
Abstract
Wilson’s disease (WD) was defined in 1912 as a rare autosomal recessive disorder that leads to defective excretion of copper from the body. Normally, copper is absorbed in the small intestine by enterocytes and transported into the blood via ATP7A proteins. Any excess copper is directed to the liver and excreted by hepatocytes via ATP7B. However, in patients with WD, this ATP7B protein is mutated, and copper accumulates within the body, causing various symptoms. Wilson’s disease can manifest in many different ways, such as neurological, fulminant hepatic fibrosis, ophthalmic, and even psychiatric presentation types, making it difficult to diagnose. Wilson’s disease is essential in the differential diagnosis of an individual expressing neurological symptoms because it is a treatable disease that can progressively increase in severity, and if left untreated, there is a risk of permanent brain impairment. In this case report, a 32-year-old female initially presents progressively worsening dystonia symptoms of the trunk, a wing-flapping tremor, cognitive dysfunction, and dysphonia. In terms of past medical history and past surgical history, no records were available, and she had no recollection of past genetic testing or brain MRIs but claimed to be taking Cuprimine® (penicillamine), Artane (trihexyphenidyl), and oxycodone. The physician performed a physical examination and ordered a brain MRI to collect supportive evidence for the WD diagnosis. This case illustrates a classic presentation of the neurologic type WD in the context of facilitating patient education, performing thorough physical examinations, and adjusting patient treatment plans.
This article is part of the Topical Collection on Medicine
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wilson’s disease (WD) is an autosomal recessive genetic disorder secondary to a mutation of the ATP7B gene on chromosome 13 (13q14.3) [1]. The ATP7B gene encodes for a transmembrane ATPase that transports copper. A homozygous mutation of this gene will cause WD with a disruption of normal copper excretion, resulting in copper accumulation. Wilson’s disease is rare and has an estimated prevalence of 1:30,000 symptomatic individuals and 1:90 carriers of the disease [1, 2].
Most WD cases are diagnosed between the ages of 5 and 35 years old, although symptoms may begin to appear at any age. Symptoms typically appear following copper accumulation due to the mutated copper transporting transmembrane ATPase. The most common clinical manifestations of WD involve the liver and the brain. Hepatic manifestations include elevated liver enzymes, acute hepatitis, acute liver failure, and compensated or decompensated cirrhosis of the liver. Neurological and neuropsychiatric manifestations include personality or mood disorders, psychosis, cognitive impairment, involuntary movements, dysphonia, drooling, and ataxia [1]. Dry mouth and anhidrosis may also be seen [3]. Other clinical manifestations found in WD include Kayser-Fleischer rings, thrombocytopenia, hemolytic anemia, leukopenia, osteoporosis, and cardiac arrhythmias [2]. Signs of systemic manifestations include dermatological findings such as hyperpigmentation of the anterior lower extremities and seborrhea [3, 4].
Neurological symptoms tend to be the first to present in WD. These symptoms include tremor, dystonia, ataxia, drooling, and dysphagia. The tremor typically begins as either unilateral or bilateral involvement of the distal upper extremities. A wing-flapping tremor may be seen in patients who also have liver failure. The most common neurological symptom found in WD patients is dysarthria. Parkinsonism is also relatively common among WD patients with neurological manifestations, and those patients present with tremor, rigidity, ataxia, and postural disturbances. On a T2-weighted brain MRI, symmetric hyperintense changes may be seen in the putamen, caudate nuclei, thalamus, midbrain, and pons, which can assist in diagnosing WD and aid in monitoring the disease progression. The “giant panda sign” is a radiologic finding seen in the midbrain of some WD patients. The panda sign is displayed as a normal intensity of the red nuclei and lateral pars reticulata with hyperintensity of the tegmentum and hypointensity of the superior colliculus [5]. The neurological changes due to excess copper deposits in the brain may lead to psychiatric manifestations as well. These include major depressive disorder, bipolar disorder, irritability, and aggression [2].
Although a rare disease, it is important for physicians to be aware of WD’s presenting signs and symptoms. An early diagnosis allows for sooner interventional treatments, preventing disease progression and improving the patient’s quality of life.
Case Report
This patient presented as a 32-year-old female in August of 2014 for evaluation of symptoms consistent with Wilson’s disease. The patient is Spanish speaking, and her husband was her primary historian. The patient was unaware of her medical history and did not present with any medical records. She has known consanguinity with parents who were first-degree cousins, but she has no known family history of WD. The patient had been experiencing symptoms as detailed below for approximately three to four years prior to being seen in August of 2014. While the patient did not have any medical records, she was prescribed multiple medications from a prior physician, which she was taking as indicated. Her medication regimen on initial presentation included Cuprimine® (250 mg, one in the morning and two at night), Atrane (2 mg, twice daily for drooling), and oxycodone (5 mg as needed for pain related to her left wrist contracture). She sought treatment with this neurologist as she reported her symptoms were rapidly worsening despite taking these medications. Her reported symptoms on her initial visit to the neurologist included dystonia of her trunk, contracture of her left arm, “wing-flapping” movements of her right hand, and dysphonic speech. She did not report any gastrointestinal or systemic complaints. It is unclear exactly when the patient first became symptomatic or the order in which her symptoms progressed as she described having symptoms for a few years prior that worsened. This patient’s history was limited. The patient did not present with any prior records of function testing, urine copper levels, brain imaging, or past medication trials.
On her initial physical examination in 2014, the patient exhibited a contracture of her left arm, profound flexion of her left wrist, and “wing-flapping” movements of her right hand. Also, she had some degree of akathisia as well as frequent standing and turning about movements. The patient displayed deep tendon reflexes of 0/4 in her left upper extremity and 1/4 in her right upper extremity and bilateral lower extremities. Cranial nerves II–XII were demonstrated to be intact; the Mini-Mental Status Examination was documented to be 30/30, and her sensations to pinprick, proprioception, vibration, and stereognosis were intact as well. She was well-nourished and well-developed.
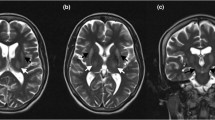
After the patient’s initial evaluation, she was scheduled to have an MRI of her brain to evaluate for metal deposits in her basal ganglia. She also had copper studies, iron levels, urinary copper studies, and liver function tests performed. The patient was recommended to have genetic testing performed. The patient underwent an MRI of her brain with and without contrast on 09/17/2014 with results indicating low signal intensities in the basal ganglia region and thalamic nuclei and in the midbrain area of the red nuclei bilaterally, which are consistent with the typical “panda sign” seen in WD (Fig. 1). Additionally, there were hyperintensities in the frontal periventricular white matter and external capsule regions bilaterally which were felt most likely to be related to chronic microvascular ischemic damage. The patient was later found to have had an MRI done previously with her referring physician on 01/09/2014, and no new changes were present. Her liver function tests were normal, so hepatic ultrasonography or further investigation was not indicated as liver involvement was not suspected. The patient’s ceruloplasmin level on 09/11/2014 was less than 3 mg/dL (reference 18–53 mg/dL), and on penicillamine, she was excreting copper in the urine as expected. She had a minimally elevated hematocrit level of 46.1% (reference 35.0–45.0%), but otherwise demonstrated no significant lab abnormalities. The patient underwent genetic testing in March 2015, which was positive for one suspected pathogenic mutation in the ATP7B gene supporting WD’s suspicion.

Axial brain MRI from 09/17/2014 obtained via high-field 3.0-Tesla Philips Intera Scanner. Sagittal T1, axial T2, axial T2 flare, axial DWI with ADC maps, and coronal T2 sequences. The blue arrow indicates the typical “panda sign” seen in Wilson’s disease
Upon reviewing all of the test results, the patient’s Cuprimine® was increased to take two pills (500 mg total) twice daily. She was not started on any additional chelation agents. Additionally, she was educated on the importance of maintaining a low copper diet and proper compliance with her medications.
As of December 2020, the patient is doing well. Her last copper level was < 5 mcg/dL (reference 70–175 mcg/dL). She continues to take Cuprimine® (250 mg, two taken twice daily) and continues to maintain a low copper diet. Her physical exam results have improved as related to her initial presentation with no continued progression of the disease.
Discussion
Since the manifestation of WD can vary between patients, the symptoms differentiating the hepatic type and the neurological type should be noted carefully. The more common hepatic pattern of WD usually presents with elevated liver enzymes, acute hepatitis, and liver cirrhosis. On the other hand, a patient with neurological type WD may have symptoms such as tremor, dystonia, ataxia, drooling, and dysphagia causing them to go undiagnosed [1]. Such a rare form of WD is at risk for delayed diagnosis and a worse outcome. Classic features initially presented in this patient that indicated neurological type WD are the wing-beating tremor and the famous “panda sign” seen on a brain MRI. Clinicians must pick up on these clues promptly to initiate treatment in these patients as soon as possible. Approximately 22–55% of patients diagnosed with WD exhibit a tremor when arms are outstretched and semi-flexed, making it a hallmark of the disease [6]. The tremor should be differentiated from the common essential tremor that is seen in parkinsonism disorders as it aids in a quick diagnosis.
The brain MRI abnormalities depict the “face of a giant panda” due to hyperintensities found in the tectal plate and signal changes in the basal ganglia, thalamus, and brainstem [7]. It is proposed that the panda sign is due to the heavy metal deposition. Even though a select number of other diseases can present with a panda sign, the physician should use this as another critical diagnostic marker for WD. Other common symptoms noted in neurological type WD include ataxia and psychiatric presentations, which may assist in early diagnosis [6].
In this particular case, the patient not only had these hallmark signs but also had been prescribed Cuprimine® previously. Cuprimine®, D-penicillamine, is a chelating agent that binds to excess copper and facilitates its excretion into the urine [2]. In the absence of medical records, the neurologist successfully confirmed the diagnosis of WD via genetic testing and monitored the patient as necessary. Considering the patient was unstable on the previous dosage of Cuprimine® and had no prior record available, this could have been a dangerous situation due to diagnosis delay. To avoid the risks of a delayed diagnosis, the patient should be educated on the physiology of the disease and the importance of medication compliance. By facilitating patient education and overcoming potential language barriers, the patient can understand their medical needs and identify their condition to a different clinician if need be. Every physician should practice this technique with their patients in order to aid in unpredictable life-threatening situations.
Other treatment options for WD include trientine, another chelator, and zinc salts. Treatment of WD is determined based on the safety considerations and the patient’s response. In asymptomatic patients, trientine may be favored over D-penicillamine due to fewer adverse side effects [2]. Zinc salts are also widely used to inhibit the absorption of copper at the intestines but may be insufficient as monotherapy [2]. Liver transplants have been successful for patients suffering from severe WD. Ongoing research may include gene therapy to restore appropriate copper metabolism.
From a physician’s perspective, early diagnosis of WD is the key to preserving the patient’s quality of life and preventing irreversible brain impairment. Penicillamine is a chelating agent with a mechanism of action that blocks copper’s intestinal absorption via zinc salts and favors the copper’s excretion [8]. The treating physician must monitor the patient, especially during the initiation of chelation therapy, because of the documented adverse effects and overtreatment potential. A few adverse effects that a prescribing physician should be aware of include but are not limited to sensitivity reactions, nephrotoxicity, and irreversible neurological damage [8]. Any clinician who is initiating chelation treatment should abide by the “start low and go slow” principle to avoid overtreatment risks [9]. A full dose chelator can mobilize free copper from other tissues and actually worsen neurological deterioration in a patient that is just beginning therapy [2]. In a study involving 143 symptomatic patients previously diagnosed with WD, 11.1% were found to experience neurological damage during the first six months of treatment, promoting the general rule of “start low and go slow” regarding therapy [9]. Clinicians must be aware of the overtreatment signs such as anemia and neutropenia to promptly discontinue the medication or switch to another agent and preserve the patient’s neurological integrity [8]. The acceptable initial dosage of Cuprimine® is 250–500 mg/day with the additional 250 mg increment every 4–7 days [8]. When the patient is stable on medication during maintenance treatment, they should be monitored every six months [8].
Other recommendations that physicians should emphasize are adhering to a strict low copper diet and supplementing with pyridoxine. Foods such as chocolate, nuts, mushrooms, crustaceans, soy, and gelatin should be avoided to limit the amount of copper in the body [8]. Supplementing with pyridoxine prevents vitamin deficiency [8].
Another element that should not be overlooked when evaluating a patient with WD is the inheritance pattern of the disease. Since WD has an autosomal recessive inheritance pattern, clinicians should meticulously document family history and carefully monitor their children’s health. In this specific case, it is especially valuable to know the inheritance pattern because of its consanguinity. Therefore, this patient is at a higher risk of developing WD and should be monitored closely.
Additional notes to consider when evaluating this particular case study is the value of documentation during a full examination. Special manifestations of Wilson’s disease, such as skin presentations, can be easily overlooked but provide subtle clues to assist in the diagnosis process. In a study analyzing 20 patients with WD, 20% of them were found to have hyperpigmentation skin abnormalities on the anterior lower extremities due to an alteration in melanin deposition [4]. Other common symptoms that have been documented in WD patients were seborrhea, dry mouth, and anhidrosis [3]. Even though these symptoms can be interpreted as vague, they may provide an essential tip-off that could establish the WD diagnosis sooner and, therefore, improve the prognosis.
Physicians must consider WD in the differential diagnosis and utilize various diagnostic clues at their fingertips to prevent treatment delay. This case report displays a neurologist that recognizes the disease’s hallmarks and monitors the patient appropriately, which all clinicians should strive to do. However, it is critical to emphasize patient education and accommodation in this scenario because it can save valuable time associated with improved neurological function. Regarding treatment and management, physicians should be aware of chelation therapy risks and schedule regular follow-ups to confirm successful maintenance therapy. The widely accepted treatment principles are establishing a diagnosis, monitoring patient compliance, detecting the complications early on, and promoting interprofessional management [8]. In the future, cases like the one presented should also include records of full examinations to assist in early diagnosis.
Conclusion
Considering Wilson’s disease in a patient’s differential diagnosis and being suspicious for the disease can streamline the diagnosis process, lead to early treatment, and potentially extend the patient’s life expectancy and quality of life. By staying updated on the presentations of neurological type WD and abiding by the treatment guidelines, any clinician should be able to manage a patient with WD. In terms of future outlooks, it would be beneficial to encourage patient education and overcome language barriers in a clinical setting to allow the patient to participate in management decisions. Other manifestations should continue to be explored and recorded in cases like the one presented to assist in the diagnosis process. A physician can provide the utmost quality of care and give the patient the best lifestyle possible by implementing all these elements in their daily practice.
Data Availability
Not applicable.
References
Sapuppo A, Pavone P, Praticò AD, Ruggieri M, Bertino G, Fiumara A. Genotype-phenotype variable correlation in Wilson disease: clinical history of two sisters with the similar genotype. BMC Med Genet. 2020;21(1):128. https://doi.org/10.1186/s12881-020-01062-6.
Mohr I, Weiss KH. Biochemical markers for the diagnosis and monitoring of Wilson disease. Clinical Biochemist Reviews. 2019;40(2):59–77. https://doi.org/10.33176/aacb-18-00014.
Chu EC, Chu N-S, Huang C-C. Autonomic involvement in Wilson’s disease: a study of sympathetic skin response and RR interval variation. J Neurol Sci. 1997;149(2):131–7. https://doi.org/10.1016/s0022-510x(97)05365-3.
Leu ML. Skin pigmentation in Wilson’s disease. JAMA. 1970;211(9):1542–3. https://doi.org/10.1001/jama.211.9.1542.
Parekh JR, Agrawal PR. Wilson’s disease: “face of giant panda” and “trident” signs together. Oxford Medical Case Reports. 2014;2014(1):16–7. https://doi.org/10.1093/omcr/omu005.
Hedera P. Wilson’s disease: a master of disguise. Parkinsonism Relat Disord. 2019;59:140–5. https://doi.org/10.1016/j.parkreldis.2019.02.016.
Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. The Lancet Neurology. 2015;14(1):103–13. https://doi.org/10.1016/s1474-4422(14)70190-5.
Rodriguez-Castro KI. Wilson’s disease: a review of what we have learned. World J Hepatol. 2015;7(29):2859–70. https://doi.org/10.4254/wjh.v7.i29.2859.
Litwin T, Dzieżyc K, Karliński M, Chabik G, Czepiel W, Członkowska A. Early neurological worsening in patients with Wilson’s disease. J Neurol Sci. 2015;355(1–2):162–7. https://doi.org/10.1016/j.jns.2015.06.010.
Code Availability
Not applicable.
Author information
Authors and Affiliations
Contributions
MES performed the treatment.
KA, MD, MP, and MSP wrote the draft.
KA and MSP were involved in the article’s conception.
MSP and MES provided the final review.
All authors read and approved the final version of the document for submission.
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable.
Informed Consent
Written informed consent was obtained from the patient.
Consent for Publication
A consent for publication was obtained prior to the article writing.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Alessi, K., DeLima, M., Pfautsch, M. et al. Neurological Type Wilson’s Disease: a Case Report. SN Compr. Clin. Med. 3, 1946–1950 (2021). https://doi.org/10.1007/s42399-021-00960-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42399-021-00960-x