
Abstract
Bimetallic Ni–Al and Ni–Fe nanoparticles catalysts derived from hydrotalcite were synthesized by co-precipitation method and applied in dry reforming of methane. The samples were calcined at 800 °C and the crystalline phases were assessed by X-ray diffraction coupled with Rietveld refinement. Other analyzes were carried out to study their textural and structural properties including, Thermogravimetric Analysis (TG), Coupled Plasma Atomic Emission Spectroscopy (ICP-AES), Brunauere-Emmette-Teller (BET), Scanning Electron Microscopy and Energy Dispersive-X-ray (SEM–EDX), Temperature Programmed Reduction (TPR), Transmission Electron Microscopy (TEM) and EDS mapping. The XRD Analyses confirmed the formation of the precursor’s layered double hydroxide structure, the formation of the γ-NiFe alloy confirmed by TEM-EDS Analysis. The specific surface area of the two samples increases after calcination, attributed to the destruction of the double-layered structure at high temperature, which produced cavities or crates resulting in larger surface areas. These catalysts were evaluated in CO2 reforming of methane under continuous flow with CH4/CO2 ratio equal to 1, at atmospheric pressure and a temperature range between 400 and 700 °C. At 700 °C, the NiAlHT catalyst displayed the best CH4 conversion (87.5%) and CO2 conversion (91.4%) compared to the conversion of CH4 (79.2%) and CO2 (84.1) for NiFeHT catalyst within 10 h stability test. The iron addition to the nickel showed improved resistance to coke deposition while a slight decrease in methane conversion was observed. The possible formation of γ-NiFe alloy observed during the study of reducibility by hydrogen was invoked to account for the catalytic behavior.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Recently, the reaction of methane with carbon dioxide (dry reforming of methane, DRM) has attracted great attention because of it converts CH4 and CO2 (Eq. 1) two major greenhouse gases into syngas (CO and H2). The H2/CO ratio is close to unity, which can be directly used as fuel, feedstock for the chemical industry or production of liquid hydrocarbons via Fischer–Tropsch synthesis [1]. Moreover, syngas produced by DRM is also can be utilized for the storage of solar or nuclear energy [2].
Two major challenges for the DRM catalyst is the active phase sintering and rapid accumulation of carbon “carbon poisoning” initiated by numerous side reactions (Eqs. 2, 3, 4 and 5) [3]. The reverse water–gas shift reaction can also occur as a side-reaction in which some of the H2 from the CH4 decomposition reacts with the CO2 to produce H2O and CO (Eq. 6) [4, 5].
In addition, the high temperature of the reaction leads to deactivation of metal catalyst due to the sintering of the active phase or the reaction between metal and support forming inactive species. Thus, a thermal stable catalyst that can resist degradation and carbon deposition is essential [5, 6].
Noble metals Rh, Ru, Pd and Pt-based catalysts are considered the most active materials for the DRM because they possess a superior coke removal ability due to the high dispersion and small particle size [1, 5]. However, the high cost of precious metals is often seen as a drawback for their application on a large scale [5].
Nickel-based catalysts are the most suitable for this reaction because of their high activity, best stability and have low-cost. Nevertheless, the main drawback of Ni-based catalysts is their deactivation by sintering of active phase and/or carbon deposition, which negatively influences catalytic performance by blocking active sites [5, 7].
Several attempts have been made to limit the formation of coke deposition, for example, intercalate the active phase in a well-defined structure for instance perovskite, zeolite, core–shell, hydrotalcite… etc. [6, 8, 9] and increasing the dispersion of active phase nanoparticles (NPs). Among these materials, the hydrotalcite-type materials were found effective as catalysts for DRM due to the low cost and high activity. In addition, the thermal decomposition of hydrotalcites leads to homogeneous and fine crystalline mixed metal oxides exhibiting high specific surface areas, strong basic character and highly dispersed active particles, which may improve their catalytic performance [10,11,12,13]. Recently, bimetallic hydrotalcite-derived catalyst with alloy structure have been widely investigated in DRM reaction to suppressed coke deposition and boost the catalytic performance. The chemical formula of LDHs can be expressed by the general formula of \({\left[{M}_{1-x}^{2+}{M}_{x}^{3+}{\left(OH\right)}_{2}\right]}^{x+}{\left[{A}_{x/n}\right]}^{n-}, m{H}_{2}O\). Where: M2+ and M3+ represent divalent and trivalent metals respectively, An– is the interlayer anion and x is the ratio of \(\frac{{M}^{3+}}{\left({M}^{2+}+{M}^{3+}\right)}\). The noble metals and transition metals such as Fe, Co, Zr, Mn, Zn and Cu, etc., have been investigated as bimetallic system with Ni-based catalysts for DRM reaction [14,15,16,17,18]. Chatla et al. [16] examined the influence of Zinc-modified NiMgAl catalysts from hydrotalcite in dry reforming of methane. The NiMgAl-3Zn catalyst has been shown higher catalytic stability at 650 °C, and negligible coke formation after 100 h on stream. This result can be explained by the formation of a Ni-Zn alloy. The modification of NiMgAl mixed oxide catalysts by Zn leads to formation of lowered particles size, improved metal-support interaction, and enhanced basicity. The 3 wt% Zn-modified NiMgAl to limit carbon deposition while rapidly any adsorbed carbon to CO, whereas the NiMgAl catalyst deactivated faster due to carbon deposition. Duan et al. [17] prepared the NiCoMgAl-HTlcs alloy catalysts by co-precipitation method with different Co/Ni molar ratios for DRM reaction. The finely dispersed Ni-Co alloy particles were obtained after calcination and reduction of NiCoMgAl HTlcs. Under the reaction conditions (T = 600 °C and SV of 120,000 mL g−1 h−1 for 25 h), the NiMgAlO produces much carbon compared to NiCoMgAlO alloy catalyst which shows relatively stable activity and effectively inhibited the carbon deposition. In our previous study [18, 19], we investigated the effect of Co-Fe alloy catalyst hydrotalcite-derived catalyst in DRM reaction. The calcination and reduction of CoAl and CoFe hydrotalcite-type materials led to the formation of Co0, Fe0 and Co-Fe alloy. The CoFe catalyst exhibited enhance the resistance to carbon formation in catalyst surface, whereas a slight decrease in methane and dioxide conversion was observed compared to CoAl catalyst. This result has been explained by the re-oxidation of the Co0 by the H2O formed via the reverse water–gas shift reaction (RWGS) favored by the presence of Fe. The formed coke on the surface of CoFe catalyst during DRM reaction was removed by the oxygen of the FeOx.
Wan et al. [20] pointed out that the bimetallic Ni–Fe/Mg(Al)O alloy catalysts obtained from NiFeMgAl hydrotalcite calcined and reduced at different temperatures (700–900 °C) show good activity and stability in DRM. In particular, the catalyst reduced at 700 °C exhibits a great improvement the resistance to carbon due to its smaller Ni particle size and formation of the Fe–Ni alloy.
The purpose of the study is to present the combined effect of the Fe in the development of Ni-based catalysts derived from hydrotalcite with high activity and stability in the dry reforming of methane. The experimental results demonstrated that, Fe presence, have a positive effect in the preparation of high active and stable catalysts in the dry reforming of methane by decreasing coke deposition and sinterization of metallic nickel significantly.
2 Experimental
2.1 Catalysts Preparation
We have synthesized catalysts based on divalent and trivalent metals (NiAlHT and NiFeHT), which will allow us to examine the effect of the in Bimetallic Ni–Al and Ni–Fe.
We opted for the basic constant pH co-precipitation method following previously described procedure by Touahra et al. [21] and which consists in carrying out a controlled precipitation reaction from a solution of nitrate salts of the divalent and trivalent metals with a Brönsted base solution (0.2 mol NaOH, 2 M) and sodium bicarbonate (0.05 mol Na2CO3, 0.5 M). The addition is done dropwise, with stirring for 5 h at room temperature to facilitate precipitation. The precipitate is stirred for one night at 70 °C under reflux. the gel subsequently washed with distilled water, and then dried in an oven at 100 °C for night. The dried solids calcined at 800 °C for 6 h to produce the corresponding mixed oxides NiAlC and NiFeC.
2.2 Catalysts Characterization
The identification of the chemical composition for the investigated samples were analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) in the presence of a multichannel Thermo Jarrel Ash ICAP 957 spectrometer. X-ray diffractometer method was used determine the Crystalline sizes and phase structure using Siemens D-5000 diffractometer. The average particle size (dhkl) were calculated by Debey-Scherer equation (Eq. 7)
where λ is the radiation's wavelength, βhkl is the half width of the peak, and θ is the Bragg diffraction angle. Thermogravimetric analysis (TG) was performed using Thermal Analyzer Setaram Set Sys 16/18 from 25 to 900 °C with a heating rate of 10 °C/min in the presence of air. N2 adsorption–desorption analysis (BET) was carried under nitrogen (− 196 °C) using Micrometrics Tristar 3000. Fourier transform infrared spectroscopy (FTIR) were obtained from in the range of 4000–400 cm−1 using the pressed KBr discs on a Perkin-Elmer spectrometer. Scanning electron microscopy (SEM–EDX) and Transmission Electron Microscopy (TEM) were effected using Quanta 250 and TEM images were taken by JEOL-JEM-1200EX at 100 kV. Temperature-programmed reduction (TPR) results were determined using a TriStar3000 V6.01A apparatus equipped with a TCD detector. 20 mg of sample was flushed with Ar in a quartz reactor tub prior to each run and heated at 120 °C for 2 h under argon flow and then cooled down to room temperature. The sample was submitted to a 5% H2/Ar up flow to 1000 °C with a 10 °C/min rate.
2.3 Catalytic Activity
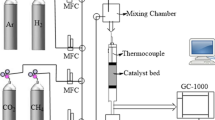
Catalytic testing experiments were carried out in a continuous flow system at atmospheric pressure using a fixed-bed tubular quartz reactor (ID = 6 mm and L = 16 cm). Prior to catalytic reaction, 100 mg of each sample was reduced in-situ under constant hydrogen flow (20 mL min−1) at 750 °C for one hour. After that, the temperature was cooled down to the initial reaction's temperature and a feed gas mixture containing CH4:CO2: Ar in a ratio of 20:20:60 was passed through. The total gas flow rate was set to 20 mL/min. The reaction products were analyzed using gas chromatograph (Delsi), equipped with a thermal conductivity detector (TCD). The TCD uses two-meters-long stainless-steel Carboseive column, in the presence of carrier gas argon. The chromatography (GC) operating conditions: Oven temperature = 100 °C; Injector and detector temperature = 100 °C; and Temperature for the accessory = 70 °C. The equations used to calculate the conversions for CH4 and CO2, hydrogen (SH2) and carbon.
monoxide (SCO), ratio of H2/CO and carbon balance (%) are shown below (Eqs. 8–11).
where F, S, R and represented the conversion, gas flow rate, selectivity and molar ratio respectively.
3 Results and Discussion
3.1 Thermogravimetric Analysis (TG/DTG)
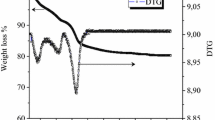
The mass losses ATG (thermogravimetry analysis) curves and the DTG signals as a function of the temperature resulting from the thermal pretreatment of the NiAlHT and NiFeHT samples are reported in supplementary Fig. S1. We followed the evolution of our samples in a temperature range of 20–1000 °C (10 °C / min). For the two samples, three obvious stages of mass loss were identified.
The first loss of mass is 4% and 8% respectively for NiAlHT and NiFeHT, which corresponds to the removel of physisorbed water molecules on the external surface of the crystallites and to the loss of inter-lamellar water without loss of structure according to the following equation [22].
A second loss of mass around 250 °C is due to dehydroxylation and nitrate removal as reported in the literature [23]. The decomposition produces is 6% and 16% respectively for NiAlHT and NiFeHT. Finaly, the last loss of mass is characteristic carbonates in the form of CO2 [24]. The well-defined plateau above 500 °C in the case of the NiFeHT solid and above 800 °C for the NiAlHT solid, is associated with the formation of the spinel phase M2+M3+2O4. The values are 22% and 6% respectively for NiAlHT and NiFeHT as the following equation indicates.
3.2 Catalyst Composition (ICP-AES)
The chemical composition and the structural formula of the samples were derived from the elemental analysis ICP (Inductively Coupled Plasma Spectrometry), EDX (energy dispersive X-ray emission spectrometry) and ATG thermogravimetry analysis. The results obtained are collated in Table 1.
The results illustrated in Table 1, allow us to observe that for the two samples, the chemical formula remains corresponding to that of hydralcites. We also note that the real molar ratio M2+/Al3+ of 2 was practically obtained for the NiAlHT sample. On the other hand, in the case of the NiFeHT sample, the actual molar ratio is 2.44. This value is probably attributed to an incomplete incorporation of Fe3+ cations within the brucite type layers.
3.3 Measurement of Specific Surface Areas and Textural Proprieties
Specific surface values (m2/g) for the calcined and non-calcined materials were determined using BET equations. The results obtained are shown in Table 1. The specific surface area of NiFeHT sample lower than that of NiAlHT sample. Following the total substitution of Al3+ cations with Fe3+ in the crystalline structure of the non-calcined solid, the specific surface area decreases from 90 to 57 m2/g. This could be explained by the agglomeration of the particles of the solid NiFeHT resulting in an increase in the size of these as observed on SEM images Fig. 4. The specific surface area of the two samples increases after calcination, this result can be can be attributed to the destruction of the double-layered structure at high temperatures. During calcination, the elevated temperatures cause thermal decomposition or transformation of the material, leading to changes in its physical and chemical properties [4, 25]. The double-layered structure typically refers to the adsorbed layers of molecules or ions (molecules of H2O and of CO32−) on the surface of a solid material [25, 26]. These layers form due to electrostatic interactions between the charged surface and the oppositely charged species in the surrounding medium. The presence of the double layer can reduce the accessible surface area for adsorption. When the samples are subjected to high temperatures during calcination, several processes occur that contribute to the increase in surface area. One of the primary mechanisms is the removal of adsorbed species and volatilization of impurities or contaminants present on the surface. This removal leads to the creation of voids, cavities, or even porous structures, which effectively increase the surface area of the material [26].
N2 adsorption/desorption isotherms of uncalcined (NiAlHT, NiFeHT) and calcined samples (NiAlC, NiFeC) are presented in Fig. 1. They exhibit typical IV isotherm with a type H1 hysteresis loop, according to the IUPAC classification which is typical of clay minerals either cationic or anionic clays such as hydrotalcite as described in the literature [27, 28]. On the other hand, the calcined samples also shown in Fig. 1 can be exhibit typical IV isotherm and hysteresis type H3 [27].

The nitrogen adsorption—desorption isotherm for samples A uncalcined and B calcined at 800 °C
3.4 X-ray Diffraction Analysis
The X-ray diffractograms of non-calcined samples are shown in Fig. 2A. In accordance with JCPDS sheet n°15-0087, the X-ray diffractogram of non-calcined samples is characterized by the presence of diffraction lines characteristic of the hydrotalcite phase. This latter crystallizes in a hexagonal mesh with principal reflections of the lattice planes (003), (006), (012), (015), (018), (110) and (113) respectively at diffraction angles 2θ = 11.6°; 23.5°; 35.3°; 39.7°; 47.2°; 61.4°; 62.8° and 66.4°. The X-ray diffractograms of the samples after heat treatment at 800 °C are shown in Fig. 2B. The calcination of samples leads to their dehydration (-H2O) and then to the dehydroxylation and decarboxylation (-CO3−2) which is accompanied by the collapse of the structure of hydrotalcite. The NiAlc show peaks of the type NiO [JCPDS file no.47-1049] and NiAl2O4 [file JCPDS n° 10-0339]. As for NiFec solid, there are peaks corresponding to spinel NiFe2O4 [JCPDS file no.10-0325] and NiO these results are in good agreement with those obtained by several researchers [29, 30].

X-ray diffractograms: A precursors samples and B their calcined products. ❖ NiO, ❑ NiAl2O4, ◯ NiFe2O4
3.5 Reducibility of Catalysts by H2
The reduction behavior of NiAlC and NiFeC catalysts was investigated using H2-TPR. The temperature profiles obtained are shown in Fig. 3. For the solid NiAlC, two reduction peaks were observed. The first peak centered at 470 °C characteristic of the reduction of NiO oxide particles (free) well dispersed and without interaction with the matrix (Eq. 16) [31, 32].

Reduction temperature profiles of the solids calcined at 800 °C
The second peak centered at 766 °C would be due to the reduction of nickel particles in the NiAl2O4 spinel structure which makes its reduction difficult (Eq. 17) [33, 34].
For the NiFeC catalyst, probably that the low intensity peak, observed at 390 °C.
in Fig. 3, could correspond to the reduction of the Ni2+ species in metallic Ni not observed by XRD because of the small size of these particles.
Thus, the most intense peaks observed at high temperatures (500–800 °C) in this case, are associated with the reduction of the Ni2+ and Fe3+ species present in the NiFe2O4 spinel structure. Generally, the Fe3+ reduction in Fe0 is done at higher temperatures in several stages following a mechanism mainly in three stages [35, 36]:
The FeO is not observed on X-ray diffractograms because, this oxide has a limited stability around 570 °C and beyond this temperature decomposes directly by giving Fe0 and Fe3O4 according to some works [37]. By analogy, to the approach proposed by Jozwiak et al. [37]
The mechanism of FeO reduction in α-FeO is relatively simple but is done at high temperatures (> 570 °C). They noticed that at 400 °C the Fe3O4 content is 97%, which reflects the dispersion of Fe2O3 and the beginning of the formation of metallic iron, while in the range of (400–560 °C) they note the progressive decrease of the Fe3O4 content in favor of the formation of metallic iron. At 580 °C, three additional phases were observed Fe3O4 (53%), FeO (7%) and Fe0 (40%).
3.6 SEM Analysis
The SEM images for the uncalcined (NiAlHT and NiFeHT) and calcined solids (NiAlC and NiFeC) are shown in Fig. 4. The images show morphologies in the form of platelets, which supposes the formation of a lamellar structure of almost hexagonal shape. We can notice that the calcination of solids leads to the disappearance of the hexagonal form in favor of a more rounded form (Fig. 4).

SEM images of uncalcined and calcined solids. a NiAlHT, b NiFeHT, c NiAlC and d NiFeC
3.7 FTIR Analysis
The infrared spectra of the uncalcined and calcined samples are shown respectively in Fig. 5A and B. The non-calcined samples Fig. 5(A) show the characteristic bands of hydrotalcite. A broad adsorption band centered at ~ 3455 cm−1, which corresponds to the vibration bands of hydroxyl groups ν(OH). This region of the infrared spectrum is characteristic of the interactions between the OH groups of the sheets, the water molecules in inter-lamellar and the physisorbed water [38]. Another band located at ~ 1628 cm−1 corresponds to the vibration of angular deformation of water δ (HOH). An intense band located around 1370 cm−1 is also observed on all spectra, corresponding to the O-C-O vibration of carbonate ions (CO3−2) in inter-lamellar form. In the low frequency region (< 1000 cm−1), the vibration bands observed are attributed to the elongation vibrations of the metal-O-metal bond (M2+-O-M3+, with M2+ = Ni and M3+ = Al or Fe) [39,40,41].

FTIR of: A precursors samples and B their calcined products
The infrared spectra of calcined solids are shown in Fig. 5B. These spectra show a significant decrease in the width and intensity of the vibration band at 3345 cm−1, due to the OH groups, as well as the disappearance of the peaks at around 1628 cm−1 probably related to dehydration since a very large part of the water and the hydroxyls are eliminated by calcination.]There is also a clear decrease in the vibration band at 1345 cm−1, corresponding to the elimination of the majority of carbonate ions following the heat treatment [21].
4 Characterization of Catalysts After Reduction
The catalysts pretreated under a flow H2, under the conditions of the reactivity hydrogen (T reduction = 750 °C, flow H2 = 20 mL/min, for 1 h) were analyzed by various techniques, i.e., DRX, BET, MEB-EDX. The reduced samples under hydrogen flow are noted below NiAlR and NiFeR.
4.1 BET Surface of Reduced Samples
The values of the specific surfaces area were summarized in Table 2. From this table, it appears that the reduction of the two catalysts leads to a decrease of the specific surfaces area. A decrease of approximately 20% is observed on the case of catalyst NiAlR and a decrease of 30% in the case of the solid NiFeR compared to calcined samples.
4.2 X-ray Diffraction After Reduction
Figure 6 illustrates the X-ray diffraction of samples reduced at 750 °C for 1 h. The diffractogram of the solid NiAlR shows the presence of species Ni0 and NiO. Furthermore, the X-ray diffractogram of the NiFeR catalyst reveals the presence of the species Ni0, NiO and γ-NiFe alloy. The average particle size of Ni0, for the reduced catalyst at 750 °C is determined from X-ray diffractograms schown in Fig. 6, using the Debye–Scherrer relation. The average size of the Ni0 particles in the NiAlR and NiFeR catalyst are 17 and 17.5 nm respectively.

X-ray diffractograms samples reduced at 750 °C: ❖NiO, ▽Ni0, × γ-NiFe and \(\uparrow\)Fe0
4.3 SEM & TEM Analyses After Reduction
The SEM–EDX images obtained by scanning electron microscopy for NiAlR and NiFeR catalysts are shown in Fig. 7. In the case of NiAlR catalyst, the SEM images shows a homogeneous distribution of the Ni0 particles on the surface of the solid. The average size of the Ni0 particles calculated from the SEM images are in good agreement with the average size calculated by the XRD method as shown in Table 2. In the case of the NiFeR catalyst, the SEM image shows a less uniform appearance than the previous one. The EDX spectra observed in Fig. 7 were obtained by targeting the electron beam on a single particle, which given us the mass percentages of the elements corresponding to these particles. EDX results showed the presence of small spherical particles, corresponding to the Ni0 and Fe0 particles and of large agglomerates corresponding to the γ-NiFe alloys of different shape.

SEM images of a NiAlR and b NiFeR and EDX of particles designated by white arrows SEM images (R = reduced samples at 750 °C)
The TEM image as well as EDS mapping of the NiFeR catalyst are presented in Fig. 8. The formation of the γ-NiFe alloy confirmed by TEM-EDS. The EDS shows that the Ni (green) one and the Fe (red) one were distributed uniformly in the sample implying the γ-NiFe alloy formation.

MET and EDS mapping for NiFeR sample. Ni (green) and Fe (red)
5 Catalytic Activity Tests
5.1 Influence of Reaction Temperature
The catalysts were tested in dry methane reforming reaction, at atmospheric pressure in the range [400–700 °C]. The catalysts, under a reaction mixture, are heated from room temperature to 700 °C with a speed of 5 °C/min and the catalytic performances are measured in a random mode from 400 to 700 °C to avoid sintering of catalysts during the reaction, according to Fig. 9.

Reaction protocol after reducing pretreatment
The results of the catalytic activity of the NiAlR and NiFeR catalysts as a function of the temperature in dry reforming reaction are illustrated in Fig. 10.

A CH4 Conversion (%), B CO2 Conversion (%) and C H2/CO versus reaction temperature
At low temperature (400 °C), the conversions obtained are 20.2% for CH4 and 25.5% for CO2 in the presence of NiAlR.
The total substitution of Al cations by Fe cations leads to a significant decrease in conversions of both CH4 and CO2. Indeed, these are only about 15.1% for CH4 conversion and 20.0% for CO2 conversion, and the selectivity H2/CO remains less than unity in the case of NiFeR catalyst.
The major advantage of the methane reforming reaction related in the value close to unity of the H2/CO ratio obtained during a catalyst working cycle. However, in the case of the two catalysts, in the temperature range [400–550 °C], the conversion rate of CO2 is greater than that of CH4 as shown in Fig. 10, resulting in an H2/CO ratio less than 1. This result suggests the participation of the reverse reaction of gas to water conversion \(\left({CO}_{2}+{H}_{2} \leftrightarrow CO+ {H}_{2}O\right)\), which tends to increase the CO2 conversion and the CO production becomes higher than that of H2.
At high temperatures [500–700 °C], the recorded conversions show a clear improvement in the presence of those two catalysts, which confirms the highly endothermic nature of the dry reforming reaction. At 700 °C, the conversions of CH4 and CO2 are respectively in the order (87.5%, 91.4%) and (79.2%, 84.1%) for NiAlR and NiFeR catalysts.
The H2/CO ratio is slightly greater than unity in the case of the NiAlR solid. This difference, with respect to the stoichiometry of the reaction, may involve the participation of secondary reactions such as the transformation of CO via the Boudouard reaction \(\left(2CO \leftrightarrow C+C{O}_{2}\right)\) or the methane decomposition reaction \(\left({CH}_{4} \leftrightarrow C+ {2H}_{2}\right)\). The results also show that at these temperatures, the CO2 conversions are slightly higher than those of CH4 and the carbon balance is thus far from 100%. In this case, the carbon deposit would come essentially from Boudouard's reaction.
In the same temperature range [500–700 °C], in the case of NiFeR catalyst, the ratio H2/CO is close to 1 and the conversion of CO2 remains higher than the conversion of CH4, which again implies the progress of the reaction. RWGS reaction favored by the presence of iron [42].
5.2 Effect of Aluminium Substitution by Iron on Stability of the Catalytic Activity of Catalysts as a Function of Time of Reaction
In this study, we examined the effect of Al substitution by Fe on stability of the catalytic activity of the catalysts during DRM reaction as a function of time at TReaction = 700 °C; TReduction = 750 °C; CH4/CO2 = 1 and P = 1 atm. The results obtained are illustrated in Fig. 11. The CH4 Conversions were 87.5% and 79.2% respectively for NiAlR and NiFeR catalysts, whereas CO2 conversions were 91.4% and 84.1% respectively for NiAlR and NiFeR catalysts. Under the experimental conditions used, the NiAlR catalyst has better performance. Let us also note that the NiFeR reaches the stationary regime from the first minutes of contact with the reaction mixture while the NiAlR reaches the stability plateau after 2 h of reaction and this step is maintained beyond 10 h of catalytic test.

A CH4 conversion (%), B CO2 conversion (%) as a function of time on stream in the DRM at 700 °C
In order to elucidate these differences in catalytic behaviors, we draw up comparative Table 3, which groups together the CH4 and CO2 conversions recorded, the H2/CO ratio obtained, andthe percentage of calculated carbon deposition, after 12 h of reaction to 700 °C. In addition, the values of the specific surfaces and the size of the nickel particles after reduction are grouped in Table 3.
The comparison of the catalytic performances of the two catalysts makes it possible to classify them in the following sequence: NiAlR > NiFeR similarly, when comparing the specific surface area, we found that they follow the same evolution. Indeed, a large specific surface area allows better access of the reagents and therefore constitutes a necessary but not sufficient condition for a better reactivity of the catalysts.
In addition, Han et al. [43] and Shang et al. [44] showed that metal nanoparticles (NPs) plays decisive roles in metal‐catalyzed DRM reactions.They reported that the use of small particles, of the order of nano can help reduce carbon deposition and thus improve the catalytic stability of the catalysts.
However, in our case, the particle sizes are similar and the formation of carbon is more important for the better catalyst (NiAlR), as indicated by the carbon balance given in Table 3. This carbon deposition in the case of NiAlR catalyst could be due either to the Boudouard reaction \(\left(2CO \to C+ {CO}_{2}\right)\) or to the decomposition of methane \(\left(C{H}_{4} \to C+{2H}_{2}\right)\). The CO2 conversion higher than that of CH4 confirms the formation of carbon by the Boudouard reaction. On the other hand, the carbon balance in the case of NiFeR catalyst close to 100% indicates a low carbon content formed on the surface of this catalyst.
We can assume that the carbon formed on the surface of the iron-based catalysts is removed by the oxygen species formed from FeOx obtained after the dissociation of Ni–Fe alloy in the presence of CO2 as reported in the literature [45, 46].
Indeed, Theofandis et al. [42, 47] report in their work that the carbon species formed at the Ni metal sites are removed by the oxygen species formed from iron oxide. The reaction mechanism proposed by these authors in this case is based on the Mars-Van Krevelen hypothesis, which assumes that iron oxidizes to FeOx in the presence of CO2 \(\left(\mathrm{Fe}+{\mathrm{xCO}}_{2} \leftrightarrow {\mathrm{FeO}}_{\mathrm{x}}+\mathrm{xCO}\right)\) and CH4 is activated on Ni sites to form hydrogen and carbon: \(\left({\mathrm{CH}}_{4}\stackrel{\mathrm{Ni}}{\to } 2{\mathrm{H}}_{2}+{\mathrm{C}}_{\mathrm{Ni}}\right)\). The CNi is re-oxidized by the oxygen of the iron oxide:\(\left({\mathrm{FeO}}_{\mathrm{x}}+ {\mathrm{C}}_{\mathrm{Ni}} \leftrightarrow \mathrm{xCO}+\mathrm{Ni}+\mathrm{Fe}\right)\).
In our case, the decrease in the catalytic performance of NiFeR catalyst compared to NiAlR catalyst is therefore not attributable to carbon deposition, it would be due to the re-oxidation of Ni0 active sites by H2O: \(\left(Ni+{H}_{2}O \leftrightarrow NiO+{H}_{2}\right)\) formed via the inverse reaction of the gas to water which promote in the presence of iron:\(\left(Fe{O}_{x}+ {H}_{2} \leftrightarrow Fe+ {H}_{2}O\right)\). As already reported in the literature [48]. Which tested NiFe2O4 catalysts in dry reforming reaction, showed that the small amount of carbon present in NiFe2O4 catalyst may be related to their low catalytic reactivity. It may also be due to the formation of Ni–Fe alloy by decomposition of the spinel structure, which seems to play an important role in inhibiting the carbon deposition through a dilution effect of Ni active sites.
5.3 Characterization of the Catalysts after 12 h of Reaction at 700 °C
The specific surfaces of the catalysts measured after 12 h of DRM reaction under a reaction mixture are summarized in Table 4. Also in this table give the average particle size Ni° (nm) before and after reaction calculated from the SEM images.
From these results, the specific surfaces area after catalytic tests decrease in the case of the two catalysts. The decrease in the surface area of NiAlR catalyst may be due to the sintering of the Ni particles during the reaction. While the decrease in the specific surface area of NiFeR catalyst after reaction, may be due to the active phase re-oxidation.
The deactivation of the catalysts in the methane dry reforming reaction has been widely studied in the literature [49,50,51]. This deactivation originates from carbon formation or sintering of metal particles. In order to highlight the causes of NiFeR catalyst activity decrease compared with NiAlR, we followed the evolution of the texture by scanning electron microscopy after 12 h of stability test at 700 °C. The images obtained are shown in Fig. 12. SEM micrographs showed the presence of filamentous carbon formed on the surface of the various catalysts during the reaction. Carbon is much more observed on the NiAlR catalyst surface than NiFeR catalyst. In order to highlight the presence or absence of the second deactivation phenomenon: “sintering”, the size of the nickel particles was measured from the SEM images after 12 h reaction. In the case of NiFeR catalyst, there is the presence of small particles always well dispersed on the surface of the catalyst. Therefore, the decrease of the catalytic activity in the case of this catalyst would not be due to the agglomeration of these particles, but would be due to the re-oxidation of the active sites by H2O formed via the inverse reaction of the gas to the water \(\left({Ni}^{0}+ {H}_{2}O \to NiO+ {H}_{2}\right)\). This result explains the reason for the decrease in catalyst catalytic activity after a total substitution of Al by the Fe reported in the reactivity part. The re-oxidation of the active sites was confirmed by MEB-EDX analysis, which shows the formation of NiO oxide after reaction.

SEM–EDX of used catalysts after reaction at 700 °C for 12 h. a NiAlR and b NiFeR
6 Conclusion
Catalysts based on Ni, Al and Fe, derived from hydrotalcites catalysts has been prepared by co-precipitation method. TEM-EDS and MEB-EDX analysis of samples reduced at 750 °C revealed the presence of γ-NiFe alloys. The catalysts were tested in dry reforming of methane with prior reduction at 750 °C for 1 h. The addition of Fe in Ni-based catalyst helps stabilize the active sites and prevents the agglomeration of metal particles, thereby maintaining a highly dispersed and active catalytic phase. This stability is crucial for continuous and long-term operation of the catalyst in the reforming process. We have also been able to show that the total substitution of aluminum by iron leads to a decrease in the catalytic performances.
This decrease has been attributed to the re-oxidation of the active phase by the water, which is formed via the reverse water–gas shift reaction (WGSR-inverse) favored by the presence of iron. During the catalytic test, the carbon formed on the surface of the catalyst was removed by the oxygen of the iron oxide (FeOx).
Data availability
Data available within the article and its supplementary materials.
References
Atsbha TA, Yoon T, Seongho P, Lee CJ (2021) A review on the catalytic conversion of CO2 using H2 for synthesis of CO methanol and hydrocarbons. J CO2 Utiliz 44:101413–101435. https://doi.org/10.1016/j.jcou.2020.101413
Gao X, Ge Z, Zhu G, Wang Z, Ashok J, Kawi S (2021) Anti-coking and anti-sintering Ni/Al2O3 catalysts in the dry reforming of methane recent progress and prospects. Catalysts 11(8):1003. https://doi.org/10.3390/catal11081003
Le Saché E, Reina TR (2022) Analysis of Dry Reforming as direct route for gas phase CO2 conversion. The past, the present and future of catalytic DRM technologies. Prog Energy Combust Sci 8:100970–101040. https://doi.org/10.1016/j.pecs.2021.100970
Djebarri B, Touahra F, Aider N, Bali F, Sehailia M, Chebout R, Bachari K, Halliche D (2020) Enhanced long-term stability and carbon resistance of Ni/MnxOy-Al2O3 catalyst in near-equilibrium CO2 reforming of methane for syngas production. Bull Chem React Eng Catal 15(2):331–347. https://doi.org/10.9767/bcrec.15.2.6983.331-347
Gao X, Ge Z, Zhu G, Wang Z, Ashok J, Kawi S (2021) Anti-coking and anti-sintering Ni/Al2O3 catalysts in the dry reforming of methane: recent progress and prospects. Catalysts 11(8):1003. https://doi.org/10.3390/catal11081003
Touahra F, Chebout R, Lerari D, Halliche D, Bachari K (2019) Role of the nanoparticles of Cu-Co alloy derived from perovskite for enhancing Carbon-Resistant in the dry reforming of methane. Energy 171:465–474. https://doi.org/10.1016/j.energy.2019.01.085
Gao N, Salisu J, Quan C, Williams P (2021) Modified nickel-based catalysts for improved steam reforming of biomass tar. A critical review. Renew Sustain Energy Rev 145:111023. https://doi.org/10.1016/j.rser.2021.111023
Sellam D, Ikkour K, Dekkar S, Messaoudi H, Belaid T, Roger AC (2019) CO2 reforming of methane over LaNiO3 perovskite supported catalysts: influence of silica support. Bull Chem React Eng Catal 14(3):568–578. https://doi.org/10.9767/bcrec.14.3.3472.568-578
Calgaro CO, Rocha AL, Perez-Lopez OW (2020) Deactivation control in CO2 reforming of methane over Ni–Mg–Al catalyst. React Kinet Mech Catal 130(1):159–178. https://doi.org/10.1007/s11144-020-01770-3
Świrk K, Grams J, Motak M, Da Costa P, Grzybek T (2020) Understanding of tri-reforming of methane over Ni/Mg/Al hydrotalcite-derived catalyst for CO2 utilization from flue gases from natural gas-fired power plants. J CO 2Utiliz 42:101317–101335. https://doi.org/10.1016/j.jcou.2020.101317
Calgaro CO, Lima DDS, Tonietto R, Perez-Lopez OW (2021) Biogas dry reforming over Ni–Mg–La–Al catalysts: influence of La/Mg ratio. Catal Lett 151(1):267–280. https://doi.org/10.1007/s10562-020-03296-8
Djebarri B, Gonzalez-Delacruz VM, Halliche D, Bachari K, Saadi A, Caballero A, Holgado JP, Cherifi O (2014) Promoting effect of Ce and Mg cations in Ni/Al catalysts prepared from hydrotalcites for the dry reforming of methane. React Kinet Mech Catal 111:259–275. https://doi.org/10.1007/s11144-013-0646-2
Djebarri B, Aider N, Touahra F, Bali F, Holgado JP, Halliche D (2021) Effect of silicates and carbonates on the structure of nickel-containing hydrotalcite-derived materials. In: Khellaf A (ed) Advances in renewable hydrogen and other sustainable energy carriers. Springer proceedings in energy. Springer, Singapore, pp 443–450. https://doi.org/10.1007/978-981-15-6595-3_57
Zambaldi P, Haug L, Penner S, Klötzer B (2022) Dry reforming of methane on NiCu and NiPd model systems: optimization of carbon chemistry. Catalysts 12:311–334. https://doi.org/10.3390/catal12030311
Touahra F, Sehailia M, Halliche D, Bachari K, Saadi A, Cherifi O (2016) (MnO/Mn3O4)-NiAl nanoparticles as smart carbon resistant catalysts for the production of syngas by means of CO2 reforming of methane: Advocating the role of concurrent carbothermic redox looping in the elimination of coke. Int J Hydrogen Energy 41:21140–21156. https://doi.org/10.1016/j.ijhydene.2016.08.194
Chatla A, Abu-Rub F, Prakash AV, Ibrahim G, Elbashir NO (2022) Highly stable and coke-resistant Zn-modified Ni-Mg-Al hydrotalcite derived catalyst for dry reforming of methane: synergistic effect of Ni and Zn. Fuel 308:122042. https://doi.org/10.1016/j.fuel.2021.122042
Duan X, Pan J, Yang X, Wan C, Lin X, Li D, Jiang L (2022) Nickel-cobalt bimetallic catalysts prepared from hydrotalcite-like compounds for dry reforming of methane. Int J Hydrogen Energy 47:24358–24373. https://doi.org/10.1016/j.ijhydene.2022.05.211
Aider N, Touahra F, Bali F, Djebarri B, Lerari D, Bachari K, Halliche D (2018) Improvement of catalytic stability and carbon resistance in the process of CO2 reforming of methane by CoAl and CoFe hydrotalcite-derived catalysts. Int J Hydrogen Energy 43:8256–8266. https://doi.org/10.1016/j.ijhydene.2018.03.118
Aider N, Touahra F, Djebarri B, Bali F, Abdelsadek Z, Halliche D (2021) Application of hydrotalcite for the dry reforming reaction of methane and reduction of greenhouse gases. In: Khellaf A (ed) Advances in renewable hydrogen and other sustainable energy carriers. Springer Proceedings in Energy. Springer, Singapore, pp 299–305. https://doi.org/10.1007/978-981-15-6595-3_39
Wan C, Song K, Pan J, Huang M, Luo R, Li D, Jiang L (2020) Ni–Fe/Mg (Al) O alloy catalyst for carbon dioxide reforming of methane: influence of reduction temperature and Ni–Fe alloying on coking. Int J Hydrogen Energy 45:33574–33585. https://doi.org/10.1016/j.ijhydene.2020.09.129
Touahra F, Sehailia M, Ketir W, Bachari K, Chebout R, Trari M, Cherifi O, Halliche D (2016) Effect of the Ni/Al ratio of hydrotalcite-type catalysts on their performance in the methane dry reforming process. Appl Petrochem Res 6:1–13. https://doi.org/10.1007/s13203-015-0109-y
LijiSobhana SS, Mehedi R, Malmivirta M, Paturi P, Lastusaari M, Dîrtu MM, Garcia Y, Fardim P (2016) Heteronuclear nanoparticles supported hydrotalcites containing Ni(II) and Fe(III) stable photocatalysts for orange II degradation. Appl Clay Sci 132:641–649. https://doi.org/10.1016/j.clay.2016.08.016
Qi C, Amphlett JC, Peppley BA (2009) H2 production via the steam reforming of methanol over NiAl-layered double hydroxide derived catalysts. Catal Surv Asia 13:16–21. https://doi.org/10.1007/s10563-009-9063-y
Braterman PS, Xu ZP, Yarberry F (2004) Layered double hydroxides (LDHs). Handbook Layer Mater 8:373–474
Aider N, Djebarri B, Touahra F, Layeb H, Halliche D (2023) Studies of the solvent-free knoevenagel condensation over commercial NiO compared with NiO drived from hydrotalcites. Bull Chem React Eng Catal 18:186–199. https://doi.org/10.9767/bcrec.17598
Silva Neto LD, Anchieta CG, Duarte JL, Meili L, Freire JT (2021) Effect of drying on the fabrication of MgAl layered double hydroxides. ACS Omega 6:21819–21829. https://doi.org/10.1021/acsomega.1c03581
Leofanti G, Padovan M, Tozzola G, Venturelli B (1998) Surface area and pore texture of catalysts. Catal Today 41:207–219. https://doi.org/10.1016/S0920-5861(98)00050-9
Othman MR, Rasid NM, Fernando WJN (2006) Mg–Al hydrotalcite coating on zeolites for improved carbon dioxide adsorption. Chem Eng Sci 61:1555–1560. https://doi.org/10.1016/j.ces.2005.09.011
Zhao X, Xu S, Wang L, Duan X, Zhang F (2010) Exchange-biased NiFe2O4/NiO nanocomposites derived from NiFe-layered double hydroxides as a single precursor. Nano Res 3:200–210. https://doi.org/10.1007/s12274-010-1023-3
Carvalho DC, Ferreira NA, Josue Filho M, Ferreira OP, Soares JM, Oliveira AC (2015) Ni–Fe and Co–Fe binary oxides derived from layered double hydroxides and their catalytic evaluation for hydrogen production. Catal Today 250:155–165. https://doi.org/10.1016/j.cattod.2014.08.010
Haipeng WU, Wenjing W, Li GUO, Yan FP, Xiufeng XU (2011) Effect of promoter species and precursors on catalytic activity of alkali metal promoted NiAl mixed oxides for N2O decomposition. Journal of Fuel Chemistry and Technology 39:550–555. https://doi.org/10.1016/S1872-5813(11)60034-0
Rudolf C, Dragoi B, Ungureanu A, Chirieac A, Royer S, Nastro A, Dumitriu E (2014) NiAl and CoAl materials derived from takovite-like LDHs and related structures as efficient chemoselective hydrogenation catalysts. Catal Sci Technol 4:4179–4189. https://doi.org/10.1039/C3CY00611E
Smoláková L, Čapek L, Botková Š, Kovanda F, Bulánek R, Pouzar M (2011) Activity of the Ni–Al Mixed oxides prepared from hydrotalcite-like precursors in the oxidative dehydrogenation of ethane and propane. Top Catal 54:1151–1162. https://doi.org/10.1007/s11244-011-9737-3
Touahra F, Rabahi A, Chebout R, Boudjemaa A, Lerari D, Sehailia M, Halliche D, Bachari K (2016) Enhanced catalytic behaviour of surface dispersed nickel on LaCuO3 perovskite in the production of syngas: an expedient approach to carbon resistance during CO2 reforming of methane. Int J Hydrogene Energy 41:2477–2486. https://doi.org/10.1016/j.ijhydene.2015.12.062
Munteanu G, Ilieva L, Andreeva D (1997) Kinetic parameters obtained from TPR data for α-Fe2O3 and Au α-FeO3 systems. Thermochim Acta 291:171–177. https://doi.org/10.1016/S0040-6031(96)03097-3
Paldey S, Gedevanishvili S, Zhang W, Rasouli F (2005) Evaluation of spinel based pigment system as CO oxidation catalyst. Appl Catal B 56:241–250. https://doi.org/10.1016/j.apcatb.2004.09.013
Jozwiak W, Kaczmarek E, Maniecki T, Ignaczak W, Maniukiewicz W (2007) Reduction behavior of iron oxides in hydrogen and carbon monoxide atmospheres. Appl Catal A 326:17–27. https://doi.org/10.1016/j.apcata.2007.03.021
Aoudjit F, Touahra F, Aoudjit L, Cherifi O, Halliche D (2020) Efficient solar heterogeneous photocatalytic degradation of metronidazole using heterojunction semiconductors hybrid nanocomposite, layered double hydroxides. Water Sci Technol 82:2837–2846. https://doi.org/10.2166/wst.2020.519
Perez-Ramirez J, Mul G, Kapteijn F, Moulijn J (2011) A spectroscopic study of the effect of the trivalent cation on the thermal decomposition behaviour of Co-based hydrotalcites. J Mater Chem 11:2529–2536. https://doi.org/10.1039/B104989P
Subhashini J, Ferdinand AC, Sagayaraj R (2022) Structural, spectroscopy and magnetic properties of copper doped nickel ferrite by the co-precipitation method. Chem Afr 5:1387–1396. https://doi.org/10.1007/s42250-022-00438-w
Masmoudi-Soussi A, Hammas-Nasri I, Horchani-Naifer K, Férid M (2019) Study of rare earths leaching after hydrothermal conversion of phosphogypsum. Chemistry Africa 2:415–422. https://doi.org/10.1007/s42250-019-00048-z
Theofanidis SA, Batchu R, Galvita VV, Poelman H, Marin GB (2016) Carbon gasification from Fe–Ni catalysts after methane dry reforming. Appl Catal B 185:42–55. https://doi.org/10.1016/j.apcatb.2015.12.006
Han JW, Park JS, Choi MS, Lee H (2017) Uncoupling the size and support effects of Ni catalysts for dry reforming of methane. Appl Catal B Environ 203:625–632. https://doi.org/10.1016/j.apcatb.2016.10.069
Shang Z, Li S, Li L, Liu G, Liang X (2017) Highly active and stable alumina supported nickel nanoparticle catalysts for dry reforming of methane. Appl Catal B 201:302–309. https://doi.org/10.1016/j.apcatb.2016.08.019
Motomura A, Nakaya Y, Sampson C, Higo T, Torimoto M, Tsuneki H, Furukawa S, Sekine Y (2022) Synergistic effects of Ni–Fe alloy catalysts on dry reforming of methane at low temperatures in an electric field. RSC Adv 12(44):28359–28363. https://doi.org/10.1039/D2RA05946K
Zhang T, Liu Z, Zhu YA, Liu Z, Sui Z, Zhu K, Zhou X (2020) Dry reforming of methane on Ni-Fe-MgO catalysts: influence of Fe on carbon-resistant property and kinetics. Appl Catal B Environ 264:118497. https://doi.org/10.1016/j.apcatb.2019.118497
Theofanidis SA, Galvita VV, Poelman H, Marin GB (2015) Enhanced carbon-resistant dry reforming Fe-Ni catalyst: role of Fe. ACS Catal 5:3028–3039. https://doi.org/10.1021/acscatal.5b00357
Benrabaa R, Boukhlouf H, Löfberg A, Rubbens A, Vannier RN, Bordes-Richard E, Barama A (2012) Nickel ferrite spinel as catalyst precursorin the dry reforming of methane: synthesis, characterization and catalytic properties. J Nat Gas Chem 21:595–604. https://doi.org/10.1016/S1003-9953(11)60408-8
Muraza O, Galadima A (2015) A review on coke management during dry reforming of methane. Int J Energy Res 39:1196–1216. https://doi.org/10.1002/er.3295
Rahbar Shamskar F, Rezaei M, Meshkani F (2017) The influence of Ni loading on the activity and coke formation of ultrasound-assisted co-precipitated Ni–Al2O3 nanocatalyst in dry reforming of methane. Int J Hydrogene Energy 42:4155–4164. https://doi.org/10.1016/j.ijhydene.2016.11.067
Xu Y, Du X, Shi L, Chen T, Wan H, Wang P, Song M (2021) Improved performance of Ni/Al2O3 catalyst deriving from the hydrotalcite precursor synthesized on Al2O3 support for dry reforming of methane. Int J Hydrogene Energy 46(27):14301–14310. https://doi.org/10.1016/j.ijhydene.2021.01.189
Acknowledgements
The authors would like to thank the Ministry of Higher Education and Scientific Research (MESRS), Algeria and the General Directorate for Scientific Research and Technological Development (DGRSDT), Algeria, for their financial help that prompted the accomplishment of this scientific material.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Djebarri, B., Aider, N., Touahra, F. et al. Synergistic Effect of Bimetallic Ni-Based Catalysts Derived from Hydrotalcite on Stability and Coke Resistance for Dry Reforming of Methane. Chemistry Africa 7, 1091–1106 (2024). https://doi.org/10.1007/s42250-023-00772-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-023-00772-7