
Abstract
Calcitonin gene-related peptide (CGRP) is known to be involved in the pathophysiology of migraine headache. CGRP may play a role in the mechanisms of somatic, visceral, neuropathic, and inflammatory pain. Currently, there are multiple new biologic therapies targeting CGRP which have been approved for the acute and preventive treatment of migraine. Studies have also shown CGRP to be involved in the pathophysiology of trigeminal neuralgia (TN), an often severe and refractory facial pain condition. This article outlines the pathophysiology of trigeminal neuralgia and the role CGRP may play in this condition. We discuss the current evidence on CGRP in trigeminal neuralgia and how modulation of CGRP may be a potential target in the treatment of trigeminal neuralgia in the future. With many similarities in the pathophysiology and efficacy of drug therapies for migraine and trigeminal neuralgia, we hypothesize that targeting CGRP may be a potential mechanism of pain relief in trigeminal neuralgia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Trigeminal neuralgia is a disabling neurological disease with an estimated annual incidence of TN in the USA (age-adjusted to the 1980 US total population) of ~5.9 per 100,000 women and ~3.4 per 100,000 men [1]. Onset occurs in adulthood, around 50 years of age, and with higher prevalence in women [2]. Trigeminal neuralgia is characterized by recurring episodes of electric shock-like, shooting, stabbing, or sharp pain along the distribution of the trigeminal nerve. Pain may be spontaneous in origin or may be triggered by minimal stimulation such as talking, chewing, drinking, or light touch [3]. Episodes typically last from seconds up to 2 min and can re-occur in clusters, ranging from several up to hundreds of episodes per day [4]. Many patients experience variable periods of pain remission, extending from several months to years [5, 6]. The trigeminal nerve gives rise to three branches: ophthalmic (V1), maxillary (V2), and mandibular (V3). Each branch supplies sensory innervation to a distinct region of the face. Paroxysmal pain of trigeminal neuralgia has been reported to occur most often along either the maxillary or mandibular branch alone or simultaneously in both branches together [2, 6]. Pain remains localized to the trigeminal dermatome without radiation, one of the diagnostic criteria for TN [3] (Fig. 1). Nearly half of the patients with classical trigeminal neuralgia report constant, dull facial pain in addition to the characteristic stabbing paroxysmal pain [7].

International Classification of Headache Disorders, 3rd Edition, (ICHD-3) diagnostic criteria for trigeminal neuralgia [3]
Classification of Trigeminal Neuralgia
The International Headache Society classifies trigeminal neuralgia into three types based on etiology. These include classical, secondary, and idiopathic TN, with the classical type contributing to the majority of cases [3]. Classical trigeminal neuralgia is caused by neurovascular compression of the trigeminal nerve root, producing morphological changes that can be detected using MRI [8]. Secondary TN is established when compression or demyelination of the trigeminal nerve is consequential to neurologic disease such as a tumor or multiple sclerosis (MS), a condition causing plaque formation, which highly increases the risk for the development of TN [8,9,10]. MRI has been found to detect up to 15% of TN cases secondary to a tumor or MS plaque, while abnormal trigeminal reflex testing has shown higher diagnostic sensitivity (87%) and specificity (94%) for identifying patients with secondary TN [11]. Cases in which diagnostic investigation fails to reveal the cause of TN are classified as idiopathic [9]. In addition to the stabbing, paroxysmal pain characteristic of TN, a cross-sectional study demonstrated that nearly 50% of patients (n=158) also experienced concomitant pain, continuous dullness, burning, or tingling in the same distribution as the paroxysmal pain [12]. The mechanism of continuous pain is not fully understood and may occur in classical, secondary, or idiopathic trigeminal neuralgia [13]. Furthermore, Maarjberg et al. suggests that concomitant pain is not a consequence of a long duration of disease nor a more severe disease course [12].
Current Accepted Pharmacological Approach in Trigeminal Neuralgia
The first-line medications for the long-term treatment of trigeminal neuralgia are carbamazepine and oxcarbazepine. Both drugs have been proven efficacious in reducing the number of pain episodes by >50% in 88% of patients [14]. Despite efficacy, drug compliance is limited by adverse effects including drowsiness, ataxia, nausea, leucopenia, rash, and GI upset [15]. More serious complications include agranulocytosis and aplastic anemia [4]. The numbers needed to harm for carbamazepine were 3.4 for minor adverse effects vs. 24 for severe adverse effects [16]. Other drugs proven effective for trigeminal neuralgia pain control include lamotrigine, baclofen, and pizonidine. However, these drugs have been studied only in single trials and are associated with adverse side effects as well [14, 17].
Surgical treatment is considered for patients who fail to respond to or cannot tolerate pharmacologic intervention. Surgical intervention includes microvascular decompression (MVD) or percutaneous procedures to ablate the trigeminal ganglion. Surgical interventions have demonstrated high post-operative pain relief, with >70% of MVD cases and about 50% of percutaneous ablation cases remaining pain-free five years post-operation [10, 14, 17, 18]. Serious long-term complications of MVD included aseptic meningitis (11%), hearing loss (10%), and sensory loss (7%). Less common (4%) but serious adverse effects of MVD included infarction, hematoma, or CSF leakage [10, 19]. The most common complication of ablation was sensory loss (10%) [17, 18].
In the long-term, MVD for treatment of TN had recurrence rates of 27.1% and 26.6% at 3 years post-operation in two studies [20, 21]. Many factors impact the prognosis of MVD in TN patients including patient age, gender, pain characterization as typical vs. atypical, duration of symptoms, and whether venous compression is the etiology for TN [22].
Emerging Treatment for Trigeminal Neuralgia
OnabotulinumtoxinA (BTXA) was FDA-approved for prophylaxis of chronic migraine in 2010. A review of BTXA for chronic migraine found BTXA to increase the number of headache-free days. Evidence suggests BTXA can block CGRP release, interfering with pain transmission [23, 24].
Botulinum toxin type A (BTXA) is an emerging medical therapy for trigeminal neuralgia as well. Metanalysis by Morra et al. found a clinically significant benefit in reducing the frequency and intensity of pain in trigeminal neuralgia patients treated with BTXA. BTXA was shown to be safe and well tolerated with minimal side effects and provided relief of symptoms for a relatively long duration (several months) [25]. Though the mechanism behind BTXA action is unclear, Zhang et al. propose that it may be associated with the inhibition of CGRP release as CGRP levels in the blood significantly decrease following treatment with BTXA in trigeminal neuralgia patients [26]. BTXA may be a promising treatment for neuralgia; however, further investigation is required to determine the optimal dose, duration of efficacy, and indications for repeated treatment [18].
Recent studies suggest a role for sumatriptan in the treatment of refractory trigeminal neuralgia. Kanai et al. found that subcutaneous sumatriptan was effective in reducing pain in 80% of patients and completely eliminating pain in 50% of patients with trigeminal neuralgia refractory to previous treatments (n=24) [27]. In another study by Kanai and colleagues, 15 patients with idiopathic trigeminal neuralgia received 3 mg of subcutaneous sumatriptan followed by a week of twice daily 50 mg of oral sumatriptan. Results were significant for immediate and continued pain relief with no serious side effects [28]. Nasal sumatriptan adjunctive to carbamazepine was also found effective in reducing visual analog scale (VAS) scores in three trigeminal neuralgia patients refractory to carbamazepine alone [29].
Triptans are the first-line treatment for migraine and have been found to decrease plasma levels of CGRP in patients experiencing acute migraine. This decrease in plasma CGRP levels directly correlates with the degree of migraine pain relief following the administration of sumatriptan [30, 31]. Triptans act as 5HT1B/1D agonists, which are receptors located within trigeminal nerve endings and the trigeminal nucleus. Stimulation of these receptors via triptans leads to inhibition of CGRP release, subsequently disrupting pain neurotransmission in the trigeminovascular system and relieving migraine pain [32]. The mechanism behind pain relief in trigeminal neuralgia following the administration of sumatriptan has not yet been established; however, findings from the above studies suggest a role for 5-HT receptors and CGRP in the pathogenesis of trigeminal neuralgia [27].
Overall, many treatments currently available for trigeminal neuralgia are not effective and can be associated with side effects and post-operative complications. Thus, there is a need for more effective treatments with less adverse effects for trigeminal neuralgia. More recently, evidence on the role of CGRP in trigeminal neuralgia has emerged. The modulation of CGRP has been suggested to be a potential target in the treatment of trigeminal neuralgia in the future.
CGRP
Calcitonin gene-related peptide was discovered in 1982 by Amara et al. as a product of alternative mRNA splicing of the calcitonin gene [33]. CGRP is a 37-amino-acid peptide involved in pain neurotransmission throughout the central and peripheral nervous systems [34]. The role of CGRP in migraine pathophysiology has been extensively studied in recent years, which led to the development of anti-CGRP therapies for both acute treatment and prevention [35]. Accumulating knowledge from CGRP’s role in migraine has sparked research for its role in cluster headache, inflammatory pain, and various other pain disorders [36, 37]. The mechanisms of CGRP in pain signaling is complex and requires further research to determine its full range of effects [34].
CGRP and Trigeminal Nociceptive System
Previous research on the anatomy of the trigeminal nociceptive pathway has elucidated how migraine pain is processed and generated. From the trigeminal ganglion arises a rich plexus of nociceptive fibers including nonmyelinated C-fibers and thinly myelinated Aδ- fibers that innervate the intracranial vasculature and dura mater [38]. Previous research using immunohistochemical staining in rat and human tissue demonstrated that CGRP expression was limited to the unmyelinated C-fibers innervating both cerebral arteries and dura mater, with nearly equal expression in each location [39]. It was found that nearly all C-fibers that expressed substance P also expressed CGRP; however, there were many more C-fibers that expressed CGRP without substance P. CGRP was also found to colocalize with pituitary adenylate cyclase-activating polypeptide (PACAP) in the dura mater and cerebral vessels [39]. Immunohistochemical staining has further identified that both Aδ-fibers and vascular smooth muscle cells express the calcitonin receptor-like receptor (CLR) and receptor activity-modifying protein 1 (RAMP1), suggesting the presence of functional CGRP receptors. These findings strengthen the notion that the release of CGRP from C-fibers may result in the sensitization of the adjacent Aδ-fibers and that CGRP signaling at cerebral and meningeal (dural and pial) arteries, via increased intracellular cAMP, results in increased blood flow [39, 40].
Primary afferent pain fibers innervating the meningeal arteries, cerebral arteries, and dura mater are pseudounipolar neurons, with cell bodies in the trigeminal ganglion, and a central branch projecting to the spinal trigeminal nucleus or upper cervical (C1–C2) spinal segments [38]. CGRP released from central terminals of primary afferents is believed to bind to CGRP receptors in the spinal trigeminal nucleus, which promotes increased glutamate release and facilitates nociceptive transmission to second-order neurons [41, 42]. Second-order neurons from the spinal trigeminal nucleus project to the thalamus, hypothalamus, locus coeruleus, and periaqueductal gray [38]. Cell bodies of third-order neurons were found mainly in the thalamus, specifically in the posterior and ventromedial thalamic nuclei. From the thalamus, Noseda et al. found that neurons projected diversely to various cortical regions—primary/secondary somatosensory and motor, parietal association, auditory, ectorhinal, and visual cortices. The complex signaling originating from the thalamus, particularly the posterior nucleus, and extending widespread throughout the CNS has been proposed to account for the diverse neurological disturbances associated with migraine [43].
CGRP receptors have also been identified on Schwann cells and satellite cells within the trigeminal ganglion, indicating that CGRP released within the ganglion can activate both neurons and glial cells [44, 45]. Satellite glial cells (SGCs) tightly surround the cell bodies of sensory neurons in the trigeminal ganglion [46]. These pseudo-unipolar neurons, with a sensory branch innervating the skin, muscles, and joints and a central terminal reaching the spinal trigeminal nucleus, have a role in transmitting somatosensory stimuli such as touch, pain, temperature, and itch from the periphery to the CNS [46, 47]. It is thought that each sensory neuron is enveloped by several SGCs that form a distinct morphological unit [48]. SGCs have been found to play a role in the regulation of the neuronal microenvironment primarily via buffering of K+, which is crucial for maintaining neuronal resting membrane potential. When there is a disturbance in the SGCs K+ buffering capacity, extracellular K+ can rise leading to neuronal hyperexcitability, which is believed to be a mechanism contributing to neuropathic pain [49].
In addition, CGRP has also been found to be involved in the neuronal transmission of pain within the trigeminal ganglion. Thalakoti et al. found that CGRP released from neuron cell bodies within the trigeminal ganglion stimulated SGCs to release inflammatory cytokines, which enhance neuronal pain transmission [50]. Furthermore, intra-ganglionic administration of CGRP was found to increase SGC and neuronal activation and increase cytokine expression in the TG, which was accompanied by an increased sensitivity to thermal pain. These findings may suggest that SGC activity within the trigeminal ganglion contributes to neurogenic inflammation and the development of orofacial pain [51].
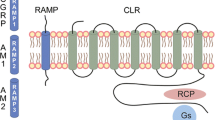
CGRP has been shown to activate multiple receptor subtypes. CGRP binds with high affinity to the GCRP receptor, a membrane receptor composed of two subunits, calcitonin receptor-like receptor (CLR) and receptor activity-modifying protein 1 (RAMP1) [52, 53]. These subunits are not unique to the CGRP receptor; they comprise components of receptors for related peptides such as adrenomedullin (AM), calcitonin (CT), and amylin (AMY). A combination of CLR with RAMP2 or RAMP3 creates the AM1 and AM2 receptors, respectively, and the combination of CT with RAMP1 creates the AMY1 receptor [54]. CGRP has demonstrated activity at both adrenomedullin and amylin receptors, particularly at AMY1, which exhibits a comparable affinity for CGRP with the CGRP receptor [55]. This evidence suggests that CGRP action may be mediated through multiple different receptor subtypes, each with differing degrees of CGRP binding affinity and unique functions [53].
Pathophysiology of Trigeminal Neuralgia
The pathophysiology of trigeminal neuralgia pain is complex, and not fully clarified, and multiple mechanisms have been proposed. The most commonly accepted cause of trigeminal neuralgia is compression of the trigeminal nerve root at the nerve root entry zone, often by a blood vessel or tumor [15]. Additionally, the transition from Schwann cell myelination to oligodendrocyte myelination within the nerve root entry zone has been proposed to increase the trigeminal nerve’s susceptibility to damaging pressure, leading to demyelination [15]. This mechanism may explain the association between demyelinating disorders, such as multiple sclerosis, with trigeminal neuralgia [56]. Furthermore, the “ignition hypothesis” explains that demyelination may contribute to hyperexcitable neurons capable of generating ectopic impulses that initiate spontaneous episodes of trigeminal neuropathic pain [56,57,58].
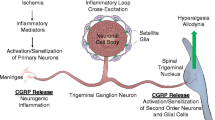
Two mechanisms are thought to be responsible for the ability of weak, localized stimuli to spread and trigger pain paroxysms, ephaptic cross-talk, and crossed after discharge [58]. Ephaptic cross-talk occurs when electrical currents from a depolarized neuron spread to demyelinated neurons in close proximity. Crossed after-discharge occurs when excitation of nearby sensory afferents induces the non-synaptic release of neurotransmitters, which diffuse to nearby trigeminal neurons, leading to their activation and inducing autorhythmic firing. This may explain how minimal sensory stimulation in the trigeminal distribution can result in widespread trigeminal excitation and trigger episodes of neuropathic pain [58,59,60] (Fig. 2).

Pathophysiology of trigeminal neuralgia
CGRP and Pathophysiology of Trigeminal Neuralgia
CGRP is a neuropeptide present within nearly 40% of trigeminal neuron cell bodies [61]. Accumulating evidence has found CGRP to be a major mediator of migraine [62]. The levels of CGRP are significantly increased in the serum (measured in external jugular blood) during acute migraine attacks and significantly increased in the cerebrospinal fluid in chronic migraine patients [63, 64]. Furthermore, intravenous infusion of CGRP was shown to induce migraine-like attacks in migraine sufferers [65]. Trigeminal nerve activation releases CGRP, which acts to facilitate pain transmission and contribute to the generation of a hyperresponsive state. CGRP also acts as a potent vasodilator, causing cerebral vascular vasodilation upon receptor binding [66,67,68]. The advancement in the understanding of CGRP in migraine pathophysiology has led to the development of migraine therapies that inhibit the action of CGRP [66]. The occurrence and abundance of CGRP in nervous tissue give this neuropeptide an important role as a neuromodulator in many parts of the nervous system, including the nociceptive system [37].
Similar to migraine, several studies have shown significantly increased levels of CGRP in blood and cerebrospinal fluid (CSF) in patients with primary trigeminal neuralgia, suggesting that CGRP may also be involved in the pathophysiology of trigeminal neuralgia [26, 68]. Qin et al. found that CGRP levels were associated with the severity of pain symptoms [68]. Interestingly, disease onset and duration of disease did not directly correlate with CGRP levels in this study. Nonetheless, these studies provide strong direct evidence suggesting the involvement of CGRP in the pathogenesis of pain in trigeminal neuralgia, likely through increasing ectopic discharges within injured trigeminal nerves [28, 69].
There is currently little data published on the use of CGRP-targeted monoclonal antibodies in patients with trigeminal neuralgia. However, several studies involving CGRP antagonism have been conducted in animal models of trigeminal neuralgia. In two separate studies, Michot and colleagues [70, 71] found that both olcegepant and MK-8825, chemically unrelated CGRP receptor antagonists, significantly reduced hyperalgesia caused by infra-orbital nerve ligation in rats. Chronic constriction injury of the infraorbital branch (CCI-ION) of the trigeminal nerve was used as a rat model of trigeminal neuralgia. Data from these studies suggest that CGRP plays a role in infra-orbital nerve constriction-mediated neuropathic pain and that CGRP antagonists should be further studied as a potential treatment for trigeminal neuralgia [70, 71].
Xiong et al. studied the effects of Emodin on pain transmission mediated by P2X3 receptors and CGRP in the CCI-ION rat model of trigeminal neuralgia. This study found a significant increase in P2X3 receptor expression and CGRP levels in the trigeminal ganglion 14 days post-CCI-ION surgery compared to the control, a group of rats which underwent a sham surgery. These increased measurements of P2X3 receptor expression and CGRP levels were associated with neuronal hyperresponsiveness as demonstrated by a lower mechanical withdrawal threshold. This experimental group of rats then received Emodin, an inhibitor of P2X3 receptors, which reduced the expression of both P2X3 receptors and CGRP levels in the trigeminal ganglion. Reduced expression of P2X3 and CGRP levels was found to interfere with pain transmission, demonstrated by an increased threshold to mechanical hypersensitivity. The findings in this study suggest that reduced levels of CGRP, through inhibition of P2X3 receptors by Emodin, may be the mechanism of pain relief in trigeminal neuralgia [72].
According to previous findings, activation of glial cells in the central nervous system results in the release of pro-inflammatory cytokines that may increase pain hypersensitivity [73]. Adenosine monophosphate-activated protein kinase (AMPK) activation is thought to inhibit the glial release of pro-inflammatory cytokines and ultimately reduce neuropathic pain. Thus, Yang et al. [74] hypothesized that activation of AMPK could suppress neuroinflammation induced by inflammatory cytokines in the spinal trigeminal nucleus to relieve pain in trigeminal neuralgia. Resveratrol, a natural compound found in red wine with anti-inflammatory properties, was employed as an activator of AMPK. The study found that administration of a single dose of resveratrol to rats 14 days post CCI of the trigeminal nerve, significantly relieved mechanical allodynia. The threshold for mechanical withdrawal increased with increasing doses of resveratrol. Subsequent administration of an AMPK inhibitor was found to reverse the effects of the resveratrol on mechanical allodynia in this group of rats. These findings were consistent with the hypothesis, suggesting that activation of AMPK by resveratrol could reduce pain in a rat model of trigeminal neuralgia. The study also found that elevated CGRP levels in the spinal trigeminal nucleus post-CCI were suppressed following the administration of resveratrol. Overall, evidence of this study suggests that resveratrol may be used for trigeminal neuralgia treatment through targeting neuroglia and CGRP [74].
All the above studies [70,71,72, 74] suggest a role for CGRP in the pathophysiology of trigeminal neuralgia. Two of the studies demonstrated significantly elevated levels of CGRP in the trigeminal ganglion [72] and spinal trigeminal nucleus [74] after CCI-ION in rats, which serves as a model of TN. In summary, inhibition of CGRP neurotransmission through various interventions was found to improve pain hypersensitivity in the rat model of trigeminal neuralgia. Similarly, current emerging therapies for TN, including botulinum toxin and triptans, have been shown to effectively decrease CGRP levels, contributing to pain relief as further described above. These findings have the potential to be translated into the clinic as more direct trigeminal neuralgia interventions.
CGRP and Human Studies
Recently, a retrospective study [75] was conducted that included 10 patients, ages 49 to 64 years, diagnosed with TN and treated with monthly self-administered injections of Erenumab for a total of 6 months. All 10 patients enrolled in the trial were either on or had failed 2 preventative medications for trigeminal neuralgia. This study assessed self-reported pain on a Numeric Pain Rating Scale (NPRS) of 1-10 at baseline and at follow-up. In addition, data was collected on side effects experienced during treatment, changes in headache frequency in patients with comorbid migraine, and global mood improvement assessment (worse/no change/improved). Results of the study revealed that 9 out of 10 patients experienced a significant decrease in pain and a significant increase in global mood improvement. Furthermore, 5 out of 10 patients reported an NPRS score of 0 at the 6-month follow-up. Out of the 5 patients with comorbid migraine, 4 reported a decrease in the number of headache days per month. These findings are highly supportive of erenumab being an effective and safe treatment for refractory TN on top of their contribution to pain management in migraine. The small sample size, lack of placebo control group, and self-reported data are limitations of the study and large-scale controlled trials are needed to determine long-term efficacy.
Andersen et al. [76] completed the first randomized, double-blind trial comparing erenumab vs, placebo in patients with classical and idiopathic trigeminal neuralgia. In this study, 80 trigeminal neuralgia patients were randomly assigned to receive injections of either Erenumab 140 mg (n=40) or placebo (n=40). Participants were assessed over the following 4 weeks for a reduction of at least 30% in mean average daily pain intensity score compared to baseline, measured over 1 week prior to the treatment period. At the end of the 4-week treatment period, no significant difference was found in the number of patients who had >30% reduction in mean average daily pain intensity score in the Erenumab (18 of 40 (45%)) vs placebo (14 of 40 (35%)) group. There was additionally no significant difference in secondary endpoints including >30% reduction in the mean number of daily pain paroxysms in the erenumab (21 of 40 (53%)) vs placebo (17 of 40 (45%)) group. The results of this trial suggest that anti-CGRP monoclonal antibodies do not play a significant role in the reduction of pain or the number of pain paroxysms in patients with trigeminal neuralgia. However, a limitation of this study may be the duration of treatment with erenumab as significant reductions in neuropathic pain may take longer to occur. Thus, additional longer-term double-blind, placebo-controlled clinical trials are necessary to assess for erenumab efficacy.
Conclusions
CGRP and its receptors are widely distributed in the peripheral and central trigeminal nociceptive system. CGRP may also have a pro-inflammatory effect on peripheral nociceptors in the trigeminal system. Moreover, while CGRP is a known mediator in migraine and cluster headache, accumulating data suggests that CGRP may play a role in the pathophysiology of trigeminal neuralgia as well. CGRP levels in the cerebrospinal fluid and blood are found to be significantly elevated in patients with trigeminal neuralgia patients during an acute pain attack. OnabotulinumtoxinA has shown some efficacy in reducing CGRP plasma levels in trigeminal neuralgia patients. There is some emerging evidence for the use of CGRP inhibitors and monoclonal antibodies in patients with trigeminal neuralgia. If CGRP inhibition is a potential target for pain relief in trigeminal neuralgia, CGRP modulator biological therapies approved in the treatment of a migraine headache may have the potential to be effective in treating trigeminal neuralgia. Further investigations on this subject are warranted.
References
Katusic S, Beard CM, Bergstralh E, Kurland LT. Incidence and clinical features of trigeminal neuralgia, Rochester, Minnesota, 1945-1984. Ann Neurol. 1990;27(1):89–95. https://doi.org/10.1002/ana.410270114.
De Toledo IP, Conti Réus J, Fernandes M, Porporatti AL, Peres MA, Takaschima A, Linhares MN, De Luca Canto G. Prevalence of trigeminal neuralgia: a systematic review. J Am Dent Assoc. 2016;147(7):570–576.e572. https://doi.org/10.1016/j.adaj.2016.02.014.
Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd. (2018). Cephalalgia., 38(1), 1-211. https://doi.org/10.1177/0333102417738202.
Jones MR, Urits I, Ehrhardt KP, Cefalu JN, Kendrick JB, Park DJ, Cornett EM, Kaye AD, Viswanath O. A comprehensive review of trigeminal neuralgia. Curr Pain Headache Rep. 2019;23(10):74. https://doi.org/10.1007/s11916-019-0810-0.
Rasmussen P. Facial pain. II. A prospective survey of 1052 patients with a view of: character of the attacks, onset, course, and character of pain. Acta Neurochir. (Wien). 1990;107(3-4):121–8. https://doi.org/10.1007/BF01405790.
Maarbjerg S, Gozalov A, Olesen J, Bendtsen L. Concomitant persistent pain in classical trigeminal neuralgia--evidence for different subtypes. Headache. 2014a;54(7):1173–83. https://doi.org/10.1111/head.12384.
Brisman R. Constant face pain in typical trigeminal neuralgia and response to γ knife radiosurgery. Stereotact Funct Neurosurg. 2013;91(2):122–8. https://doi.org/10.1159/000343206.
Cruccu G, Finnerup NB, Jensen TS, Scholz J, Sindou M, Svensson P, Treede RD, Nurmikko T. Trigeminal neuralgia: new classification and diagnostic grading for practice and research. Neurology. 2016;87(2):220–8. https://doi.org/10.1212/WNL.0000000000002840.
Brisman R. Trigeminal neuralgia and multiple sclerosis. Arch Neurol. 1987;44(4):379–81. https://doi.org/10.1001/archneur.1987.00520160021008.
Di Stefano G, Maarbjerg S, Truini A. Trigeminal neuralgia secondary to multiple sclerosis: from the clinical picture to the treatment options. J Headache Pain. 2019;20(1):20. https://doi.org/10.1186/s10194-019-0969-0.
Gronseth G, Cruccu G, Alksne J, Argoff C, Brainin M, Burchiel K, Nurmikko T, Zakrzewska JM. Practice parameter: the diagnostic evaluation and treatment of trigeminal neuralgia (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the European Federation of Neurological Societies. Neurology. 2008;71(15):1183–90. https://doi.org/10.1212/01.wnl.0000326598.83183.04.
Maarbjerg S, Gozalov A, Olesen J, Bendtsen L. Trigeminal neuralgia--a prospective systematic study of clinical characteristics in 158 patients. Headache. 2014b;54(10):1574–82. https://doi.org/10.1111/head.12441.
Cruccu G. Trigeminal neuralgia. CONTINUUM: Lifelong Learn. in Neurol. 2017;23(2):396–420. https://doi.org/10.1212/CON.0000000000000451.
Cruccu G, Gronseth G, Alksne J, Argoff C, Brainin M, Burchiel K, Nurmikko T, Zakrzewska JM. AAN-EFNS guidelines on trigeminal neuralgia management. Eur J Neurol. 2008;15(10):1013–28. https://doi.org/10.1111/j.1468-1331.2008.02185.x.
Bendtsen L, Zakrzewska JM, Heinskou TB, Hodaie M, Leal PRL, Nurmikko T, Obermann M, Cruccu G, Maarbjerg S. Advances in diagnosis, classification, pathophysiology, and management of trigeminal neuralgia. Lancet Neurol. 2020;19(9):784–96. https://doi.org/10.1016/S1474-4422(20)30233-7.
McQuay HJ, Moore RA, Eccleston C, Morley S, Williams AC. Systematic review of outpatient services for chronic pain control. Health Technol Assess. 1997;1(6):i–iv. https://doi.org/10.3310/hta1060.
Al-Quliti KW. Update on neuropathic pain treatment for trigeminal neuralgia: The pharmacological and surgical options. Neurosciences. (Riyadh). 2015;20(2):107–14. https://doi.org/10.17712/nsj.2015.2.20140501.
Montano N, Conforti G, Di Bonaventura R, Meglio M, Fernandez E, Papacci F. Advances in diagnosis and treatment of trigeminal neuralgia. Ther Clin Risk Manag. 2015;11:289–99. https://doi.org/10.2147/TCRM.S37592.
Obermann M. Treatment options in trigeminal neuralgia. Ther Adv Neurol Disord. 2010;3(2):107–15. https://doi.org/10.1177/1756285609359317.
Zhang WB, Min LZ, Tao BB, Sun QY, Li ST, Wang XQ. Prognosis comparison of different branches of trigeminal neuralgia. World Neurosurg. 2020;133:e1–5. https://doi.org/10.1016/j.wneu.2019.06.115.
Zhang WB, Zeng YY, Chang BW, Min LZ, Sun QY, Li B, Tao BB, Wang XQ. Prognostic nomogram for microvascular decompression-treated trigeminal neuralgia. Neurosurg Rev. 2021;44(1):571–7. https://doi.org/10.1007/s10143-020-01251-0.
Zheng JH, Sun K, Zhang HT, Xie YJ, Wang-Yang LX, Chen HY, Wang C. A study on the recurrence rate of trigeminal neuralgia after MVD and the related factors. J Neurol Surg B Skull Base. 2020;81(5):572–8. https://doi.org/10.1055/s-0039-1692687.
Mimeh H, Fenech Magrin AM, Myers S, Ghanem AM. A critical review of botulinum toxin type a in the prophylactic treatment of chronic migraine in adults. Aesthet Surg J. 2019;39(8):898–907. https://doi.org/10.1093/asj/sjy224.
Tassorelli C, Sances G, Avenali M, De Icco R, Martinelli D, Bitetto V, Nappi G, Sandrini G. Botulinum toxin for chronic migraine: clinical trials and technical aspects. Toxicon. 2018;147:111–5. https://doi.org/10.1016/j.toxicon.2017.08.026.
Morra ME, Elgebaly A, Elmaraezy A, Khalil AM, Altibi AM, Vu TL, Mostafa MR, Huy NT, Hirayama K. Therapeutic efficacy and safety of botulinum toxin A therapy in trigeminal neuralgia: a systematic review and meta-analysis of randomized controlled trials. J Headache Pain. 2016;17(1):63. https://doi.org/10.1186/s10194-016-0651-8.
Zhang Y, Lian Y, Zhang H, Xie N, Chen Y. CGRP plasma levels decrease in classical trigeminal neuralgia patients treated with botulinum toxin type A: a pilot study. Pain Med. 2020;21(8):1611–5. https://doi.org/10.1093/pm/pnaa028.
Kanai A, Saito M, Hoka S. Subcutaneous sumatriptan for refractory trigeminal neuralgia. Headache. 2006;46(4):577–82. https://doi.org/10.1111/j.1526-4610.2006.00405.x.
Kanai A, Suzuki A, Osawa S, Hoka S. Sumatriptan alleviates pain in patients with trigeminal neuralgia. Clin J Pain. 2006;22(8):677–80.
Shimohata K, Shimohata T, Motegi R, Miyashita K. Nasal sumatriptan as adjunctive therapy for idiopathic trigeminal neuralgia: report of three cases. Headache. 2009;49(5):768–70. https://doi.org/10.1111/j.1526-4610.2008.01254.x.
Juhasz G, Zsombok T, Jakab B, Nemeth J, Szolcsanyi J, Bagdy G. Sumatriptan causes parallel decrease in plasma calcitonin gene-related peptide (CGRP) concentration and migraine headache during nitroglycerin induced migraine attack. Cephalalgia. 2005;25(3):179–83. https://doi.org/10.1111/j.1468-2982.2005.00836.x.
Halker RB, Vargas BB. Pathophysiology of migraine & triptan mechanism of action. In: Triptans for Migraine. London, England: Future Medicine Ltd; 2012. p. 6–15.
Benemei S, Cortese F, Labastida-Ramírez A, Marchese F, Pellesi L, Romoli M, Vollesen AL, Lampl C, Ashina M. Triptans and CGRP blockade - impact on the cranial vasculature. J Headache Pain. 2017;18(1):103. https://doi.org/10.1186/s10194-017-0811-5.
Amara SG, Jonas V, Rosenfeld MG, Ong ES, Evans RM. Alternative RNA processing in calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature. 1982;298(5871):240–4. https://doi.org/10.1038/298240a0.
Ho TW, Edvinsson L, Goadsby PJ. CGRP and its receptors provide new insights into migraine pathophysiology. Nat Rev Neurol. 2010;6(10):573–82. https://doi.org/10.1038/nrneurol.2010.127.
Tepper SJ. History and review of anti-calcitonin gene-related peptide (CGRP) therapies: from translational research to treatment. Headache. 2018;58(Suppl 3):238–75. https://doi.org/10.1111/head.13379.
Yuan H, Spare NM, Silberstein SD. Targeting CGRP for the prevention of migraine and cluster headache: a narrative review. Headache. 2019;59(Suppl 2):20–32. https://doi.org/10.1111/head.13583.
Schou WS, Ashina S, Amin FM, Goadsby PJ, Ashina M. Calcitonin gene-related peptide and pain: a systematic review. J Headache Pain. 2017;18(1):34. https://doi.org/10.1186/s10194-017-0741-2.
Goadsby PJ, Holland PR, Martins-Oliveira M, Hoffmann J, Schankin C, Akerman S. Pathophysiology of migraine: a disorder of sensory processing. Physiol Rev. 2017;97(2):553–622. https://doi.org/10.1152/physrev.00034.2015.
Eftekhari S, Warfvinge K, Blixt FW, Edvinsson L. Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J Pain. 2013;14(11):1289–303. https://doi.org/10.1016/j.jpain.2013.03.010.
Edvinsson L, Fredholm BB, Hamel E, Jansen I, Verrecchia C. Perivascular peptides relax cerebral arteries concomitant with stimulation of cyclic adenosine monophosphate accumulation or release of an endothelium-derived relaxing factor in the cat. Neurosci Lett. 1985;58(2):213–7. https://doi.org/10.1016/0304-3940(85)90166-1.
Messlinger K, Lennerz JK, Eberhardt M, Fischer MJ. CGRP and NO in the trigeminal system: mechanisms and role in headache generation. Headache. 2012;52(9):1411–27. https://doi.org/10.1111/j.1526-4610.2012.02212.x.
Messlinger K. The big CGRP flood - sources, sinks and signalling sites in the trigeminovascular system. J Headache Pain. 2018;19(1):22. https://doi.org/10.1186/s10194-018-0848-0.
Noseda R, Jakubowski M, Kainz V, Borsook D, Burstein R. Cortical projections of functionally identified thalamic trigeminovascular neurons: implications for migraine headache and its associated symptoms. J Neurosci. 2011;31(40):14204–17. https://doi.org/10.1523/JNEUROSCI.3285-11.2011.
Lennerz JK, Rühle V, Ceppa EP, Neuhuber WL, Bunnett NW, Grady EF, Messlinger K. Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: differences between peripheral and central CGRP receptor distribution. J Comp Neurol. 2008;507(3):1277–99. https://doi.org/10.1002/cne.21607.
Eftekhari S, Salvatore CA, Johansson S, Chen TB, Zeng Z, Edvinsson L. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood-brain barrier. Brain Res. 2015;1600:93–109. https://doi.org/10.1016/j.brainres.2014.11.031.
Pannese E. The satellite cells of the sensory ganglia. Adv Anat Embryol Cell Biol. 1981;65:1–111. https://doi.org/10.1007/978-3-642-67750-2.
Pannese E, Ledda M, Cherkas PS, Huang TY, Hanani M. Satellite cell reactions to axon injury of sensory ganglion neurons: increase in number of gap junctions and formation of bridges connecting previously separate perineuronal sheaths. Anat Embryol. (Berl). 2003;206(5):337–47. https://doi.org/10.1007/s00429-002-0301-6.
Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev. 2005;48(3):457–76. https://doi.org/10.1016/j.brainresrev.2004.09.001.
Vit JP, Jasmin L, Bhargava A, Ohara PT. Satellite glial cells in the trigeminal ganglion as a determinant of orofacial neuropathic pain. Neuron Glia Biol. 2006;2(4):247–57. https://doi.org/10.1017/s1740925x07000427.
Thalakoti S, Patil VV, Damodaram S, Vause CV, Langford LE, Freeman SE, Durham PL. Neuron-glia signaling in trigeminal ganglion: implications for migraine pathology. Headache. 2007;47(7):1008–23. https://doi.org/10.1111/j.1526-4610.2007.00854.x.
Afroz S, Arakaki R, Iwasa T, Oshima M, Hosoki M, Inoue M, Baba O, Okayama Y, Matsuka Y. CGRP induces differential regulation of cytokines from satellite glial cells in trigeminal ganglia and orofacial nociception. Int J Mol Sci. 2019;20(3):711. https://doi.org/10.3390/ijms20030711.
Hay DL, Poyner DR, Sexton PM. GPCR modulation by RAMPs. Pharmacol Ther. 2006;109(1-2):173–97. https://doi.org/10.1016/j.pharmthera.2005.06.015.
Walker CS, Hay DL. CGRP in the trigeminovascular system: a role for CGRP, adrenomedullin and amylin receptors? Br J Pharmacol. 2013;170(7):1293–307. https://doi.org/10.1111/bph.12129.
Christopoulos A, Christopoulos G, Morfis M, Udawela M, Laburthe M, Couvineau A, Kuwasako K, Tilakaratne N, Sexton PM. Novel receptor partners and function of receptor activity-modifying proteins. J Biol Chem. 2003;278(5):3293–7. https://doi.org/10.1074/jbc.C200629200.
Walker CS, Eftekhari S, Bower RL, Wilderman A, Insel PA, Edvinsson L, Waldvogel HJ, Jamaluddin MA, Russo AF, Hay DL. A second trigeminal CGRP receptor: function and expression of the AMY1 receptor. Ann Clin Transl Neurol. 2015;2(6):595–608. https://doi.org/10.1002/acn3.197.
Love S, Coakham HB. Trigeminal neuralgia: pathology and pathogenesis. Brain. 2001;124(Pt 12):2347–60. https://doi.org/10.1093/brain/124.12.2347.
Maarbjerg S, Di Stefano G, Bendtsen L, Cruccu G. Trigeminal neuralgia - diagnosis and treatment. Cephalalgia. 2017;37(7):648–57. https://doi.org/10.1177/0333102416687280.
Rappaport HZ, Devor M. Trigeminal neuralgia: the role of self-sustaining discharge in the trigeminal ganglion. Pain. 1994;56(2):127–38. https://doi.org/10.1016/0304-3959(94)90086-8.
Devor M, Amir R, Rappaport ZH. Pathophysiology of trigeminal neuralgia: the ignition hypothesis. Clin J Pain. 2002;18(1):4–13. https://doi.org/10.1097/00002508-200201000-00002.
Cruccu G, Di Stefano G, Truini A. Trigeminal neuralgia. N Engl J Med. 2020;383(8):754–62. https://doi.org/10.1056/NEJMra1914484.
Tajti J, Uddman R, Möller S, Sundler F, Edvinsson L. Messenger molecules and receptor mRNA in the human trigeminal ganglion. J Auton Nerv Syst. 1999;76(2-3):176–83. https://doi.org/10.1016/s0165-1838(99)00024-7.
Ashina M. Migraine. N Engl J Med. 2020;383(19):1866–76. https://doi.org/10.1056/NEJMra1915327.
Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28(2):183–7. https://doi.org/10.1002/ana.410280213.
van Dongen RM, Zielman R, Noga M, Dekkers OM, Hankemeier T, van den Maagdenberg AM, Terwindt GM, Ferrari MD. Migraine biomarkers in cerebrospinal fluid: a systematic review and meta-analysis. Cephalalgia. 2017;37(1):49–63. https://doi.org/10.1177/0333102415625614.
Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CGRP may play a causative role in migraine. Cephalalgia. 2002;22(1):54–61. https://doi.org/10.1046/j.1468-2982.2002.00310.x.
Edvinsson L. The trigeminovascular pathway: role of CGRP and CGRP receptors in migraine. Headache. 2017;57(Suppl 2):47–55. https://doi.org/10.1111/head.13081.
Limmroth V, Katsarava Z, Liedert B, Guehring H, Schmitz K, Diener HC, Michel MC. An in vivo rat model to study calcitonin gene related peptide release following activation of the trigeminal vascular system. Pain. 2001;92(1-2):101–6. https://doi.org/10.1016/s0304-3959(00)00475-9.
Iyengar S, Ossipov MH, Johnson KW. The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain. 2017;158(4):543–59. https://doi.org/10.1097/j.pain.0000000000000831.
Qin ZL, Yang LQ, Li N, Yue JN, Wu BS, Tang YZ, Guo YN, Lai GH, Ni JX. Clinical study of cerebrospinal fluid neuropeptides in patients with primary trigeminal neuralgia. Clin Neurol Neurosurg. 2016;143:111–5. https://doi.org/10.1016/j.clineuro.2016.02.012.
Michot B, Bourgoin S, Viguier F, Hamon M, Kayser V. Differential effects of calcitonin gene-related peptide receptor blockade by olcegepant on mechanical allodynia induced by ligation of the infraorbital nerve vs the sciatic nerve in the rat. Pain. 2012;153(9):1939–48. https://doi.org/10.1016/j.pain.2012.06.009.
Michot B, Kayser V, Hamon M, Bourgoin S. CGRP receptor blockade by MK-8825 alleviates allodynia in infraorbital nerve-ligated rats. Eur J Pain. 2015;19(2):281–90. https://doi.org/10.1002/ejp.616.
Xiong W, Wu RP, Tan MX, Tong ZJ, He LK, Guan S, Liu LJ, Yin CC, Shen YL, Ge HX, Gao Y. Emodin inhibits the expression of receptor and calcitonin-gene-related peptide release in trigeminal ganglia of trigeminal neuralgia rats. Int J Clin Exp Pathol. 2017;10(11):11317–25.
Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24(8):450–5. https://doi.org/10.1016/s0166-2236(00)01854-3.
Yang YJ, Hu L, Xia YP, Jiang CY, Miao C, Yang CQ, Yuan M, Wang L. Resveratrol suppresses glial activation and alleviates trigeminal neuralgia via activation of AMPK. J Neuroinflammation. 2016;13(1):84. https://doi.org/10.1186/s12974-016-0550-6.
Parascandolo E, Levinson K, Rizzoli P, Sharon R. Efficacy of erenumab in the treatment of trigeminal neuralgia: a retrospective case series. Neurol: Clin Pract. 2021;11(3):227–31. https://doi.org/10.1212/cpj.0000000000001075.
Schott Andersen AS, Maarbjerg S, Noory N, Heinskou TB, Forman JL, Cruccu G, Ashina M, Bendtsen L. Safety and efficacy of erenumab in patients with trigeminal neuralgia in Denmark: a double-blind, randomised, placebo-controlled, proof-of-concept study. Lancet Neurol. 2022;21(11):994–1003. https://doi.org/10.1016/S1474-4422(22)00294-0.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Code Availability
Not applicable.
Author information
Authors and Affiliations
Contributions
Roni Sharon, M. D., identified the idea for this work. Rachel Retsky, B. S., conducted the literature search and wrote the first draft of the manuscript. Roni Sharon, M. D.; Sait Ashina, M. D.; Daniel Oved, M. D.; and Rachel Retsky, B. S., edited the previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
Sait Ashina (S. A.) received honoraria for consulting from Allergan/AbbVie, Amgen, Biohaven, Eli Lilly, Impel, NeuroPharma, Novartis, Satsuma, Supernus, Theranica, and Percept. Roni Sharon has been a paid speaker or consultant for the following companies: Novartis, Abbvie, Pfizer, Teva, Eli Lilly, Theranica, Neurolief, BOL, GLG, Guidepoint. Rachel Retsky and Daniel Oved declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Medicine
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Retsky, R., Ashina, S., Oved, D. et al. Calcitonin Gene-Related Peptide and Trigeminal Neuralgia. SN Compr. Clin. Med. 5, 75 (2023). https://doi.org/10.1007/s42399-023-01407-1
Accepted:
Published:
DOI: https://doi.org/10.1007/s42399-023-01407-1