
Abstract
The epidemic of COVID-19 brought on by SARS-CoV-2 (coronavirus related to SARS) as well as its variants is still a life-threatening and critical risk to public health around the world. Due to the COVID-19 virus’s propensity for mutation, the pandemic is still spreading havoc compared to SARS-CoV-2. Several phytochemicals are now undergoing in silico analysis for their anti-SARS-CoV-2 efficacy. Scientists all over the world are on the lookout for novel lead phytochemicals that can effectively block SARS-CoV-2 entrance and infection in host cells. Piper longum phytomolecules suggest that it may be more effective to use natural remedies and herbal extracts to improve immunity and fight the infection. In this current in silico study of different Piper longum phytochemicals like sesamin, piperine, gamma-sitosterol, and epizonarene, using Lipinski's rule, the druggability guidelines and oral bioavailability of these compounds are studied. Four putative SARS-CoV-2 therapeutic targets were explored using the top four ligands in a comparative study. Our finding shows that sesamin, piperine, gamma-sitostenone, and epizonarene all displayed binding affinity values of − 8.9, − 7.8, − 7.4, and − 7.2 kcal/mol, respectively. Sesamin was found to have a greater negative binding energy toward the SARS-CoV-2 targets, which is a significant finding. We concluded that sesamin may be effective as a treatment for SARS-CoV-2.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
One of the deadliest and most contagious viruses ever was the coronavirus, and deadliest viruses in recent years have killed millions of people worldwide (Dong et al. 2020). SARS-CoV-2 was a novel coronavirus variant that emerged in China in December 2019 (Guo et al. 2020).
SARS-CoV-2 is a ss-RNA virus with a nearly 30-kb non-segmented genome (Andersen et al. 2020). It has a spherical form and is a member of the betacoronavirus genus (Lu et al. 2020). The spike protein structure of SARS-CoV and SARS-CoV-2 is connected to the viral membrane. It recognizes and binds to the host cell's protease enzyme and angiotensin-converting enzyme (ACE2) (Zhou et al. 2020). SARS-CoV-2 has four structural proteins and fourteen open reading frames (ORFs), including membrane (M), spike (S), nucleocapsid (N), and envelope (E) (Li et al. 2020). In addition, the virus has a 5′ poly (A) tail, an untranslated region (UTR) at the 3′ end, and a large number of undiscovered non-structural ORFs. S protein, which is 150 kD, is essential for virus attachment to receptors on recipient cell surfaces, allowing the virus to enter the recipient (Kirchdoerfer et al. 2016).
The protein that can fuse with the RNA genome is the N protein. The complete virion transformation by virus occurs through the budding process (Garg and Roy 2020). The E protein (8–12 kDa) is also involved in the assembly and budding of viruses (Suryanarayanan et al. 2015). Two more polyproteins that are components of the complex are generally required for viral replication and are encoded by the ORF gene. They replicate into more viral particles after being proteolyzed by the coronavirus major protease (Mpro) and the papain-like protease (PLpro) enzymes (Hilgenfeld 2014). Mpro is a very important and critical coronavirus enzyme necessary for viral transcription and replication. SARS-CoV-2 Mpro and SARS-CoV-2 Mpro have a 96% structural similarity (Ullrich and Nitsche 2020). Research investigations, therefore, seek therapy options that directly target this enzyme to limit the transmission of viruses (Li et al. 2020). Due to the virus's pathogenicity and effectiveness at mutation, no specific medicine or effective treatment has yet been identified. The current findings and research point in the direction of searching for or formulating powerful antiviral medications that can be approved as treatment options for SARS-CoV-2 (Jin et al. 2020). The majority of the bioactive molecules used to make potential medications are derived from plants (Roy 2018). Ayurveda has arisen as a rapidly rising field of medicine for the treatment of various ailments because of its benefits, including affordability, effectiveness without side effects, and capacity to handle an exponentially growing population (Mirza and Froeyen 2020).
Piper longum, commonly known as "Indian long pepper" or "pipli", is an Ayurvedic herb from the Piperaceae family. The humid regions of Peninsular India, including Maharashtra, Kerala, Tamil Nadu, and Karnataka, are native to this plant. This native plant from the western part of the world has been shown to have anti-inflammatory qualities, help with congestion brought on by the common cold and flu, function as a disinfectant, and even reduce stress and anxiety. This herb's biological activity comes from several different bioactive chemicals, including a large class of sterols, alkaloids, terpenes, and phenolic compounds. In silico analysis using molecular docking makes a substantial contribution to the process of developing new drugs since it can screen a large library of compounds and lessen the amount of work involved in conducting in vitro trials. Such technologies investigate docking interactions between receptor and ligand/drug molecules using built-in scientific computation approaches.
In the current study, bioactive compounds from Piper longum are tested for their ability to inhibit the SARS-CoV-2 Mpro enzyme using a computational approach. The interactions of the selected drugs with Mpro were ascertained using computational techniques such as molecular docking analysis and ADME analysis.
Materials and methods
Selection and prediction of protein
The 3D structure of the SARS-CoV-2 Mpro enzyme was obtained from the RCSB Protein Data Bank, with the corresponding PDB identification code being 6LU7. Table 1 presents a comparison of various structures, including a homodimer structure with 2 chain-A proteins that have 306 amino acids and an inhibitory N3 molecule, in addition to the SARS-CoV-2 chimeric receptor-binding domain (PDB id: 6VW1), spike glycoprotein (6VXX), and perfusion spike protein (6VSB).
As shown in Table 1, both 6VSB (open state) with a single receptor-binding domain up and 6VXX (closed state) are glycoproteins. 6VSB SARS-CoV-2 uses spike (S) glycoprotein for entry into the host cell, whereas 6VXX SARS-CoV-2 spike glycoprotein binds to ACE2 to enter the host cell (Wrapp et al. 2020; Walls et al. 2020).
There were about 100 chemicals in Piper longum; however, only 27 were considered to be bioactive. A '.pdb' file containing 3D structures was used for docking. All the data on phytocompounds, plants, phytochemicals, and pharmacological and therapeutics databases were compiled through the Indian Medicinal Plants Database, often known as IMPPAT (Mohanraj et al. 2018).
ADME analysis
The characteristics of Absorption, Distribution, Metabolism, and Excretion (ADME) of compounds were ascertained when evaluating them as potential ligands (Table 2). It made use of the Lipinski rule. Using the SwissADME internet application (available at https://www.swissadme.ch/) (https://www.swissadme.ch/), the characteristics of the compounds were evaluated. The SwissADME is a free online application that provides a straightforward method to evaluate data in a system for computer-aided drug design. The online resource is quite informative on lipophilicity, water solubility, physiochemical properties, and drug-likeness. To demonstrate the ligand's ability to traverse the digestive tract and digestive system, the molecule is also shown within a boiled egg. The ADME profiling of the chosen phytomolecules was investigated. Lipinski's criterion was broken by any ligands, and they were rejected from further analysis.
Swiss Target Prediction
The Swiss Target Prediction is a web-based simulation application that aids in the discovery of bioactive compounds with comparable designs that have related or identical biological targets (http://www.swisstargetprediction.ch). It is essential for selecting alternative targets for known compounds and filtering the database of potential drug targets. It is necessary to do a molecular characterization of the bioactive chemicals and their mechanism of action to comprehend the phenotypes that have been seen, make preparations for the future, and enhance the efficiency of the treatments that are currently available. The SwissTargetPrediction virtual server was utilized in the present investigation to estimate the percentage activity of the six different phytochemical substances that were selected. These targets include oxidoreductases, membrane receptors, kinases, phosphodiesterases, ligases, secreted proteins, cytochrome P450, proteases, voltage, gated ion channels, hydrolases, phosphatases, G-protein-coupled receptors, and main active transporters. These are all examples of proteins that are involved in this process.
Evaluation of bioactivity using the Molinspiration network
Based on several descriptors, Molinspiration (http://www.molinspiration.com/cgi-bin/properties) predicts a compound's drug similarity characteristics. Drugs that enter the body should bind to a biological molecule to convey their movement. Molinspiration provided a biological function score of phytocompounds against receptors found in humans such as G-protein-coupled receptors, ion channels, kinases, and other receptors, as well as proteases and proteins, providing an opportunity for the forecasting of the biological action of substances over time (Table 3). According to (Verma 2012), the bioactivity score of a complex is considered high if it is greater than 0.0, moderate when it ranges from 5.0 and 0.0, and low if it is 5.0.
Molecular docking
During the preliminary phases of the process of developing a drug, molecular docking is an essential computational approach for screening potential candidates (Table 4). Determining the interaction affinity of the protein–ligand complex aids in choosing and screening prospective inhibitors for targeted drug development. Protein and ligand preparation was done before the docking procedure. Auto Dock 4.2 was used. To prepare the optimal structure of proteins, antagonist N3 and H2O molecules have to be eliminated. Kollman and Gasteiger charges as well as the addition of polar hydrogens were calculated. Protein energy was reduced by utilizing the Swiss PDB Viewer.
The combined action of ellagic acid and ( +)-sesamin has been investigated in the catalytic amino acid regions present in SARS-CoV-2 3CL pro. The findings indicate a noteworthy affinity for binding to the catalytic cavity. The study demonstrated that three distinct ligands, namely 3WL, ellagic acid, and ( +)-sesamin, can form stable configurations adjacent to the effective interaction domain of the SARS-CoV-2 3CL Pro characteristics. Sesamin has been observed to enhance the production of the immune-responsive cytokine interleukin-2 (IL-2) while concurrently reducing the production of the inflammatory cytokines interleukin-1 (IL-1) and tumor necrosis factor-alpha (TNF-α) (Umar et al. 2022). This effect is achieved through the inhibition of the MAPK signaling pathways, specifically the phosphorylation of JNK, p38, and ERK1/2. The findings of this study corroborate prior research indicating that sesamin effectively diminishes p38 and JNK MAPK signaling pathways through IL-1-induced human articular. In addition, the study conducted revealed that BR13 exhibited interaction with four distinct amino acid residues situated within the catalytic cavity of SARS-CoV-2 3CL pro, namely HIS41, CYS145, ASN142, and GLU166 (Bossman et al. 2022). Sesamin has been observed to promote the production of the immune-responsive cytokine interleukin-2 (IL-2), while concurrently decreasing the expression of pro-inflammatory cytokines, namely interleukin-1 (IL-1) and tumor necrosis factor (TNF), through the inhibition of mitogen-activated protein kinase (MAPK) signaling pathways such as p-JNK, p-p38, and p-ERK1/2. The present study's results corroborated previous research indicating that sesamin mitigated IL-1-induced human articular p38 and JNK MAPK, as demonstrated by Phitak (Phitak et al. 2012).
Bioavailability radar
The bioavailability radar provides a quick assessment of a molecule's potential as a medicine and provides insight into how well it will be absorbed orally. SwissADME, which focuses on bioavailability, has obtained radar for molecules that meet Lipinski's requirements. The radar's colored area, which stands for the physicochemical space, represents oral bioavailability. Large deviations from these criteria indicate that the ligand is not suitable for oral administration (Mohanraj et al. 2018).
Results and discussion
ADME assessment
Before docking analysis, all compounds were analyzed based on their physical and chemical characteristics using ADME assessment. Then molecules that did not deviate from two or even more of Lipinski's rule criteria were chosen for additional examination. The molecular weight of a molecule must be less than 500 Da considering Lipinski's standard, and it must also contain fewer than five H-bond acceptors and a log P value that is less than 10 (Table 5).
Molecular docking analysis
The chosen ligands were used in a molecular docking technique once an ADME evaluation was finished. The binding energies of the ligands were ranked, with one having the highest. The docked molecule was the most stable whenever the lowest binding energy was given at the top of the list (Table 4).
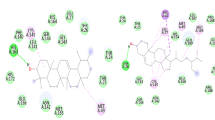
Sesamin displayed a binding energy of − 8.9 kcal/mol, and two distinct bonding interactions, a traditional hydrogen bond and a pi-alkyl, was noted—pi-alkyl bonds between ALA1020 and ARG1039 and conventional hydrogen bonds between them. Van der Waals interactions weakly linked the last five residues (Fig. 1).

A Interaction of sesamin with Mpro, B interaction of piperine with Mpro, C interaction of gamma-sitostenone with Mpro, D interaction of epizonarene with Mpro
The binding energy of piperine was − 7.8 kcal/mol. There were only three different kinds of bonding interactions found: carbon-hydrogen bonds, alkyl, and pi-alkyl. Through van der Waal interactions, the ligand is joined to the five remaining residues, except for LEU861 which is bound through a pi-alkyl, ARG765 which is bound through an alkyl, and LYS733 which is bound through a traditional hydrogen bond (Fig. 1).
The docked complex of gamma-sitostenone and Mpro had a binding energy of − 7.4 kcal/mol. The alkyl bonding involved ILE1013. Van der Waals forces were used by four additional residues to keep the complex stable (Fig. 1).
The binding energy of epizonarene was − 7.2 kcal/mol. There were found to be two different kinds of bonds between ligand atoms and amino acid residues. Pi-alkyl bonding involved TRP886 and ALA890. Through van der Waal forces, the other two nearby residues help to stabilize the complex (Figs. 1).
Figure 2 depict the physicochemical properties of phytoconstituents viz .Sesamin, gamma-sitostenone, Piperine, and Epizonarene, whereas gamma-sitostenone had physicochemical properties that fell within the pink zone.
Figure 3 depicts the % bioactivity of selected phytoconstituents with respect to selected protein targets viz. enzymes, kinases, oxidoreductases, phosphatases, proteases, phosphodiesterases and lyases. Result show that Epizonarene displayed maximum activity towards nuclear receptor (60.0%) and towards family AG protein-coupled receptor (26.7%).

Bioavailability radar and score prediction of selected phytoconstituents using SwissADME

Computation of the Swiss target for particular phytoconstituents
To evaluate the reliability and accuracy of the three-dimensional model of the Mpro 6VSB protein, the ProSA method was applied, and the resulting Z-score was 9.7. When comparing protein structures to the energy distribution seen in random configurations, the Z-score is used to quantify the degree of departure. Figure 4 displays the full ProSA analysis results. Different colors are used to denote the various group structures obtained from various techniques (NMR, X-ray). Plots smaller than 1000 residues are used to depict those chains. The central residue is shown by a plot of the average energy of residue lying over a sliding window shown in Fig. 4—(A) all protein chains' ProSA-web z-scores are shown here, (B) a plot of energy, and (C) the protein states with the highest and lowest energies.

Z-score for the SARS-CoV-2 receptor (6VSB), together with a plot of residue scores. A The z-scores on ProSA-web for each and every protein chain. B A plot of energy. C The protein states with the highest and lowest energies
Analysis of SARS-CoV-2 spike proteins about specific ligands
The process of drug discovery is contingent upon the identification of the active affinities and conformations of protein ligands (Table 6). The utilization of blind docking is a valuable methodology for obtaining relevant data. Blind docking is a common feature in both inverse docking and high-throughput screening, as noted (Sánchez-Linares et al. 2012). Efficient blind docking methods are deemed essential, hence the need for research in this area. The widely recognized AutoDock Vina molecular docking software enabled us to focus on the most significant aspects. Enhancing the process of blind docking necessitates the formulation of techniques for the identification of cavities and the optimization of docking parameters. CB-Dock is a novel blind docking software that incorporates guided cavity detection. It was created utilizing AutoDock Vina, a widely used Vina-based tool. Further information about AutoDock Vina can be found at http://vina.scripps.edu/manual.html#faq. The study assessed the binding energies of the top five compounds that were docked with Mpro. The compounds with the highest binding energies were then subjected to docking with the SARS-CoV-2 protein receptors 6LU7, 6VSB, 6VXX, and 6VW1, respectively, using the AutoDock comparative analysis (Table 7). Subsequently, the outcomes were verified utilizing the CB-Dock service, a no-cost software application that shares comparable molecular geometry software known as CB-Dock. The availability of ligand molecules can be accessed through the online platform cao.labshare.cn/drugrep/. The theoretical framework relies on the complementary geometry of the docking criterion, as presented (Table 8). The CB-Dock software is utilized to assess the attributes of the complexes that have been docked. This statement elucidates the sterically hindered region of the interface that is most extensive and is covered by the ligand. This region is a significant indicator of the strongest molecular interaction between the protein and the ligand. Multiple docking analyses were performed utilizing the CB-Dock online platform. In a recent study, cavity monitoring was analyzed as a means of facilitating the blind docking of proteins and ligands in a computer simulation (Liu et al. 2020). While the antiviral properties of ( +)-sesamin have not been established, it has demonstrated its potential to reduce the production of cytokines in the context of an influenza a H1N1 infection (Fanhchaksai et al. 2016). Several studies have shown that sesamin has the top interacting molecules with the main protease enzyme (Kumar et al. 2021; Majdalawieh et al. 2021). Drug-likeness properties of screened phytochemicals of drug molecules (Table 9). The Molinspiration Chemoinformatics website serves as a valuable resource for acquiring knowledge on the inhibition of proteases, as well as other biological targets such as kinases, nuclear receptors, and ion channels. The discussion solely pertained to the drugs' capacity to impede proteases. Given that this investigation also encompasses a protease (SARS-CoV-2 Mpro), the similarities are apparent. The rest of the information is provided (Table 1).
Conclusion
Most studies have been focused on finding new uses for existing drugs, which is the progression of finding novel uses for previously used or chemically synthesized drugs to treat SARS-CoV-2. Because bioactive molecules have shown remarkable efficacy in treating a variety of diseases, using phytocompounds for therapeutic expansion can support the effort to eradicate COVID-19. As it turns out, according to the findings of the current study, the top four ligands, i.e., the four names mentioned, provide higher binding energies against Mpro. The affinities of the top four ligands were compared using AutoDock and CB-Dock. In this study, sesamin shows highest binding affinity with open state glycoprotein (6vsb) of SARS-Cov-2 shown in (Table 4) out of 27 compounds. Apart from that, sesamin also follows Lipinski rule of five as well as Ghose, Veber, Egan, Muegge rule (Table 9) and show high gastrointestinal absorption when evaluating pharmacokinetics, drug-likeness, and medicinal chemistry through SwissADME web tools (Table 2). A more in vitro study is required to prove sesamin's potential as a therapeutic therapy for COVID-19.
Data availability
The data required to validate the conclusions of this study can be found in public databases.
Change history
08 January 2024
A Correction to this paper has been published: https://doi.org/10.1007/s42485-023-00127-3
References
Andersen KG, Rambaut A, Lipkin WI et al (2020) The proximal origin of SARS-CoV-2. Nat Med 26:450–452. https://doi.org/10.1038/s41591-020-0820-9
Bossman A, Umar Z, Teplova T (2022) Modelling the asymmetric effect of COVID-19 on REIT returns: a quantile-on-quantile regression analysis. J Econo Asymmetries 26:e00257. https://doi.org/10.1016/j.jeca.2022.e00257
Dong E, Du H, Gardner L (2020) An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis 20:533–534. https://doi.org/10.1016/S1473-3099(20)30120-1
Fanhchaksai K, Kodchakorn K, Pothacharoen P, Kongtawelert P (2016) Effect of sesamin against cytokine production from influenza type A H1N1-induced peripheral blood mononuclear cells: computational and experimental studies. In Vitro Cell DevBiol-Animal 52:107–119. https://doi.org/10.1007/s11626-015-9950-7
Garg S, Roy A (2020) In silico analysis of selected alkaloids against main protease (Mpro) of SARS-CoV-2. Chem Biol Interact 332:109309. https://doi.org/10.1016/j.cbi.2020.109309
Guo Y-R, Cao Q-D, Hong Z-S et al (2020) The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—an update on the status. Military Med Res 7:11. https://doi.org/10.1186/s40779-020-00240-0
Hilgenfeld R (2014) From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J 281:4085–4096. https://doi.org/10.1111/febs.12936
Jin Z, Du X, Xu Y et al (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582:289–293. https://doi.org/10.1038/s41586-020-2223-y
Kirchdoerfer RN, Cottrell CA, Wang N et al (2016) Pre-fusion structure of a human coronavirus spike protein. Nature 531:118–121. https://doi.org/10.1038/nature17200
Kumar A, Mishra DC, Angadi UB, Yadav R, Rai A, Kumar D (2021) Inhibition potencies of phytochemicals derived from sesame against SARS-CoV-2 main protease: a molecular docking and simulation study. Front Chem 9:744376. https://doi.org/10.3389/fchem.2021.744376
Li X, Geng M, Peng Y et al (2020) Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal 10:102–108. https://doi.org/10.1016/j.jpha.2020.03.001
Liu Y, Grimm M, Dai W et al (2020) CB-Dock: a web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacol Sin 41:138–144. https://doi.org/10.1038/s41401-019-0228-6
Lu R, Zhao X, Li J et al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet 395:565–574. https://doi.org/10.1016/S0140-6736(20)30251-8
Majdalawieh A et al (2021) Potential immunomodulatory role of sesamin in combating immune dysregulation associated with COVID-19. Asian Pac J Trop Biomed 11(10):421
Mirza MU, Froeyen M (2020) Structural elucidation of SARS-CoV-2 vital proteins: computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J Pharm Anal 10:320–328. https://doi.org/10.1016/j.jpha.2020.04.008
Mohanraj K, Karthikeyan BS, Vivek-Ananth RP et al (2018) IMPPAT: a curated database of Indian medicinal plants phytochemistry and therapeutics. Sci Rep 8:4329. https://doi.org/10.1038/s41598-018-22631-z
Phitak T, Pothacharoen P, Settakorn J et al (2012) Chondroprotective and anti-inflammatory effects of sesamin. Phytochemistry 80:77–88. https://doi.org/10.1016/j.phytochem.2012.05.016
Roy A (2018) Role of medicinal plants against Alzheimer’s disease. IJCAM. https://doi.org/10.15406/ijcam.2018.11.00398
Sánchez-Linares I, Pérez-Sánchez H, Cecilia JM, García JM (2012) High-throughput parallel blind virtual screening using BINDSURF. BMC Bioinform 13:S13. https://doi.org/10.1186/1471-2105-13-S14-S13
Suryanarayanan TS, Gopalan V, Sahal D, Sanyal K (2015) Establishing a national fungal genetic resource to build a major cog for the bioeconomy. Curr Sci 109:1033. https://doi.org/10.18520/v109/i6/1033-1037
Ullrich S, Nitsche C (2020) The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett 30:127377. https://doi.org/10.1016/j.bmcl.2020.127377
Umar AK, Zothantluanga JH, Aswin K, Maulana S, Sulaiman Zubair M, Lalhlenmawia H, Rudrapal M, Chetia D (2022) Antiviral phytocompounds “ellagic acid” and “(+)-sesamin” of Bridelia retusa identified as potential inhibitors of SARS-CoV-2 3CL pro using extensive molecular docking, molecular dynamics simulation studies, binding free energy calculations, and bioactivity prediction. Struct Chem 33(5):1445–1465. https://doi.org/10.1007/s11224-022-01959-3
Verma A (2012) Lead finding from Phyllanthus debelis with hepatoprotective potentials. Asian Pac J Trop Biomed 2:S1735–S1737. https://doi.org/10.1016/S2221-1691(12)60486-9
Walls A, Park Y, Tortorici M, Wall A, McGuire A, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020; 181:281–292.e6.https://doi.org/10.1016/j.cell.2020.02.058
Wrapp D, Wang N, Corbett K, Goldsmith J, Hsieh C, Abiona O, Graham BS, McLellan JS. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020; 367:1260–1263. https://doi.org/10.1126/science.abb2507
Zhou Y, Hou Y, Shen J et al (2020) Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov 6:14. https://doi.org/10.1038/s41421-020-0153-3
Funding
This research was unable to secure any financial support.
Author information
Authors and Affiliations
Contributions
IS and AK developed the concept and design of the work. IS, AK, and JI contributed to data collection, analysis, and interpretation of results All authors revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Authors have no conflict of interest.
Ethical approval
This study did not involve the use of any human or animal model.
Informed consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: In this article authors Indrajeet Singh and Juveriya Israr were missing from the author list.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Singh, I., Israr, J. & Kumar, A. An in silico investigation of the phytomolecules present in Piper longum roots as a potential treatment for SARS-CoV-2. J Proteins Proteom 14, 237–251 (2023). https://doi.org/10.1007/s42485-023-00117-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42485-023-00117-5