of interaction parameters and simulation time
Molecular Behavior by Self-Diffusion Coefficient and Time Scale Factor
Diffusivity should also be considered to reproduce realistic molecular behaviors via CGMD. Diffusivity is defined as the displacement of molecules by thermal energy. In particular, the SDC is a quantitative physical property that indicates the speed of particles. SDC can be calculated from MSD, as shown below:
where ∆t is time interval. This calculation limits the differentiation of MSD by ∆t when sending ∆t to infinity, which indicates the normalized value for molecular diffusion in a system. In a CG system, the time scale is treated delicately due to reduced degrees of freedom. In CG systems, beads move faster than single molecule, which can affect interpretations related to the properties of movement [27]. Time scale factors, which are multiplied by the simulation time (t), address the gap of time that occurs by coarse-graining of molecules. Diffusivity derived from MSD is calculated based on the behavior of the bead, which behaved as a single particle. However, beads contain more molecules, so the correction for the time scale is needed to reflect the moving distance of all individual molecules included in the beads. The time scale factor is empirically determined in a range from 4 to 10. The value of time scale factor was determined that the value was matched the self-diffusion coefficient of the experimental results when the time scale factor was multiplied by the simulation time in SDC calculation. In addition, the MSD is multiplied by the number of molecules per bead to represent the unified motion of all molecules comprising a single bead. Figure 4 shows the dependence of SDC on the time scale factor, which was calculated using CG bead including four water molecules. In Fig. 4, SDC relatively more influenced from the energy parameter, epsilon, than the distance parameter, sigma. CG bead with larger epsilon had lower SDCs when applying the same time scale factor due to strong inter-bead interactions.

Self-diffusion coefficients (SDCs) of water beads containing four water molecules as a function of interaction parameters and simulation time. The gray dashed line represents experimental SDCs [28]
In addition, we calculated the SDCs of water and ethylene glycol with various numbers of molecules per bead as a function of the time scale factor. The data are shown in Fig. 5. An epsilon value of 1.0 kcal/mol was used to compare SDCs across systems, regardless of the depth of the potential well. Based on comparisons with experimentally derived SDCs, we found that time scale factors greater than 3 were required for beads containing four molecules of water. For both water and ethylene glycol, longer time scale factors were required to accurately describe the experimental SDC depending on the number of molecules per bead. Ethylene glycol required larger time scale factors than water due to lower SDCs at a given bead mass. Intermolecular friction, which is relatively high in all-atom models, is reduced as the number of molecules per bead increases, which then requires longer time scale factors to accurately describe the system. To investigate the effect of the time scale factor on the SDC value, we calculated SDCs by applying the time scale factor to be 10 (Table 2). All-atom model has relatively high intermolecular friction than simplified CG model, so when the number of molecules in a bead increases, the neglected intermolecular friction is increased. Therefore, to compensate the neglected frictional forces of CG models, larger time scale factor should be applied to the CG model contained larger number of molecules. The maximum number of water molecules that can accurately represent the experimental SDC was 24, and the timescale factor was 10. With the same time scale factor, the maximum of two ethylene glycol molecules per bead were deemed appropriate number. These parameters satisfied the observed physical behavior of each system.

SDCs of a water and b ethylene glycol as a function of the number of molecules in a single bead. Note that the value of epsilon was fixed at 1.0 kcal/mol and only the values of sigma were adjusted. Gray dashed lines represent experimental SDCs
In conclusion, the CG models contained 10 water and 2 ethylene glycol molecules, respectively, most properly represented the density and self-diffusion coefficients of water and EG simultaneously. The epsilon/sigma for water and EG beads were 1.0 kcal/mol/6.46 Å and 1.0 kcal/mol/5.49 Å, respectively. A single bead can be represented by various numbers of molecules. This number, however, is limited because the coarse graining step during a CGMD simulation requires compensation for time and space. Eventually, CGMD simulations must be carried out with modified interaction parameters, obtained from parametric studies, to accurately describe molecular behavior.
Parameters Applicable for Prediction of Physical Property
To establish the proper CG parameters, it is important to determine the physical properties comparable to experimental or all-atom MD results. The CG parameters developed in this study are optimized for reproducing the density and SDC characteristics, so they are suitable for studying the phase of the solvent, which is mainly determined by the intermolecular distance and structure. Many researchers have reported the CGMD parameters applicable to the other physical properties. Go et al. [29] reported the mechanism of heat transfer in nano-scale Newtonian fluid by using the CGMD simulation of which parameters are adjusted to describe the density, SDC, and thermal conductivity of the system. In addition, Moon et al. [30] investigated the mechanical behavior of polymer via CGMD simulation considering molecular structure, radial distribution function (RDF), and density of the system. However, it is hard to match all physical properties simultaneously due to the limitation of simplified CG beads even the CGMD parameters were well optimized. Therefore, the desired physical properties should be selected carefully to avoid the mutually exclusive relation.
To apply the CG parameters developed in this study to the mixture system, additional CG parameterization between water and ethylene glycol beads is required. Based on our results, the CG bead models contained 8 waters and 2 ethylene glycols are consequently suited for the mixture system, which considered the maximum number of molecules in a bead and the bead mass. To find the LJ parameters for water-ethylene glycol beads, density and self-diffusion coefficients of the mixture system should be compared with the experimental results, as shown in this study.
Conclusions
To overcome the limits in both simulation time and system size that compromise MD simulations, the concept of a “bead”, i.e., several atoms grouped together, has been suggested in order to perform simulations by interactions in a mesoscopic range. In this study, we performed a parametric study of water and ethylene glycol as represented by LJ parameters to describe the physical behavior in CGMD simulations. In particular, we investigated the LJ parameters for each bead representing water and ethylene glycol by adjusting physical properties such as densities and SDCs. The resulting map of paired energy and distance parameters shows the possible range of parameters that can accurately describe the physical behaviors of these materials, preventing the crystallization. Although the CGMD parameters were well optimized, it is hard to match all physical properties simultaneously due to the limitation of simplified CG beads. Thus, the desired physical properties should be selected carefully to develop the most suitable CG force field considering the properties to be evaluated from the simulation system. In addition, this study found the maximum number of molecules for a single bead, which were 24 and 2 molecules for water and ethylene glycol, respectively., with a time scale factor of 10.
References
F. Laborda, E. Bolea, G. Cepriá, M.T. Gómez, M.S. Jiménez, J. Pérez-Arantegui, J.R. Castillo, Detection, characterization and quantification of inorganic engineered nanomaterials: a review of techniques and methodological approaches for the analysis of complex samples. Anal. Chim. Acta. 904, 10–32 (2016)
J. Cha, W. Kyoung, Molecular dynamics simulation of the effects of affinity of functional groups and particle-size on the behavior of a graphene sheet in nanofluid. Comput. Mater. Sci. 139, 202–208 (2017)
J. Cha, W. Hong, S. Noh, S. Cho, Pore-size effects on thermal conductivity of SiO2 quartz using non-equilibrium molecular dynamics simulations. J. Theor. Comput. Chem. 17, 1850010 (2018)
J. Cha, W. Kyoung, K. Song, Molecular dynamics simulation of dispersion improvement of graphene sheets in nanofluids by steric hindrance resulting from functional groups. Mol. Simul. 43, 228–233 (2017)
G. Choi, H. Kim, K. Lee, S.H. Park, J. Cha, I. Chung, W.B. Lee, Study on thermal conductivity and electrical resistivity of Al-Cu alloys obtained by Boltzmann transport equation and first-principles simulation: semi-empirical approach. J. Alloys Compd. 727, 1237–1242 (2017)
J. Cha, W. Kyoung, K. Song, S. Park, T. Lim, J. Lee, H. Kang, Quantitative evaluation of the dispersion of graphene sheets with and without functional groups using molecular dynamics simulations. Nanoscale Res. Lett. 11, 136 (2016)
J.R. Perilla, B.C. Goh, C.K. Cassidy, B. Liu, R.C. Bernardi, T. Rudack, H. Yu, Z. Wu, K. Schulten, Molecular dynamics simulations of large macromolecular complexes. Curr. Opin. Struct. Biol. 31, 64–74 (2015)
L.E. Ratcliff, S. Mohr, G. Huhs, T. Deutsch, M. Masella, L. Genovese, Challenges in large scale quantum mechanical calculations. WIREs Comput. Mol. Sci. 7, 1–24 (2017)
D. Fritz, K. Koschke, V.A. Harmandaris, N.F.A. van der Vegt, K. Kremer, Multiscale modeling of soft matter: scaling of dynamics. Phys. Chem. Chem. Phys. 13(22), 10412 (2011)
S. Kmiecik, D. Gront, M. Kolinski, L. Wieteska, A.E. Dawid, A. Kolinski, Coarse-grained protein models and their applications. Chem. Rev. 116, 7898–7936 (2016)
F. Müller-Plathe, Scale-hopping in computer simulations of polymers. Soft Mater. 1, 1–31 (2002)
G.A. Voth, Coarse-graining of condensed phase and biomolecular systems (CRC Press, Boca Raton, 2009)
M. Baaden, S.J. Marrink, Coarse-grain modelling of protein–protein interactions. Curr. Opin. Struct. Biol. 23, 878–886 (2013)
E.M. Go, T.K. Lee, S.H. Min, W.C. Jeon, B. Kim, M.S. Yeom, S.K. Kwak, Theoretical study on enhancement of sensing capability of plasmonic dimer Au nanoparticles with amphiphilic polymer brushes. J. Phys. Chem. C 120, 11068–11077 (2016)
G.E. Son, N. Sugartseren, W. Yoon, S.K. Kwak, Phase behavior of ternary mixtures of water–vanillin–ethanol for vanillin extraction via dissipative particle dynamics. J. Chem. Eng. Data 59, 3036–3040 (2014)
J.J. Uusitalo, H.I. Ingólfsson, S.J. Marrink, I. Faustino, Martini coarse-grained force field: extension to RNA. Biophys. J. 113, 246–256 (2017)
S.P. Fu, Z. Peng, H. Yuan, R. Kfoury, Y.N. Young, Lennard–Jones type pair-potential method for coarse-grained lipid bilayer membrane simulations in LAMMPS. Comput. Phys. Commun. 210, 193–203 (2017)
P. Wang, Z. Li, Y. Ma, Z. Liu, J. Zhang, The coarse-grained model for a water/oil/solid system: based on the correlation of water/air and water/oil contact angles. RSC Adv. 5, 51135–51141 (2015)
K.R. Hadley, C. McCabe, Coarse-grained molecular models of water: a review. Mol. Simul. 38, 671–681 (2012)
S. Izvekov, G.A. Voth, A multiscale coarse-graining method for biomolecular systems. J. Phys. Chem. B. 109, 2469–2473 (2005)
G. Tóth, Effective potentials from complex simulations: a potential-matching algorithm and remarks on coarse-grained potentials. J. Phys. Condens. Matter. 19(33), 335222 (2007)
G. Rossi, P.F.J. Fuchs, J. Barnoud, L. Monticelli, A coarse-grained MARTINI model of polyethylene glycol and of polyoxyethylene alkyl ether surfactants. J. Phys. Chem. B 116(49), 14353–14362 (2012)
J.R. Allison, S. Riniker, W.F. van Gunsteren, Coarse-grained models for the solvents dimethyl sulfoxide, chloroform, and methanol. J. Chem. Phys. 136, 054505 (2012)
K.Y. Leong, R. Saidur, S.N. Kazi, A.H. Mamun, Performance investigation of an automotive car radiator operated with nanofluid-based coolants (nanofluid as a coolant in a radiator). Appl. Therm. Eng. 30, 2685–2692 (2010)
R. Saidur, K.Y. Leong, H.A. Mohammad, A review on applications and challenges of nanofluids. Renew. Sustain. Energy Rev. 15, 1646–1668 (2011)
H.J.C. Berendsen, J.P.M. Postma, W.F. van Gunsteren, A. DiNola, J.R. Haak, Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684 (1984)
L. Monticelli, S.K. Kandasamy, X. Periole, R.G. Larson, D.P. Tieleman, S. Marrink, The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 4, 819–834 (2008)
K. Krynicki, C.D. Green, D.W. Sawyer, Pressure and temperature dependence of self-diffusion in water. Faraday Discuss. Chem. Soc. 66, 199–208 (1978)
E.M. Go, E. Shin, J. Cha, S.K. Kwak, Estimation of heat transfer coefficient of water and ethylene glycol mixture in nanopipe via non-equilibrium coarse-grained molecular dynamics. J. Ind. Eng. Chem. 77, 128–134 (2019)
J. Moon, B. Kim, J. Choi, M. Cho, Multiscale study of the relationship between photoisomerization and mechanical behavior of azo-polymer based on the coarse-grained molecular dynamics simulation. Macromolecules 52, 2033–2049 (2019)
Acknowledgements
This work was supported by Hyundai Motor Company.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cha, J., Lee, W., Shin, E. et al. Parametric Study of Lennard–Jones Potentials to Predict Physical Behavior via Coarse-Grained Molecular Dynamics Simulations of Water and Ethylene Glycol Over Wide Temporal and Spatial Scales. Multiscale Sci. Eng. 2, 153–160 (2020). https://doi.org/10.1007/s42493-020-00046-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42493-020-00046-1