
Abstract
Catalytic partial oxidation of methane to produce syngas were studied over calcined hydrotalcites of varying Mg/Al ratios supported Rh catalysts. Hydrotalcites of varying Mg/Al ratios were prepared using their hydroxide precursors and intercalating them with amino acid lysine. Upon calcination, these hydrotalcites converted into their mixed metal oxides having mesoporosity and used directly as support materials for the synthesis of rhodium catalysts. Rh dispersion, size of the nanoparticles, and metal-support interactions were found to strongly influence the activity of the catalyst and their stability. Feed composition, gas hourly space velocity, and temperature were found to influence the catalyst activity, CO/H2 ratio, and CO/CO2 selectivity. Alumina and alumina-rich calcined hydrotalcites exhibit higher catalytic activity, but carbon formation and aggregation were prominent. In contrast, MgO and MgO-rich calcined hydrotalcite supported Rh catalysts exhibit moderate activity; however, they were resistant against particle sintering and carbon formation. Long-term testings of these catalysts were carried out, and it was observed that calcined hydrotalcites of varying Mg/Al ratios supported Rh catalysts were promising candidates as stable and active catalysts toward catalytic partial oxidation of methane.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Syngas production from the catalyst-mediated partial oxidation (CPOx) is an exothermic reaction, in contrast to other energy-intensive endothermic reforming processes. The syngas produced from the CPOx reaction consists of a mixture of H2:CO with ratio of 2, a desirable syngas composition for downstream processes including methanol and DME synthesis [1,2,3]. Since CPOx reaction involves methane oxidation as one of the reaction step, the resultant syngas consists of traces of CO2, which is very important during the syngas conversion into methanol [4,5,6]. Another major advantage of this reaction is the possibility of operating this reaction at very high gas hourly space velocity that enables the intensification of this reaction into very small scale [7,8,9,10,11]. Due to the exothermic nature of this reaction, the catalyst bed temperature increases rapidly, which gives rise to the development of hotspots on the surface of catalyst. These types of hot spot formations generally promote the sintering of active metal, which increases the particle size and ultimately deactivates the catalyst [12,13,14,15,16]. The reactant feed composition is generally maintained at 2:1 mol ratio of CH4 to O2, to maintain catalytic partial oxidation, and this ratio generally leads to the formation of carbon on the active sites of the catalyst. Carbon deposition-mediated deactivation is also a major disadvantage during dry and steam reforming of methane, and the phenomenon of carbon deposition was known to affect catalyst stability during the reaction. Therefore, significant efforts were made to identify stable catalysts that are resistant to hot spot formation and carbon formation, which remain challenging. To develop such stable and coke-resistant catalysts, a molecular level understanding of the reaction mechanism, hot spot and carbon formation mechanism is essential. Two kinds of reaction mechanism were proposed to explain the methane’s catalytic partial oxidation. The first mechanism involves a two-step pathway, and the first step involves a complete combustion of a fraction of methane by the total oxygen present in the reactant feed, which produces CO2 and H2O. The second step is very much similar to combined dry and steam reforming of methane, and the combustion products from the first step CO2 and H2O reform the remaining unreacted methane into syngas. The first step is exothermic and is responsible for the formation of hot spots, while the second step is endothermic and is responsible for the carbon formation. Both reaction steps occur at the different domains of the same catalyst bed; thus, temperature gradients were formed between the sites that promote the combustion and sites that catalyze reforming of methane [14, 15, 17,18,19,20,21,22]. As the CPOx reaction is generally carried out using a fixed bed reactor, the front end of the catalyst bed that promotes the oxidation and combustion stays at relatively higher temperature, as compared with the temperature of the catalyst bed region where the reforming step occurs. A second mechanism or direct mechanism was also proposed, which produces syngas in one step directly without involving combustion-reforming steps. However, a two-step mechanism to form syngas has been widely accepted. Therefore, a stable and active catalyst must have the uniform dispersion of active sites, strong metal-support interaction as well as resistance toward coking to make this process efficient and stable. In the category of non-precious metal catalysts, supported nickel catalysts have been demonstrated as active catalysts toward CPOx reaction to produce syngas, but it was observed that different oxidation states of nickel perform different parts in steps of surface reaction [14, 21, 23,24,25]. Syngas selectivity was observed on the zerovalent Ni active sites, while divalent Ni sites promote combustion [26]. Distribution of the active nickel sites between the surface and bulk were also found to influence the reaction; thus, synthetic procedures play a vital role in controlling this distribution, which eventually dictate the catalyst activity. Moreover, the nickel active sites tend to have poor oxygen tolerance at high temperature; therefore, oxidation of the metallic nickel sites was difficult to control. Another disadvantage is that the reforming step on nickel active site tends to promote carbon formation and to deactivate the site rapidly.
In contrast, supported precious metal catalysts including Rh catalysts were demonstrated as highly reactive toward CPOx to form syngas [27,28,29,30]. Hot spot formation can be reduced, and activity can be increased by uniform dispersion of Rh on suitable supports, and moreover, it can refine metal support interaction and varying oxidation states. Alkaline earth supports were known to reduce the carbon formation due to their basicity, In contrast, uniform dispersion of rhodium can be achieved on acidic sites; thus, alumina supports were used mainly to improve the dispersion. Mixed metal oxide supports obtained from the calcination of hydrotalcites [17, 31] have both kinds of alkaline earth oxides and acidic alumina sites. Rh dispersion, particle size, reducibility of active metal and product selectivity, can be further improved as these bifunctional mesoporous metal oxides and monolith supports were used as catalytic supports [32, 33]. Therefore, this present work aims to develop rhodium catalysts supported on hydrotalcites, derived from mixed metal oxides of varying ratios of Mg/Al (HT1, HT2, HT3, HT4), and compare their performance against Rh/MgO and Rh/Al2O3. Because of its enhanced activity over nickel catalyst even with low weight percentage, rhodium was opted, and moreover, the reducibility of rhodium can be promoted by supports that have different Mg/Al ratios. To demonstrate this phenomenon, the synthesized catalysts were not reduced under H2 stream and used as synthesized. Therefore, the present study aims to analyze the role of support characteristics, reducibility of rhodium catalyst, and their variable interactions with the calcined hydrotalcites for regulating their activity in syngas production by CPOx of methane.
2 Experimental section
2.1 Catalyst synthesis
Magnesium hydroxide [Sigma-Aldrich, Mg(OH)2] and aluminum hydroxide [Sigma-Aldrich Al(OH)3] precursors were used by calcining at 550 °C in static air for getting the supports MgO and Al2O3. Calcined hydrotalcites consist of MgO:Al2O3 in the ratios of (4.5:0.5, HT1), (3:2 HT2), (1.5:3.5 HT3), and (0.5:4.5 HT4) and were prepared using the following procedure and used as catalyst supports. The hydroxides [Mg(OH)2 and Al(OH)3] were mixed in a specific Mg/Al ratio and dispersed in 50 mL of water. To this dispersion, lysine amino acid (50 mg) was added, and this mixture was kept under magnetic stirrer for 2 h, for obtaining hydrotalcites of varying Mg/Al ratios. Subsequently, to remove water content, these dispersions were transferred to a hot air oven and dried in air. These dried materials were further calcined in a muffle furnace at 550 °C and were used as catalyst supports without any further processing. Modified homogenous wet impregnation method was used for depositing 1 wt% of rhodium on these supports. A total of 50 mg urea and 3 g support were added to the aqueous solution containing (RhCl3) 1 wt%, and this slurry was stirred for few hours, and these materials were transferred to hot air oven for drying. After complete drying of these materials, these dried catalyst materials were calcined at 550 °C in a furnace for 4 h. Thus, the synthesized catalysts are referred to as Rh/HT1, Rh/HT2, Rh/HT3, Rh/HT4, Rh/Al2O3, and Rh/MgO, hereafter. ICP-MPES technique was used for estimating the Rh content in these calcined materials. Around 1 wt% of Rh was estimated to be found from all these samples from the ICP-MPES analysis.
2.2 Catalyst screening procedure and characterization procedures
To screen the synthesized catalysts for the CPOx of methane, a fixed bed reactor setup was used. All the catalysts were pelletized, and 300 mg of each pelletized catalyst (sieved to obtain 0.3–0.5 μm) was used for the screening study and loaded into the fixed bed reactor. As mentioned in the introduction, these catalysts were not activated or pre-reduced by hydrogen before the screening. CPOx of methane over these catalysts were performed in a temperature range (700–850 °C), GHSVs (66,000–1,10,000 mLh−1 g−1), and three different feed compositions. The feed composition of CH4/O2/N2 was varied to 2/1/8, 2.2/1/7.8, and 1.8/1/8.2 different compositions to study the influence of feed composition on the catalyst activity.
Long-time testing and the catalyst stability were studied by testing all the catalysts at 750 °C. Gas products and unreacted reactant gases were analyzed by the online Shimadzu GC equipped with TCD and FID detectors using the Varian CP-PoraPLOT Q column and Varian CP-Molsieve 5A column, respectively, to calculate methane conversion. CO and H2 ratios were estimated from the product gas analysis, and each catalyst performance was estimated from these methane conversion and syngas composition.
X-ray photoemission spectroscopic (XPS) measurements of the supported Rh catalysts were obtained using a Thermo K-5 Alpha XPS instrument, and the core spectral levels were aligned with C 1 s binding energy of 285 eV. Powder XRD patterns of these supported Rh catalysts were collected using Bruker D8 machine operated at 40 kV accelerating voltage. The Micrometrics ASAP2010 instrument was used to obtain the nitrogen adsorption-desorption isotherms at 77 K, and surface area, pore volume, and pore diameter of all the catalyst materials were calculated from these isotherms. Transmission electron microscopic (TEM) imaging of all these samples was carried out using the JEOL 1010 electron microscope operated at an accelerating voltage of 100 kV.
3 Results and discussion
Figure 1 shows N2-sorption isotherms obtained from the MgO and Al2O3 and calcined hydrotalcite supported Rh catalysts at 77 K. Textural properties including, pore diameter, pore volume, and surface area were estimated from these sorption isotherms. The adsorption/desorption isotherms of these materials and presence of hysteresis in all the materials demonstrated the presence of mesopores. Calcination of Mg(OH)2, Al(OH)3, hydrotalcites of varying MgO:Al2O3 ratios led to the formation of slit-like mesopores due to the removal of interlayer hydroxides as well as the removal of amino acid lysine, which was used during the synthesis of catalyst support materials. Mesopore fillings were observed at high relative pressures in the Rh/MgO, Rh/HT1, and Rh/HT2 materials, which have high MgO:Al2O3 ratio.

Nitrogen adsorption-desorption isotherms of Rh/MgO, Rh/HT1, Rh/HT2, Rh/HT3, and Rh/HT4 materials
In contrast, mesopore fillings in the adsorption branch were observed at lower relative pressures in the case of Rh/Al2O3, Rh/HT3, and Rh/HT4 materials, which had smaller MgO:Al2O3 ratio. Table 1 shows materials’ textural properties, and significant changes by varied MgO:Al2O3 ratios were seen. The obtained BET surface area of the materials improved as the MgO:Al2O3 ratio of the support decreased. Addition of aluminum hydroxide to magnesium hydroxide was known to increase the cationic charge density in each layer, and the amount of hydroxide ions present in the interlayer also increases with the increase in Al(OH)3 concentration. Therefore, calcination of these alumina-rich hydrotalcites led to the removal of more hydroxyl groups, which was the main reason of high specific surface area of these materials. In addition, the estimated pore volume and average pore diameter were observed to decrease as the MgO/Al2O3 ratio decreased in the case of calcined hydrotalcite supported Rh catalysts. Another interesting aspect that was observed was that the surface areas of the calcined hydrotalcites were found to increase after the impregnation of Rh onto these supports. Because of internalization of rhodium with the structural framework of calcined hydrotalcite, the surface area would have been reduced if the rhodium was settled on the surface as seen to be the same as Rh/MgO and Rh/Al2O3.
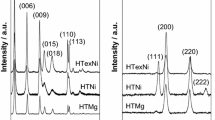
The nature of crystalline phases present in the samples were studied using the powder XRD patterns obtained from all the supported Rh catalysts, and the results are given in Fig. 2. Calcined hydrotalcites (Rh/HT1, Rh/HT2) supported Rh catalysts rich in MgO phase were crystalline, while Rh/Al2O and Rh/HT3, and Rh/HT4 were amorphous and less crystalline. The intense X-ray reflections were assigned to the cubic phase of MgO, and calcined hydrotalcites also exhibit patterns of XRD regarding MgO phase. MgO crystalline faces observed at 2θ 36.8, 42.7, 61.7, and 78.2° were broadened as the aluminum oxide content became higher in the calcined hydrotalcite XRD patterns of the support. After impregnation with Rh catalysts, similar broadening of peaks were observed. But the diffraction peaks regarding metallic rhodium or rhodium oxide were absent. Following Rh impregnation, it was found that that d value of supports and its crystalline size ere slightly changed as shown in Table 1. From the results, it was clearly observed that rhodium was uniformly distributed throughout the supports. This uniform dispersion of Rh precursors on the support materials was mainly due to the addition of urea during the synthesis of Rh impregnated catalysts. During the calcination process, the tendency of Rh in forming mixed oxides phases with MgO, oxides of Mg and Al in the calcined hydrotalcites and Al2O3, was considered as main reason of the disappearance of crystalline phase related to the rhodium oxides. This clearly demonstrates the highly uniform dispersion of rhodium oxides within the support.

Powder X-ray diffraction patterns of Rh/MgO, Rh/HT1, Rh/HT2, Rh/HT3, Rh/HT4, and Rh/Al2O3
Any kind of metal oxide formation that consists of Rh, MgO, and Al2O3, as well as uniform dispersion of Rh, was observed by change in diffraction peak broadening and absence of diffraction peaks corresponding to metallic rhodium. In the present work, XRD analysis of these materials showed the absence of metallic rhodium peaks, and peak width broadening in the diffraction patterns evidently indicate the Rh dispersion uniformity as well as formation of the any mixed metal oxide phase. This was the clear evidence that calcination temperature and method of synthesis were found to play key role in altering the nature of interaction between the rhodium phase and the support oxides. CPOx of methane and the rate of this reaction are expected to have higher influence over the nature of Rh interaction with various phase of support, when the composition of the support materials changes from pure crystalline MgO phase to amorphous phase of alumina and also by having variable fractions of these oxides. Particle size, support morphology, and formation of any phase separated rhodium oxide can be identified with the help of transmission electron microscopic (TEM) imaging of Rh/MgO, Rh/HT1, Rh/HT2, Rh/HT3, Rh/HT4, and Rh/Al2O3. In Fig. 3, It was clearly observed that the morphology of supports was clearly distinct from each other, evidently noticed from the TEM images. It was observed that the size of the particles was significantly different. Rh/MgO only consists of a particulate type of morphology, while all the other calcined hydrotalcites and alumina supported Rh catalysts exhibit sheet-like morphology. MgO and MgO-rich calcined hydrotalcites have smaller sized support particles.

TEM images of Rh/MgO, Rh/HT1, Rh/HT2, Rh/HT3, Rh/HT4, and Rh/Al2O3
Increasing the Al2O3 fraction (Rh/HT3, Rh/HT4) led to transformation of irregular small-sized particles into long planar sheets and was probably due to the calcination of layered hydrotalcite containing higher amount of amorphous alumina phase formation. The clear absence of any segregated Rh phase demonstrates the uniform mixing of rhodium onto these supports. These outcomes were clearly in agreement with the XRD results; therefore, rhodium was uniformly dispersed within oxide phase of the support.
To understand the chemical state of oxygen, aluminum, rhodium, and magnesium in all such supports and its role in modifying the interaction with rhodium metal, XPS core level spectra of all the supported catalysts were collected. Figure 4 shows O1s, Al2p, Mg1s, and Rh3d core level spectra that were collected from the Rh/MgO, Rh/HT1, Rh/HT2, Rh/HT3, RH/HT4, and Rh/Al2O3 materials. O1s core level spectra of each catalyst were considerably different, which is a clear indication that support composition have a major role in modulating the surface oxygen chemical states. All the catalysts exhibit three kinds of chemically distinct oxides, and the BE observed around 529.5 eV was assigned to the oxygen bind to the rhodium. The other two oxide components were assigned to the bulk oxide phase from the MgO phase and surface hydroxide. Rh/HT3, Rh/HT4, and Rh/Al2O3 materials exhibit less intense peaks corresponding to the oxide of rhodium, which was probably due to the very high dispersion of Rh onto these supports. These results indicated that the presence of alumina enhances the dispersion of Rh onto the calcined hydrotalcites.

XPS analysis of Al2p, Mg1s O1s, and Rh3d core level spectra obtained from Rh/MgO, Rh/HT1, Rh/HT2, Rh/HT3, Rh/HT4, and Rh/Al2O3
Al2p and Mg1s core level spectra also exhibit similar a pattern that indicates the dispersion of rhodium was dependent upon the chemical composition of the support. Mg1s spectral profiles observed in the case of Rh/Al2O3, Rh/HT3, and Rh/HT4 were found to be significantly different from the Mg 1s spectral profile of Rh/HT1, Rh/HT2 and Rh/MgO. In a similar manner, Rh/HT1 and Rh/HT2 exhibit similar Al2p core level spectral, as compared to the Al2p spectral profile of Rh/Al2O3, Rh/HT3 and Rh/HT4. This was probably due to the presence of alumina content in the calcined hydrotalcite has strong role in achieving the dispersion of Rh. Moreover, the strong propensity of Rh to form mixed metal oxide phase with MgO, HT1, and HT2 led to the different kinds of metal-support interaction. This was clearly demonstrated from the Mg1s core level spectra, which exhibited an additional chemical component that was assigned to the mixed metal oxide phase corresponding to Rh/MgO, Rh/HT1, and Rh/HT2. This phenomenon was not observed in the case of Rh/Al2O3, Rh/HT4, and Rh/HT3; rather, higher fractions of alumina enable uniform dispersion. Al2p core level displayed prominent variations with respect to Rh deposition in comparison with MgO core level variation. This was perhaps because of the strong interaction of alumina support with Rh than the MgO supports interaction with Rh and due to its high dispersion.
It has been observed that strong overlaps between Mg KLL Auger peaks and the Rh3d core level spectra mask the Rh 3d spectra due to highly intense and powerful Mg KLL lines. These Auger lines that correspond to the Mg KLL level were observed between 300 and 306 eV that exhibit larger chemical shifts and provide insight into the different chemical states of Mg. Similar to the above results, chemical shifts of Mg KLL Auger lines observed in the case of Rh/HT4 and Rh/HT3 were found to be different from the Mg KLL Auger lines observed in the case of Rh/HT2, Rh/HT1, and Rh/MgO. From these detailed XPS core level spectral analysis, Rh was found to form mixed metal oxide phase with Rh/MgO and MgO-rich calcined hydrotalcites (Rh/ HT1 and Rh/HT2). However, Rh/Al2O3 and calcined alumina-rich hydrotalcites (Rh/HT3, Rh/HT4) enhance the dispersion of Rh rather than forming a mixed oxide phase with the support materials. In this study, the variable interaction of Rh with MgO or Al2O3 phases will therefore influence their catalytic activity toward methane’s partial oxidation.
3.1 Catalyst screening for partial oxidation of methane
Catalytic partial oxidation of methane over the six catalysts including Rh/MgO, Rh/HT1, Rh/HT2, Rh/HT3, Rh/HT4, and Rh/Al2O3 were studied to evaluate their catalytic activity and presented in Fig. 5. As mentioned in the experimental conditions, these catalysts were not activated by hydrogen and used after the calcination.

Methane conversion over Rh/MgO, Rh/HT1, Rh/HT2, Rh/HT3, Rh/HT4, and Rh/Al2O3 as a function of temperature
As mentioned in the introduction, catalytic partial oxidation occurs via a two-step mechanism that combines the catalytic combustion and catalytic reforming. However, the rate of combustion was faster than the rate of reforming, and catalyst efficiency can be assessed based on the enhanced CO selectivity over the CO2 selectivity. CO2 was the product of exothermic combustion, while CO was the product of endothermic reforming; therefore, selectivity comparison between CO and CO2 can provide insight into the catalyst activity at different temperatures. Figure 6 shows the CO and CO2 selectivity of all the six catalysts at different temperatures. Rh/Al2O3 showed very high selectivity toward CO formation, while Rh/MgO showed high selectivity toward CO2 formation. Methane conversion was found to be 90% and 54% over Rh/Al2O3 and Rh/MgO, respectively, at 850 °C. Rh/HT1, Rh/HT2, Rh/HT3, and Rh/HT4 showed also high CO selectivity, and this selectivity was found to increase as the catalyst bed temperature increased. Methane conversion over the calcined hydrotalcite supported Rh catalysts such as Rh/HT1 and Rh/HT4 showed around 75% at 850 °C, next to Rh/Al2O3. Rh/HT2 and Rh/HT3 showed 70% and 67% methane conversion, but higher than Rh/MgO. At higher temperatures (850 °C), the catalyst activity of Rh/Al2O3, Rh/HT1, and Rh/HT4 were found to be the same, while at 700 °C, the activity of all the catalysts were significantly different and much lower. Syngas selectivity and methane conversion were found to increase with the increase in the catalyst bed temperature, because the high temperature tends to promote the endothermic reforming reaction.

CO and CO2 selectivity estimated from the catalytic partial oxidation of methane over Rh/MgO, Rh/HT1, Rh/HT2, Rh/HT3, Rh/HT4, and Rh/Al2O3 as a function of temperature
As the oxides of Rh are considered as the dynamic active sites for the catalytic oxidation of methane, the activity measured at 700 °C was not high enough to access the dynamic active sites of Rh. The total weight percentage of rhodium was maintained constant in all the six catalysts; therefore, the measured catalyst activity was associated with the surface rhodium concentration. Higher conversion of methane was observed in the following order Rh/Al2O3 > Rh/HT4 > Rh/HT1. XPS analysis showed that alumina and alumina-rich calcined hydrotalcites promote the Rh dispersion on the surface, while MgO and MgO-rich calcined hydrotalcites promote the mixed metal oxide formation with Rh. Support materials that can enhance the Rh dispersion can increase the surface concentration of active oxides of Rh. In contrast, support materials that promote the formation of mixed metal oxide phase reduce the concentration and might be the plausible explanation behind the lesser activity observed in the case of Rh/MgO and Rh/HT2.
Syngas composition, i.e., the H2/CO ratio, obtained from the catalytic partial oxidation of methane is considered as an important parameter to compare the catalyst efficiency. H2/CO ratio was calculated for each catalyst at different reaction temperatures and is shown in Fig. 7. It is evident from the figure that the average H2/CO ratio was found to vary between 2.5 and 3 except in the case of Rh/MgO. H2/CO ratio was found to be higher in the case of Rh/Al2O3, Rh/HT4, and Rh/HT3. Rh/HT1 and Rh/HT2 showed lower H2/CO ratio due to their high conversion of CO2, as a result of their basicity. Another possible explanation for this minor change of ratio as a function of temperature was probably due to the competing reactions such as shift reactions.

H2/CO ratio estimated from the catalytic partial oxidation of methane over Rh/MgO, Rh/HT1, Rh/HT2, Rh/HT3, Rh/HT4, and Rh/Al2O3 as a function of temperature
The feed composition, i.e., ratio of CH4/O2/N2, is also considered to influence the methane conversion and product selectivity. CPOx reactions over Rh/HT1, Rh/HT2, Rh/HT3 and Rh/HT were carried out in three different feed ratios, and the results are presented in Fig. 8 (left panel). Major changes in methane conversion were observed when the feed composition was varied. Rh/HT4, Rh/HT3, and Rh/HT1 showed very high methane conversion, when the feed ratio was maintained at 1.8/1/8.2. However, Rh/HT1 only showed high conversion when the other two feed ratios were used, wherein the methane/oxygen ratio was high. With less oxygen in the feed, Rh/HT1 demonstrated enhanced action, whereas Rh/HT4 and Rh/HT1 showed moderate methane conversion. This was probably due to the involvement of lattice oxygen (Rh/HT1) in promoting the methane conversion.

Effect of feed composition (left) and GHSV mlh−1 g−1 (right) on catalytic partial oxidation of methane over Rh/HT1, Rh/HT2, Rh/HT3, and Rh/HT4
As mentioned in the introduction, catalytic partial oxidation of methane can be operated at very high space velocities; therefore, catalytic partial oxidations of methane over Rh/HT1, Rh/HT2, Rh/HT3, and Rh/HT4 were studied at two different GHSVs, and the results are presented in the right panel of the Fig. 8. Rh/HT4, Rh/HT3, and Rh/HT2 did not show much changes when the GHSV was varied. In contrast, Rh/HT1 showed high conversion at lower GHSV and higher than all the other calcined HT supported Rh catalysts.
Hot spot formation and carbon formation were the two major factors that are responsible for the catalyst deactivation and affect the longevity of the catalyst used for catalytic partial oxidation of methane. Figure 9 shows the stability of all the catalysts including Rh/HT1, Rh/HT2, Rh/HT3, and Rh/HT4 over CPOx reaction that were assessed for the period of 10 h. Among all the catalysts, Rh/HT1 showed higher conversion as compared with the other catalysts for the period of 10 h without any drop in methane conversion.

Time on stream of the catalytic partial oxidation of methane over Rh/HT1, Rh/HT2, Rh/HT3, and Rh/HT4
3.2 Spent catalyst characterization
All the spent catalysts after the CPOx reaction were characterized by XRD, TGA, XPS, and TEM to identify the structural and chemical changes on the catalyst active sites as well as the nature of carbon deposited on the surface of the catalyst.
Powder XRD patterns of all six spent catalysts and the calcined catalysts are presented in Fig. 10, and the XRD patterns of the spent catalysts of Rh/MgO, Rh/HT1, and Rh/HT2 did not show much changes to the XRD patterns corresponding to the fresh catalyst. MgO and MgO-rich calcined hydrotalcites are known for their thermal stability as well as not promoting the carbon formation on its surface. These results clearly demonstrate that these catalysts were strongly resistant against high temperature hot spot-mediated sintering and carbon deposition. Rh/HT1 showed long-term stability and did not show any kind of crystalline carbon formation on its surface. In contrast, carbon formation on the catalyst surface was prominent in the case of Rh/Al2O3, Rh/HT3, and Rh/HT4. Characteristic X-ray reflections (marked with *) corresponding to the graphitic carbon (PCPDF#752078) observed in these three spent catalysts indicated the carbon deposition was difficult to control on these catalysts. An intense peak observed at 2θ 44.03 was assigned to the (010) plane of the graphite phase of carbon, and this carbon type of carbon formation generally lead to the deactivation of the catalyst. This was probably due to less thermal stability and higher amount of carbon formation on Rh/Al2O3, Rh/HT4, and Rh/HT3. To study the structural changes in detail, inter-planar spacing (d111) and crystallite size were calculated and given in Table 2. Except for Rh/MgO, the estimated crystallite size was found to be higher at Rh/Al2O3, Rh/HT4, RH/HT3, Rh/HT2, and Rh/HT1.

XRD patterns of the spent catalysts Rh/MgOs, Rh/HT1s, Rh/HT2s, Rh/HT3s, Rh/HT4s, and Rh/Al2O3s (thick lines) obtained after the catalytic partial oxidation of methane that were overlaid with XRD patterns of fresh catalysts (thin lines)
TEM imaging of all the spent catalysts was carried out to study the changes in catalyst morphology, type of deposited carbon, and sintering of catalyst particles after they were used for catalytic partial oxidation of methane. Figure 11 shows the representative TEM images, and it was clearly seen that carbon formation was prominent in the case of Rh/HT2, Rh/HT3, Rh/HT4, and Rh/Al2O3. Spent catalysts including Rh/HT1 and Rh/MgO did not show much carbon formation as well as not much morphological changes due to their thermal stability, which was in agreement with the XRD results. Carbon nanotubes were formed on the catalyst surface in all the spent catalysts including Rh/Al2O3, Rh/HT4, Rh/HT3, and Rh/HT2, indicating that the alumina contents of the supports tend to promote the carbon formation. This was probably due to the acidic active sites of alumina supports, which are known to promote carbon formation.

TEM images of the spent catalysts Rh/MgOs, Rh/HT1s, Rh/HT2s, Rh/HT3s, Rh/HT4s, and Rh/Al2O3s obtained after the catalytic partial oxidation of methane
XPS studies of all the spent catalysts were carried out to investigate the chemical state of oxygen, aluminum, magnesium, and rhodium during the catalytic partial oxidation of methane.
To investigate the changes in the chemical state and oxidation of the elements, Al2p, Mg1s, O1s, and Rh3d core level spectra were collected from the spent catalysts and are presented in Fig. 12. The peak binding energies of these core levels are provided in Table 2. Significant changes in the XPS spectral profile were observed in the case of spent catalysts from the spectral profile of fresh catalysts. Uniform dispersion of Rh onto the alumina supports was observed from the presence of rhodium oxide components, which was not present in the case of Rh/Al2O3 spent catalyst. This was probably due to the formation of carbon on the active oxide species of rhodium. Spent catalysts including Rh/MgO and Rh/HT1 exhibit O1s core level spectra which were significantly different from the O1s spectral profile of the other spent catalysts. This was probably due to the formation of mixed metal oxide phase between Rh and Rh/HT1 (and Rh/MgO), which restricts the presence of Rh within the structural frame work of MgO.

XPS core level spectra of the spent catalysts Rh/MgOs, Rh/HT1s, Rh/HT2s, Rh/HT3s, Rh/HT4s, and Rh/Al2O3s obtained after the catalytic partial oxidation of methane
Overall, the nature of interaction between Rh with MgO (Rh/MgO) and Rh with MgO-rich calcined hydrotalcites (Rh/HT1, Rh/HT2) was found to be different from the nature of interaction between Rh with Al2O3 (Rh/Al2O3) and Rh with Al2O3-rich calcined hydrotalcites (Rh/HT3 and Rh/HT4).The peak binding energies of Mg1s core level shifted to lower BEs as the alumina content in the calcined hydrotalcite was increased. This was probably due to the migration of Rh from the oxide framework into the surface of the alumina. Mg KLL Auger lines overlap with the Rh3d core level spectra that poses a difficulty in identifying its oxidation state in these materials reducibility of the metal, their dispersion and control the size of these metals during CPOx process.
Thermogravimetric analyses of all the spent catalysts were carried to calculate the amount of carbon deposited on each catalyst, during catalytic partial oxidation of methane. Figure 13 shows the TG profile of all the spent catalysts using air as carrier gas, and Table 2 lists the amount of carbon deposited on each catalyst. Rh/HT1 showed less carbon deposition among all catalysts, while other materials showed higher carbon content ranging from 8 to 9%. TGA profile of the Rh/Al2O3 was found to be different from the TG profile of calcined hydrotalcite supported Rh catalysts. Carbon desorption occurs at much lower temperatures in the case of Rh/HT1, Rh/HT2, Rh/HT3, and Rh/HT4, which indicated that the carbon formed on these catalysts can be easily gasified at lower temperatures; therefore, carbon formation may not be a strong factor to deactivate the catalysts.

TG analysis of the spent catalysts spent catalysts obtained after the catalytic partial oxidation of methane over Rh/MgOs, Rh/HT1s, Rh/HT2s, Rh/HT3s, Rh/HT4s, and Rh/Al2O3s
4 Conclusions
To summarize, catalytic partial oxidations of methane were studied over calcined hydrotalcites of varying Mg/Al ratios supported Rh catalyst. Hydrotalcites of varying Mg/Al ratios were prepared by mixing hydroxides of magnesium and aluminum in the presence of lysine amino acid and subsequent calcination. Impregnation of Rh (1 wt%) on these supports (Rh/HT1, Rh/HT2, Rh/HT3, and Rh/HT4) were done using homogeneous wet impregnation followed by calcination. Rh/Al2O3 and Rh/MgO were also prepared as reference catalysts to compare the catalyst activities of the calcined hydrotalcite supported Rh catalysts. The influence of support materials Mg/Al ratio in controlling the dispersion of rhodium and their main role in controlling the catalyst activity toward catalytic partial oxidation of methane were studied in detail. The performances of all these supported Rh catalysts were strongly affected by the support, and Mg/Al ratios were found to control the activity, coking, and syngas composition. MgO-rich calcined hydrotalcite supported Rh catalysts (Rh/MgO, Rh/HT2) were found to have less activity but more resistance toward carbon formation. The probable reason was the mixed metal oxide formation which facilitates the migration of Rh content to the bulk and restricts its accessibility. It also decreases its ease of reducibility and the number of active rhodium sites which leads to the reduction of catalytic activity, which eventually promotes the combustion reaction. MgO-supported Rh was a very stable catalyst because of the strong interactions between rhodium and magnesium oxides. Rh/Al2O3, Rh/HT4. and Rh/HT3 were found to enable uniform dispersion of Rh, which was the main reason that these catalysts exhibit much higher activity among all catalysts. However, carbon formation, particle sintering, and morphology changes in the support particles were found to reduce its activity and stability. By varying the feed variation and GHSV, Rh/HT1 was optimized to exhibit higher activity and catalyst stability against coking and less sintering. These results demonstrated that using calcined hydrotalcites of varying Mg/Al ratios as supports to Rh catalyst can increase the stability of Rh catalysts against the coking and sintering as well as obtaining high conversion of methane and high syngas selectivity during the CPOx reaction.
References
B.C. Enger, R. Loedeng, A. Holmen, A review of catalytic partial oxidation of methane to synthesis gas with emphasis on reaction mechanisms over transition metal catalysts. Appl. Catal. A 346(1–2), 1–27 (2008). https://doi.org/10.1016/j.apcata.2008.05.018
S. Liu, Y. Chen, H. Xu, Y. Wang, W. Li, Review on the reactors for the production of synthesis gas by catalytic partial oxidation of methane. Shiyou Yu Tianranqi Huagong 37(2), 105–109, 114 (2008)
H.-T. Wang, S.-X. Tian, Z.-H. Li, Catalytic partial oxidation of methane to syngas. Tianjin Gongye Daxue Xuebao 23(1), 43–45 (2004)
M. Baerns, O.V. Buyevskaya, L. Mleczko, D. Wolf, Catalytic partial oxidation of methane to synthesis gas - catalysis and reaction engineering. Stud. Surf. Sci. Catal. 107(Natural Gas Conversion IV), 421–428 (1997)
A. Bitsch-Larsen, Catalytic partial oxidation of methane at industrially relevant conditions. PhD thesis, University of Minnesota Minneapolis (2008)
W.-J. Choi, J.-Y. Park, M.-S. Kim, H.-S. Park, H.-S. Hahm, Catalytic partial oxidation of methane to methanol. J. Ind. Eng. Chem. (Seoul, Repub. Korea) 7(4), 187–192 (2001)
M. Bizzi, L. Basini, G. Saracco, V. Specchia, Short contact time catalytic partial oxidation of methane: analysis of transport phenomena effects. Chem. Eng. J. (Amsterdam, Neth.) 90(1–2), 97–106 (2002). https://doi.org/10.1016/S1385-8947(02)00071-2
W.-H. Chen, S.-C. Lin, Reaction phenomena of catalytic partial oxidation of methane under the impact of carbon dioxide addition and heat recirculation. Energy (Oxford, U. K.) 82, 206–217 (2015). https://doi.org/10.1016/j.energy.2015.01.031
A.M. De Groote, G.F. Froment, Simulation of the catalytic partial oxidation of methane to synthesis gas. Appl. Catal. A 138(2), 245–264 (1996). https://doi.org/10.1016/0926-860X(95)00299-5
K.L. Hohn, Catalytic partial oxidation of methane at high flow rates: production of syngas and acetylene. PhD thesis, University of Minnesota Minneapolis (1999)
C. Hurt, M. Brandt, S.S. Priya, T. Bhatelia, J. Patel, P.R. Selvakannan, S. Bhargava, Combining additive manufacturing and catalysis: a review. Catal. Sci. Technol. 7(16), 3421–3439 (2017). https://doi.org/10.1039/C7CY00615B
N. Burke, D. Trimm, Coke formation during high pressure catalytic partial oxidation of methane to syngas. React. Kinet. Catal. Lett. 84(1), 137–142 (2005)
P.J. Fleming, W. Cossutta, P.J. Jackson, Carbon deposition in the catalytic partial oxidation of methane to synthesis gas. Stud. Surf. Sci. Catal. 81(Natural Gas Conversion II), 321–324 (1994)
R. Jin, Y. Chen, W. Cui, W. Li, C. Yu, Y. Jiang, Mechanism of catalytic partial oxidation of methane to synthesis gas on nickel catalyst. Wuli Huaxue Xuebao 15(4), 313–318 (1999)
R. Jin, Y. Chen, W. Li, W. Cui, Y. Ji, C. Yu, Y. Jiang, Mechanism for catalytic partial oxidation of methane to syngas over a Ni/Al2O3 catalyst. Appl. Catal., A 201(1), 71–80 (2000). https://doi.org/10.1016/S0926-860X(00)00424-5
T. Liu, G. Veser, Temperature dynamics during catalytic partial oxidation of methane in a reverse-flow reactor. ACS Fuel Chemistry Preprints 50 (2005)
Z. Jiang, J. Su, M.O. Jones, H. Shi, T. Xiao, P.P. Edwards, Catalytic partial oxidation of methane over Ni-based catalysts derived from Ni-mg/Al ternary Hydrotalcites. Energy Fuel 23(3), 1634–1639 (2009). https://doi.org/10.1021/ef800933j
M.H. Rafiq, H.A. Jakobsen, J.E. Hustad, Modeling and simulation of catalytic partial oxidation of methane to synthesis gas by using a plasma-assisted gliding arc reactor. Fuel Process. Technol. 101, 44–57 (2012). https://doi.org/10.1016/j.fuproc.2011.12.044
R.C. Ramaswamy, P.A. Ramachandran, M.P. Dudukovic, Modeling catalytic partial oxidation of methane to syngas in short-contact-time packed-bed reactors. Ind. Eng. Chem. Res. 46(25), 8638–8651 (2007). https://doi.org/10.1021/ie070084l
J. Yu, L. Zhang, Z. Yu, Study on the catalytic partial oxidation of methane to syngas. II. Study on Pt-doped Ni/Al2O3 catalysts. Tianranqi Huagong 21(5), 23–25 (1996)
J. Yu, L. Zhang, Z. Yu, Study on catalytic partial oxidation of methane to syngas. I. Supported nickel/α-alumina catalysts. Tianranqi Huagong 21(4), 5–8 (1996)
M. Prettre, C. Eichner, M. Perrin, The catalytic oxidation of methane to carbon monoxide and hydrogen. Trans. Faraday Soc. 42(0), 335b–3339b (1946). https://doi.org/10.1039/TF946420335B
L.J.I. Coleman, E. Croiset, W. Epling, M. Fowler, R.R. Hudgins, Evaluation of foam nickel for the catalytic partial oxidation of methane. Catal. Lett. 128(1–2), 144–153 (2009). https://doi.org/10.1007/s10562-008-9707-y
J.A. Velasco, C. Fernandez, L. Lopez, S. Cabrera, M. Boutonnet, S. Jaeraas, Catalytic partial oxidation of methane over nickel and ruthenium based catalysts under low O2/CH4 ratios and with addition of steam. Fuel 153, 192–201 (2015). https://doi.org/10.1016/j.fuel.2015.03.009
L.D. Vella, S. Specchia, Alumina-supported nickel catalysts for catalytic partial oxidation of methane in short-contact time reactors. Catal. Today 176(1), 340–346 (2011). https://doi.org/10.1016/j.cattod.2010.11.068
V.R. Choudhary, B.S. Uphade, A.S. Mamman, Oxidative conversion of methane to syngas over nickel supported on commercial low surface area porous catalyst carriers precoated with alkaline and rare earth oxides. J. Catal. 172(2), 281–293 (1997). https://doi.org/10.1006/jcat.1997.1838
F. Basile, P. Benito, G. Fornasari, M. Monti, E. Scavetta, D. Tonelli, A. Vaccari, Novel Rh-based structured catalysts for the catalytic partial oxidation of methane. Catal. Today 157(1–4), 183–190 (2010). https://doi.org/10.1016/j.cattod.2010.04.039
J.-D. Grunwaldt, L. Basini, B.S. Clausen, In situ EXAFS study of Rh/Al2O3 catalysts for catalytic partial oxidation of methane. J. Catal. 200(2), 321–329 (2001). https://doi.org/10.1006/jcat.2001.3211
D. Li, S. Sakai, Y. Nakagawa, K. Tomishige, FTIR study of CO adsorption on Rh/MgO modified with co, Ni, Fe, or CeO2 for the catalytic partial oxidation of methane. Phys. Chem. Chem. Phys. 14(25), 9204–9213 (2012). https://doi.org/10.1039/c2cp41050h
S. Mandal, P.R. Selvakannan, D. Roy, R.V. Chaudhari, M. Sastry, A new method for the synthesis of hydrophobized, catalytically active Pt nanoparticles. Chem. Commun. (Cambridge, U. K.) 24, 3002–3003 (2002). https://doi.org/10.1039/b209050c
A. Ballarini, P. Benito, G. Fornasari, O. Scelza, A. Vaccari, Role of the composition and preparation method in the activity of hydrotalcite-derived Ru catalysts in the catalytic partial oxidation of methane. Int. J. Hydrog. Energy 38(35), 15128–15139 (2013). https://doi.org/10.1016/j.ijhydene.2013.08.135
F. Basile, G. Fornasari, V. Rosetti, F. Trifiro, A. Vaccari, Effect of the mg/Al ratio of the hydrotalcite-type precursor on the dispersion and activity of Rh and Ru catalysts for the partial oxidation of methane. Catal. Today 91-92, 293–297 (2004). https://doi.org/10.1016/j.cattod.2004.03.047
J.K. Hochmuth, Catalytic partial oxidation of methane over a monolith supported catalyst. Appl. Catal. B 1(2), 89–100 (1992). https://doi.org/10.1016/0926-3373(92)80035-X
Acknowledgments
The statements made herein are solely the responsibility of the authors. Tibra Mozammel sincerely acknowledge RMIT for the PhD fellowship and Dr. Jim Patel (CSIRO, Gas processing facilities) for providing permission to use the catalyst testing facilities.
Funding
This work was carried by the NPRP grant # NPRP11S-1221-170116 from the Qatar National Research Fund (a member of Qatar Foundation).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest
Rights and permissions
About this article

Cite this article
Mozammel, T., Dumbre, D., Selvakannan, P. et al. Calcined hydrotalcites of varying Mg/Al ratios supported Rh catalysts: highly active mesoporous and stable catalysts toward catalytic partial oxidation of methane. emergent mater. 4, 469–481 (2021). https://doi.org/10.1007/s42247-020-00158-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42247-020-00158-2