
Abstract
The sieving and enrichment of rare tumor cells from large-volume pleural effusion (PE) samples is a promising technique for cell-based lung tumor diagnosis and drug tests, which features high throughput and recovery, purification, as well as viability rates of rare target cells as the prerequisites for high sensitivity, specificity, and accuracy of tumor cell analysis. In this paper, we propose a three-dimensional (3D) sieving method for rare tumor cell enrichment, which effectively eliminates the "dead zones" in traditional two-dimensional (2D) cell filters with a dimension-raising strategy to satisfy the requirements mentioned above. The prototype device was combined with a funnel-shaped holder, a flexible micropore membrane in the middle, and a 3D spiral fluid channel covered on the membrane as a three-layer ice-creaming cone composite structure. Driven by gravity alone, the device performed as follows: (1) 20-fold throughput compared with the 2D commercial plane cell filter, which was up to 20 mL/min for a threefold dilution of whole blood sample; (2) high recovery rates of 84.5% ± 21%, 86% ± 25%, 83% ± 14% for 100, 1000, and 10 000 cells/mL, respectively, in 30 mL phosphate buffer saline (PBS) sample, and a 100% positive detection rate in the case of ≤ 5 A549 cells in 1 mL PBS; (3) a typical purification rate of 85.5% ± 9.1%; and (4) a viability rate of > 93%. In the demonstration application, this device effectively enriched rare target cells from large volumes (> 25 mL) of clinical pleural effusions. The following results indicated that tumor cells were easy-to-discover in the enriched PE samples, and the proliferation capability of purified cells was (> 4.6 times) significantly stronger than that of unprocessed cells in the subsequent 6-day culture. The above evaluation indicates that the proposed easily reproducible method for the effective execution of rare cell enrichments and assays is expected to become a practical technique for clinical cell-based tumor diagnosis.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Most cases of malignant pleural effusion (PE) are caused by lung cancer, breast cancer, and lymphoma; 15% of lung cancer patients will initially present a malignant pleural effusion, and up to 50% will have a malignant pleural effusion throughout the course of the disease [1]. The enrichment and purification of rare malignant tumor cells from PE samples has an important value for the diagnosis of cancer, guiding the surgical staging, surgical prognosis, and diagnostic tracking [2,3,4]. However, the enrichment of rare tumor cells with high recovery and purity has been a great challenge due to the low concentration of such cells and interference from blood cells. Furthermore, liquid biopsy samples are highly recommended for analysis within 6–8 h after collection from patients to maintain viability [5,6,7]. Therefore, there is a pressing need to develop rapid and efficient point-of-care rare tumor cell isolation technologies that could be broadly deployed in clinical settings.
Size-based methods, which have the main advantages of being label-free, simple, and fast compared to biological enrichment, are more appropriate for tumor cell analysis and thus have been widely proposed [8]. The typical techniques presented include density gradient centrifugation, mechanical filtering, deterministic lateral displacement (DLD), Dean flow fractionation (DFF), dielectrophoresis (DEP), and acoustophoresis [6,7,8,9,10,11,12,13,14,15,16]. Among these techniques, the cell sieving method has been considered the most promising approach for clinical applications due to its high throughput potential and simple operation [17,18,19]. With the advantage of being a label-free process, the above methods enable a general purpose application in laboratories or clinical application scenarios for the size-based sorting of different cells (e.g., peripheral blood lymphocytes [20], exfoliated tumor cells from bronchoalveolar lavage fluid [9], etc.).
However, traditional cell sieving techniques can hardly meet the requirements of the high-throughput, high recovery and purification, and viability rates of rare target cells enrichment at the same time owing to their inherent weakness, which is the so-called dead zone placed beside the sieving micropores, inducing cell residue on the micropore membrane. One of the optimizing strategies is to increase the micropore membrane's porosity to improve the throughput and reduce this "dead zone" to improve the recovery, purification rate, and throughput [21]. To pursue this goal, considerable efforts have been directed to develop micropore membranes based on existing technologies for size-based cell sieving to form cell filters by various micromachining fabrication techniques and materials, such as silicon [22], parylene polymer [23, 24], SU-8 photoresist [25], or palladium [26]. With the above optimizing strategies, the typical throughput of existing micropore-arrayed filtration methods has reached about 2 mL/min [27]. One of the best solutions is the parylene-based cell filters reported by Wang [9] and named as "PERFECT," which eventually achieved the above requirements (> 17 mL/min throughput and about 85% recovery rate of the blood sample) thanks to the high porosity (> 40.2%) of their membrane with a skillfully designed structure and complex micromachining fabrication processes.
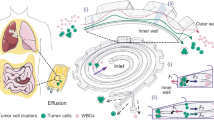
Another approach for optimizing the cell sieving technique is improving throughput by enlarging the commercial micropore membrane and inducing easily fabricated accessories to overcome its weakness so that it can be broadly reproduced by users to perform effectively. The commonly used commercial polymer filtration membrane in biological studies and clinical practice for cell enrichment [28,29,30] is the track-etched micropore membrane, whose inherent disadvantage is low porosity (2–7% or lower [8]). As illustrated in Fig. 1a, when using the small area of the two-dimensional (2D) sieving membrane, the sieving process frequently causes micropore clogging due to the high concentration of background cells and thus compromises its performance in clinical applications. In traditional 2D sieving methods, enlarging the micropore membrane area would increase not only the throughput but also the amount of background cell residue above the membrane caused by the "dead zone," broadening with the decreasing flow velocity beside the micropores, as shown in Figs. 1b and 1c. Using the 2D cell sieving methodologies, it seems impossible to solve the trilemma of increasing the throughput by enlarging the membrane area without suffering from the drawback of decreased recovery and purification rates.

The proposed 3D sieving methodology to improve the sieving performance based on the dimension raising strategy. a Schematic of the 2D plane sieving device, where the background cells are resided on the membrane and the target cells are hard to recover. b The simulated results show that the “dead zone” is placed beside the micropores on the 2D plane sieves’ membrane. c A large proportion of background cells are resided on the surface of the 2D plane sieve after cell sieving. d Schematic of the 3D sieving device with a coned three-layer structure for effective, high-throughput cell enrichment. e The simulated results show that the inclined 3D spiral channel can eliminate the “dead zones,” and simultaneously increase the chance of flowing background cells to be filtered out, and finally improve the purity of enriched cells. f The coned structure was designed to improve the recovery rate due to the enriched cells ultimately gathering at the conical tip of the coned micropore membrane
With the strategy of dimension raising, we herein propose a three-dimensional (3D) cell sieving method that enables the high-throughput and high-efficiency enrichment of rare cells. As shown in Fig. 1d, the sieving device was designed as a three-layer ice cream cone composite structure, whose interlayer is a large-area conical polymer micropore membrane coupled with the 3D spiral channels carved on the bottom surface of the 3D printed conical cover and supported by a 3D-printed funnel holder structure. When sieving rare cells, the cell samples only need to be poured over the entrance and the target cells are subsequently recovered from the colonial tip of the membrane. To eliminate the "dead zone" on the micropore membrane featured by 2D cell sieving methods, we designed a 3D spiral channel and coupled it onto a cone-shaped micropore membrane to guide the fluid driven by gravity for continuous flushing the membrane. As shown in Fig. 1e, the simulated streamline in the inclined flow channel effectively reduces the dead zone on the filter membrane. Thus, this novel structure effectively increases the throughput and avoids cell clogging or residue on the sieve membrane. As illustrated in Fig. 1f, the enriched target (large) cells are guided along with the spiral channel down to the colonial tip, where they can be further collected in a small volume. Meanwhile, the background (small) cells in the spiral flow can directly pass through the micropore on the sieve membrane and flow to the waste tube. With this dimension-raising design, the 3D cell sieving method is expected to meet the requirement of rapid rare target cell enrichment from clinical samples at a high recovery rate, purity, and viability.
Based on the proposed 3D sieving method, a prototype platform was realized (shown in Fig. 2) and evaluated as follows. Initially, the cell enrichment performance, which was characterized as the combination of throughput, recovery rate, purity, and viability, was evaluated by analyzing the recovered cells from large-volume samples spiked with target cells. Furthermore, as a demonstration, the rare tumor cells were enriched from clinical PE samples and assayed.

a Setup of the prototype platform, which has a 3D sieving device prototype, peristaltic pumps, and a waste container. The 3D sieving device has three layers and is composed by the b 3D spiral channel, c micropore membrane (SEM image, with 10 μm diameter micropores), and d membrane holder. The micropore membrane was fabricated by the e track etching technique, and coned by the f steps of folding, heat sealing and tailoring
Materials and methods
Reagents
Unless otherwise stated, all reagents for cell culture were purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA). Specifically, relevant materials used for the cell cultures and treatments included RPMI-1640 Media (GIBCO, Life Technologies Corp., USA), DMEM Media (GIBCO, Life Technologies Corp., USA), Fetal Bovine Serum (GIBCO, Life Technologies Corp., USA), Penicillin–Streptomycin (GIBCO, Life Technologies Corp., USA), 0.25% Trypsin (GIBCO, Life Technologies Corp., USA), Phosphate Buffer Saline (PBS) (GIBCO, Life Technologies Corp., USA), CCK-8 Cell Proliferation and Cytotoxicity Assay Kit (Glpbio, USA), Transforming Growth Factor-Beta (GIBCO, Life Technologies Corp, USA) and BSA (Thermo Fisher, USA). Calcein AM (Thermo Fisher, USA), propidium iodide (PI) (Thermo Fisher, USA), Hoechst 33,342 (Thermo Fisher, USA) and 2-NBDG (2-[N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose) (Thermo Fisher, USA) were applied as indicators of cell viability and metabolism.
Design and fabrication of the prototype sieving device
As shown in Fig. 2a, the setup of the prototype platform included the 3D sieving prototype device, the pumping components, and other parts. The prototype device was developed following the schematic shown in Fig. 1f, which was combined with 3D spiral channels (shown in Fig. 2b), coned polymer micropore membrane (shown in Fig. 2c), and the holder (shown in Fig. 2d) top to bottom. Besides, the peristaltic pumps were set for the injection of cell samples and washing buffers.
The structural parameters of the device were designed for handling liquid samples with 100 mL volume. Specifically, the micropore membrane was coned in the vertical section angle of 60° (determined by the folding and sealing process, as shown in Fig. S2) with a surface area of 56.5 cm2. The spiral channel was designed with a 2 mm × 1.5 mm square in the channel's cross-section and 6 mm in the pitch of screw, and 50 mm in the channel's height. The contact gap distance between the spiral channel edges and the micropore membrane was considered an important parameter for the filtering effect. It was controlled as < 0.05 mm (much lower than the 1.5 mm of the fluid channel height) by employing the precise 3D printing fabrication method with closely matching shapes to ensure the guided mainstream flow along the spiral channels. In addition, three different channel heights of 30, 40, and 50 mm were designed to verify the rising throughput when enlarging the membranes' effective area in the next section.
The 3D spiral channels, the holder, and the platform shell were fabricated from photosensitive resin (DSM SOMOS 8000) using a 3D printing machine (Lite 300, UnionTech, China).
The polymer micropore membrane was fabricated from polycarbonate (PC) material using a high-speed heavy ion bombardment and sodium hydroxide etching fabrication process (Fig. 2e) at the Heavy Ion Research Facility in Lanzhou (HIRFL), China. The membranes were approximately 10 μm in thickness and about 15%/10% in porosity for 10/8 μm diameter micropores, respectively. The membrane was formed into a cone shape by going through the folding, cutting, and heat sealing steps (Fig. 2f). In addition, it was surface modified (> 5 h before use) with 1% BSA to avoid cell adhesion.
Evaluation of cell enrichment performance
Based on the device prototype, the performance evaluation process was split into two steps, as detailed below. Firstly, the level of throughput, recovery rate, and viability were separately validated using the target cells only. Secondly, the level of purity, viability, and throughput were simultaneously validated using the simulated PE, which was created by adding the rare target cells to the diluted whole blood cell samples in different cell proportions.
In the following procedure, unless otherwise indicated, the THP-1 (human monocytic cell line) cells, which are slightly larger than most white blood cells, were employed as rare target cells. Besides, A549 cells (human lung tumor cells) were used to test the positive rate of very rare (≤ 5 per mL) cell detection and viability evaluation. Both of the THP-1 and A549 cell lines were purchased from the Chinese National Infrastructure of Cell Line Resource (NICR) and cultured in a cell incubator (Thermo Scientific, USA) at 37 ℃ under 5% CO2. Meanwhile, the whole blood cells were donated by healthy people. For the enrichment of THP-1 and A549 cells, the device was equipped with an 8-μm and 10-μm micropore membrane, respectively.
Before the tests, except for the viability evaluation tests, the target cells were pre-stained with live cell fluorescence indicator (Calcein-AM, green fluorescence) for convenient observation and counting under a fluorescence microscope.
Separate validation of high throughput, recovery rate, and viability
During the test procedure, the cell samples were supplied to the entrances of the spiral channel in the device by a pump or Eppendorf tubes (except for the tests of very rare (≤ 5 cells/mL) cell detection). Driven by gravity, the smaller cells and liquid passed through the membrane and flowed down to the lower flasks, and rare target cells were recovered from the micropore membrane's cone tip. For subsequent cell culture and analysis, the upper spiral channel scaffold needed to be removed from the device, and the membrane surface was rinsed with PBS for the recovery of enriched cells in the colonial tip. The recovered solution was transferred into a 5-mL centrifuge tube or microplate by pipetting.
For the throughput measurement, each sample of 20 mL PBS solution was directly poured to the spiral channel's entrance, and the waste was collected by a container placed on a precision electronic scale (accuracy: 0.001 g, Mettler Toledo-ME203E, Switzerland), which was employed to record the weight increment of the collected liquid over time (Fig. S3). The throughputs were calculated from the real-time recorded weight increment over time divided by the liquid's density (1.0 g/mL). To compare the throughput among the 3D and 2D sieving methods, the amount of solution after adding the same volume of PBS to the devices, including the 3D spiral sieving device with different spiral channel heights (shorted as the H) of 30, 40, and 50 mm and the 2D cell sieve formed with the same micropore membrane, was measured and analyzed.
For the viability evaluation, each 10 mL cell sample doped separately with 10 000 and 1000 A549 cells was sieved using the 3D sieving device and then analyzed. Once the cell sample was sieved, the recovered cells were stained with the viable (Calcein-AM, green fluorescence) and dead (PI, red fluorescence) cell indicators and observed under a fluorescence microscope (IX-73, Olympus) for counting the number of living and dead cells. The short-term viability of recovered cells was further analyzed based on the counting results. Moreover, the partially recovered cells were cultured to analyze their long-term viability.
For the evaluation of cell recovery, 30 mL of PBS samples spiked with target cells (THP-1) in the concentrations of 100, 1000, and 10 000 cells/mL were sieved using the 3D sieving device. Once the cell samples were sieved, the proportions of target cells in the recovered cell populations were considered as the recovery rates. Furthermore, additional tests were conducted to verify the device's ability to detect very rare tumor cells. Two sequences of repeated trials were performed: one with the coned micropore membrane coupled with 3D spiral channels and the other without these channels. These tests were repeated with the same sample preparation procedure 10 times by doping ≤ 5 cells into 1 mL PBS and performing the sieving procedure. For preparing ≤ 5 cells for each test, diluted A549 cells were transferred to the wells of a 384-well plate (Agilent Technologies, USA); next, ≤ 5 cells were sucked into a 1-mL micropipe tip to dilute to 1 mL and then delivered to the device for sieving. The reduced number of cells in the wells after pipetting was counted as the actual number of delivered cells. After the cell sieving step, in the first 5 tests, cells were counted on the membrane in situ without cell recovery. Meanwhile, in the rest of the tests, cells were recovered into the wells and counted. The cell numbers before and after cell sieving were compared among each test and calculated as the positive cell discovery rates.
Validation of throughput, purity, and viability together
For cell sample preparation, the target cells were tagged with the live cell indicator (Calcein-AM) and doped into 10 mL of diluted whole blood samples (× 5, × 50, and × 500) with blood and viable THP-1 cells in proportions of 100:1, 1000:1, and 10 000:1, respectively. During cell sieving using the 3D sieving device, the throughput was measured in the same way as mentioned above. The target cells were then observed and counted.
The proportions of recovered cells were calculated and considered as purity. Furthermore, the proportions of target cells before and after enrichment were compared and noted as enrichment efficiency.
Cell enrichments and assays from clinical PE samples
The clinical PE samples were obtained from four patients with advanced non-small cell lung cancer at Beijing Chest Hospital. The clinical PE collection and experiment were carried out in accordance with the guidelines and protocols (BJXKYYFADL-V2) approved by the Ethics and Scientific Committees of the Hospital. Once cell samples were sieved, two of the recovered samples were used to detect and enumerate tumor cells immediately, and the other two were used for proliferation and subsequent testing.
The filtration of tumor cells in PE samples was performed under the 3D sieve, which had 10 μm pore size. The enrichment steps were the same as those for the spiked samples. The concentration and distribution of the primitive and the recovered PE cells were calculated before and after enrichment. Following enrichment, the primitive and recovered PE cells were inoculated into the microplate and cultured in a CO2 incubator (37 ℃, 5% CO2) after adding the complete culture medium. The cells in the plates were observed under a microscope every day to observe their growth. The cell proliferation factors were characterized by measuring the absorbance at 450 nm under CCK-8 (Glpbio, USA) staining.
Once PE samples were sieved through the 3D sieve device, 50 000 of the recovered cells were gently resuspended in 100 μL PBS and mixed with 100 μL cell fixative (Cytellgen). The cell mixture was smeared on the formatted and coated circulating tumor cell (CTC) slides (Cytelligen) and then dried for subsequent iFISH processing. In this procedure, EpCAM/Vimentin-iFISH cell staining was performed according to the manufacture's updated protocol (Cytelligen) [31]. Briefly, the dried monolayer cells on the coated slides were rinsed and incubated with PBS at room temperature for 3 min, followed by hybridization with Vysis chromosome 8 centromere probe (CEP8) SpectrumOrange (Abbott Laboratories, Chicago, IL, USA), approved by the U.S. Food and Drug Administration to identify aneuploid solid tumor cells, for 4 h using an S500 StatSpin ThermoBrite Slide Hybridization/Denaturation System (Abbott Molecular, Abbott Park, IL, USA). The samples were subsequently incubated with the indicated post-fluorescence labeled monoclonal antibodies at 1:200 dilution, including Alexa Fluor AF594-CD45, AF488-EpCAM, and Cy7-Vimentin (Cytelligen) at room temperature for 20 min in the dark [32]. After washing, samples were mounted with mounting media containing DAPI (blue) (Vector Laboratories, Burlingame, CA, USA) and subjected to automated CTC image scanning and analyses.
Following the 3D sieve enrichment, 1 million of the recovered cells of each sample were stained with the nucleus staining DAPI, leukocyte marker (CD45), epithelial marker (EpCAM), mesenchymal markers (vimentin), and Vysis chromosome 8 centromere probe (CEP8) for identification. The tumor cells were identified as epithelial positive (DAPI + /CD45 − /EpCAM +), mesenchymal positive (DAPI + /CD45 − /Vim +), or both epithelial and mesenchymal positive (DAPI + /CD45 − /EpCAM + /Vim +), while white blood cells (WBCs) were identified as DAPI + / CD45 − /EpCAM-/Vim-.
Tumor cells have different energy metabolism pathways than normal cells. Elevated glucose uptake is a hallmark of cancer cells and has been used in positron-emission tomography to detect malignant tumors in vivo [2]. Cells with enhanced glucose uptake in PE or peripheral blood samples are highly likely to be malignant [2, 33]. Fluorescent markers can quickly detect suspected tumor cells with particularly high metabolic values. The PE cells before and after filtration were incubated separately with the fluorescent glucose analog 2-NBDG. After labeling with 2-NBDG, the images of scattered cells were captured by a fluorescent microscope in a fluorescent color.
The cells enriched from PE cells were sub-cultured for four weeks and then stained with hematoxylin–eosin (HE). The procedure of HE staining included fixation with 95% ethanol, sequential immersions in hematoxylin (Dako, USA) for 2–3 min, bluing buffer (Dako, USA) for 10–15 s, and eosin (Dako, USA) for 10–15 s. Rinsing with distilled water was conducted after each immersion. Then, cells were sequentially dehydrated with 70% ethanol, 95% ethanol, absolute ethanol, and dimethyl benzene. After those, they were mounted with neutral balsam for long-term stable storage.
Data analysis
During the testing of 3D sieve with A549/THP-1 and PE cells, Image J software, as recommended by the National Institutes of Health (NIH), was used to enumerate cells on the bright-field (BF) and fluorescence images. All data in this study were expressed as mean ± SD from no less than three independent assays.
Results and discussion
Performance of rare target cell enrichment from spiked samples
Throughput
The real-time throughput vs. filtration time plot for a typical 20 mL PBS filtration is shown in Fig. 3a. Initially, the throughput increased mainly because the fluid needed to break through the surface tension of the filter membrane; then, as the pressure of the liquid sample on the membrane surface decreased, the real-time throughput declined over the filtration time; finally, each of 20 mL PBS could be sieved within 40, 55, 70 s for the three 3D spiral channels with heights of 50, 40, and 30 mm, respectively, and within 82 s for the 2D panel sieve (still with > 10 mL residual volume). For the initial trials, the 2D plane micropore membrane area was chosen as 7.5 cm × 7.5 cm = 56.25 cm2 (approximately the same as the 3D filter's surface area of 56.5 cm2), where the 20 mL of liquid wholly resided on the membrane (see Fig. S3a). In subsequent repeated trials, 6 cm2 was selected instead of the 56.25 cm2 as the membrane's area to properly package the membrane into a commercial 2D filter holder (BD Falcon cell strainer, USA, with its own membrane removed) for a more convenient comparative study (see Fig. S4a). With the membrane's area reduced, about 7 mL liquid passed through the micropores in a much lower throughput than the 3D's, but about 13 mL still rested on the membrane. Both of the 2D filter's results showed that the fluid hardly passed through the micropores while only driven by gravity, no matter whether through an area of 56.25 or 6 cm2.

a The throughput changed over time and b averaged when sieving 20 mL PBS solution using a 2D plane sieve and three 3D sieving devices with different height of the spiral channel, respectively, all of which were using the same membrane 10 μm diameter micropore. c The viability of the target cells (A549) with two groups of primitive cell concentrations (105 and 104 cells/mL) before and after enrichment. d, e The recovery rates of the recovered very rare (≤ 5, A549) target cells which were spiked in 1 mL PBS solution, then recovered d on the membrane and e to the well plate, respectively. f The recovery rate of the recovered target cells (THP-1) that were spiked in 30 mL PBS at concentrations of 100, 1000, and 10 000 cells/mL
Figure 3b shows the average throughputs of the 3D sieve device and 2D sieve. Compared to the 2D plane sieve with a throughput of about 1.5 mL/min, the throughputs of the 3D device with spiral channel heights of 30, 40, and 50 mm significantly improved, which were recorded as 29, 24, and 17 mL/min, respectively. It's worth noting that the 3D cell sieve used in the subsequent sections has an average throughput of 29 mL/min, which is remarkably (20-fold) higher than that of the 2D cell sieve. The comparative study also indicated that, with the spiral channels' height rising, the throughput increased due to the enlarged active area of the micropore membrane.
The high throughput performance of the proposed method shown in the tests proved that it has a promising capacity for collecting rare cells from large-volume samples. Compared with previous microfluidic cell separation devices [16, 34] featuring a throughput of about 1 mL/h, the design strategy of dimension raising demonstrated an obvious throughput advantage.
Viability
The short-term cell viability results obtained via the live/dead assay after cell enrichment were shown in Fig. S5. The viability of the primitive cells was 98% ± 0.6% and 97.4% ± 4.1% at 105 per mL and 104 per mL cell concentrations, respectively, while that of the recovered cells only reduced to 94.5% ± 0.9% and 93.4% ± 5.2%, respectively, as shown in Fig. 3c. Each set of experiments was independently repeated three times. In the figure, the error bars stand for the standard deviations between different filtrations. The results show no statistically significant difference with P > 0.0001 and P > 0.01 for the two sets of experiments, respectively, as evaluated by a Student's t test. The relative deviations for the two sets of experiments were 4.06% and 4.11%, respectively. Moreover, the recovered cells were transferred to the culture dish and were successfully cultured with adherent morphology (Fig. S6). The comparative cell viability results between before and after filtration confirmed that the sustainable culture and analysis could be implemented for cells recovered with the proposed 3D sieving device.
Recovery rate
As shown in Fig. 3f, the achieved recovery rates were 84.5% ± 21%, 86% ± 25%, and 83% ± 14% for THP-1 cell concentrations of 100, 1000, and 10 000 per mL, respectively. Theoretically, it is difficult to recover the cells diffused on the 2D plane cell sieve after filtration. The results shown in Fig. 3f indicate that, given the membrane's cone-shaped structure, about 85% of target cells flowed into the conical tip of the membrane and recovered. Consequently, the 3D sieving device has a promising ability for cell recovery.
In order to verify the proposed device's benefits for very rare cell recovery by the coned structure, we performed two sequences of repeated trials with samples of ≤ 5 spiked A549 cells in 1 mL PBS (n = 5 for each trial). As demonstrated by the results shown in Fig. 3d, the 100% positive rate for cell rediscovery on the membrane indicated that the coned membrane contributed to the collection of cells in the conical tip, along with an average recovery rate of 77.8%. Moreover, as shown in Fig. 3e, the 80% positive rate of cell detection in the microplate and the average recovery rate of 53% indicated that several cells were lost; however, most of the cells were recovered successfully in the recovery process. The micrographs produced in these trials were shown as supporting information (Fig. S7).
Commercially available 2D size-based filtration methods, such as ThinPrep cytologic tests (TCTs), can only achieve a recovery rate of less than 15% [17]. Our method is therefore competitive in terms of recovery rate and comparable with the high-porosity cell sieve "PERFECT" [9, 27].
Simultaneous evaluation of purity, throughput, and viability performance
After sieving the simulated PE samples at a throughput rate of about 20 mL/min (as shown in video S1), the viable target cells (tagged with live cell indicator) were enriched, recovered, and observed. Figure S4 shows the contrast between the cell concentrations of the recovered cells under the 2D plane sieve and the 3D sieve. Figures 4a-i, 4b-i, and 4c-i indicate that the proportions of blood to viable THP-1 cells were 100:1, 1000:1, and 10 000:1, respectively, before sieving. Figures 4a-ii, 4b-ii, and 4c-ii present the corresponding cell composites after cell enrichment, where the purities of recovered viable THP-1 cells were recorded as 85.5% ± 9.1%, 64.5% ± 8.5%, and 48.3% ± 10.2%, respectively (Fig. 4d). Compared to the ratio of target cells before and after filtration, the enrichment efficiency reached about 60, 500, and 4000 times at blood to THP-1 cell ratios of 100:1, 1000:1, and 10 000:1, respectively (Fig. 4e). Here, it is worth noting that when the cell proportion was lower at 1000:1 and 10000:1, the purity of viable target cells could be affected by the monocytic blood cells, whose size is comparable to these target cells.

a–c The images of the background and viable (tagged in green) target cells before (each i) and after (each ii) cell enrichment from 10 mL of cell samples (× 5, × 50, and × 500 dilution of whole blood cells, which were spiked with target cells (THP-1) in the proportions of 1:100 (a), 1:1000 (b), and 1:10,000 (c), respectively) were observed and calculated into (d) purity and (e) enrichment efficiency
The above results show that, with the design strategy of dimension raising, during the high-throughput sieving procedure, cell residue problems induced by the "dead zone" were satisfactorily solved, and the recovered cells had high purity and viability.
Enrichment of rare target cells from clinical PE samples
For this test, primitive PEs were obtained from different patients diagnosed with lung adenocarcinoma. The volume and cell concentration of primitive PE samples were first determined (Table S1).
The samples from all four patients were 3D-sieved for target cell enrichment. Figures 5a and 5b show the micrographs of cells sampled from patients 1 and 2, respectively. Figures 5a-i/ii and 5b-i/ii present the micrographs of the primitive PE samples from two patients and the recovered cells after 3D cell sieving, respectively. Compared with the size distributions observed in the micrographs, the proportion of large cells and cell clusters was significantly increased. Moreover, after cell culture for 6 days, the micrographs shown in Figs. 5a-iii and 5b-iii indicate that the recovered cells were significantly proliferated.

The observed images of cell populations of (a-i, b-i) primitive PE cells sampled from patients (a, b for patients 1, 2, respectively), (a-ii, b-ii) recovered after cell sieving through 10 μm diameter micropores, and (a-iii, b-iii) 6-days of culture. c Diameter histogram (for patient 2) and proportions (for patients 1–4) of the primitive and recovered cells. d The proliferation factors of primitive and recovered cells in 6-day culture (for patient 3 and patient 4) were obtained. After labeling with the cellular glucose metabolism tracker (2-NBDG, green), e compared to the primitive PE samples (e-i, ii for bright-field and fluorescent, respectively), the proportion of suspected tumor cells (green) were significantly increased among (e-iii, iv) the recovered cells. f Through immunofluorescence tests, individual tumor cells, tumor cell clusters, and white blood cells enriched from the PE sample were assayed among the recovered cells (from patients 1, 2). g The suspected tumor cells (in red circles) were discovered by HE staining among the recovered cells and then the cells with 4 weeks of sub-culture (from patients 3, 4)
Primitive and recovered cells from patient 3 were imaged at random locations through a microscope under bright field (BF), and then the diameter of PE cells was measured through image analysis using the software Image J. The cumulative size distribution is plotted in Fig. 5c. After sieving through 10 μm size micropores, the proportion of large-size cells (> 10 μm) increased from 2.4% ± 0.5% to 43.2% ± 6.3%.
The process of primary tumor cell proliferation in PE played an essential role in the exploration of the tumor growth mechanism, thus improving the sensitivity and specificity of the diagnosis of malignant PE [35]. Meanwhile, a large population of proliferating cells is crucial for future drug-sensitive tests. To verify the proliferative ability of recovered cells sampled from patients 3 and 4, we compared their growth curves with the unfiltered PE cells' curves. All of the cells were successfully cultured for 6 days, while the most rapidly proliferating cells covered 95% of the culture dish bottom. After 6 days of culture, the enriched cells had obviously proliferated, as shown in Figs. 5a-ii/iii and 5b-ii/iii. The two pairs of primitive and enriched cells' growth curves are shown in Fig. 5d, indicating that the enriched cells proliferated 4.6 and 8.3 times faster than the unfiltered PE cells. Prior to target cell enrichment, the primary tumor cells in the pleural effusion are difficult to culture in vitro due to the complex cell composition of raw PE [36, 37]. After sieving, the proportion of tumor cells was effectively increased by our method, which was regarded as the main reason behind the increased proliferation rate of enriched cells.
Analysis of tumor cells from recovered PE cells
The comparison of bright-field and in situ 2-NBDG fluorescence images before and after filtration is shown in Figs. 5e and S8, where it is obvious that the number of metabolically active cells was increased. It was also verified that the size-based cell enrichment has practical significance for collecting tumor cells from clinical samples.
In order to further explore the fraction of recovered PE cells, immunofluorescence was used to identify the candidate tumor cells from the recovered PE cells of patient 1 and patient 2, as presented in Fig. 5f. The abnormal expressions of chromosome 8, which are also common in tumor cells of patients with lung cancer, show a higher proportion of aneuploidy. In the enriched cells from patients 1 and 2, 89 versus 2910 and 29 versus 2220 tumor cell clusters were discovered, respectively, by sampling 5 × 104 assayed cells (Table S1). Here, it is worth noting that the proportion of tumor cells and clusters was only about 0.25% versus 10% of enriched cells. Moreover, considering that the purity improved tens of times after cell enrichment, the raw proportion of tumor cells should be about only about 0.0068 versus 0.12%, separately, in whole PE cells from 25 versus 65 mL of clinical PE samples before cell enrichment. These results indicate that, once the background cells were effectively filtered out, rare tumor cells in the clinical PE became more distinct to be discovered.
In the other sequence of trials, cells were enriched from PE samples of patients 3 and 4 and then sub-cultured for four weeks. After being assayed by HE staining, tumor cells were also detected, as noted by the red circles in Fig. 5g. The results indicated that the tumor cells were successfully cultured after several passages, which also verified that the proposed 3D sieve provides a potential tool for tumor studies.
Conclusions
As clearly shown by the existing clinical applications, preprocessed by a size-based cell sieving technique, filtered background cells are mainly in the size of 4–8 μm, and the target tumor cells can be obviously detected and assayed. However, traditional cell sieving methods applying a 2D plane structure need to be upgraded to overcome their inherent drawback called the "dead zone." Although some attempts have been reported to reduce the negative effects of this drawback, they cannot fundamentally solve the problem because of the nature of the 2D plane structures.
This paper proposed a new design strategy, named "dimension rising strategy," to address the critical challenge. In the details of this strategy, we fabricated a 3D sieving system with a special 3D spiral channel and a coned sieving membrane to guide the rapid flow of cell samples on the sieving membrane to eliminate the "dead zone effectively." Based on the proposed design strategy of dimension raising, the 3D sieving system demonstrated several other advantages as follows. First, with the 3D spiral channel covered on the coned membrane, the 3D sieving device's throughput was theoretically tens of times higher than that of 2D plane sieves, since the highest pressure at the conical tip was 10 000 times higher than that in 2D sieves with the same surface area of micropore membrane while driven by gravity alone (for simulations, see Fig. S1). Meanwhile, the ridges on the holder's internal surface help the liquid above the membrane easily pass through the micropores to improve the throughput because the gating threshold (the pressure needed to open the pores) was reduced with the structure according to the gating mechanism theory [38]. Second, the coned structure was conducive for cell recovery, since the target cells were mainly flushed down along the 3D spiral channel and collected at the coned membrane's tip. Third, the coned structure was highly scalable for a sample volume from 100 μL to 100 mL and a cell count from 1 to 109. Fourth, the coned structure could efficiently prevent fluid leakage without the need to tightly couple those three layers, so users just need to put the cover with the spiral channel on to sieve the sample and then put it aside to recover the target cells. In addition, the membrane is detachable for easy transportation for downstream analysis. Fifth, the 3D sieving system is convenient to reproduce by users because the core components are fabricated by commercial 3D printing service providers, while the micropore membrane as a consumable item is easy to obtain. Sixth, the system was made cost-effective by using commercial services, the reusable core components cost less than $10, and the consumable part cost less than $0.1 per test.
In this initial proof-of-concept demonstration, we developed a device prototype and verified its promising capability for enriching pure and viable rare target cells from large-volume samples at high throughput and recovery rates. Specifically, the 3D sieving platform's throughput was 20 times higher compared with the 2D plane sieve using the same micropore membrane. Relatively competitive results were obtained based on three primary considerations (viability, purity, and recovery rate) based on the assessment of the efficacy of cell separation. From diluted whole blood spiked by the target cells (THP-1), we could detect viable target cells with a purity of 85.5% ± 9.1% (in the proportion of 1:100) and a recovery rate of 84.5% ± 21% (in the concentration of 100/mL) in repetitive tests.
For clinical application, PE samples from four lung cancer patients were sieved, cell clusters were successfully enriched and the purity of target cells was increased typically from 2.4 to 43.2%. We successfully cultured the recovered PE cells and found that the PE cell proliferation factor increased 8.3 times after enrichment. From the perspective of glucose metabolism, we realized the rapid identification of tumor cells through 2-NBDG staining. The enhanced glucose uptake and proliferation factor after enrichment also validated the clinical relevance of this metabolically active subset.
In comparison with commercial 2D plane cell filters, we could demonstrate the great potential of the design strategy of dimension raising supported by additive manufacturing techniques to upgrade and reform traditional cell sieving techniques. Moreover, the performance of our device prototype was comparable with the best existing well-optimized and precisely micro-fabricated cell sieves. We believe that our strategy can help to raise the performance of 2D cell filters to a new level. Given the reproducibility of the proposed methodology, we can expect great application prospects of the presented 3D cell sieving device.
Supporting Information
Model simulation; simulation of fluid pressure; fabrication procedure of the 3D membrane; platform for throughput measurement; comparison of unpassed blood cells; viability of recovered A549 cells; growth of recovered A549 cells; recovered rare target cells on membrane and microplate; Hoechst/2-NBDG fluorescence staining of recovered PE cells; parameters of clinical PE cell enrichment (PDF).
References
Heffner JE, Nietert PJ, Barbieri C (2000) Pleural fluid pH as a predictor of survival for patients with malignant pleural effusions. Chest 117(1):79–86. https://doi.org/10.1378/chest.117.1.79
Tang Y, Wang Z, Li Z et al (2017) High-throughput screening of rare metabolically active tumor cells in pleural effusion and peripheral blood of lung cancer patients. Proc Natl Acad Sci USA 114(10):2544–2549. https://doi.org/10.1073/pnas.1612229114
Krebs MG, Sloane R, Priest L et al (2011) Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J Clin Oncol 29(12):1556–1563. https://doi.org/10.1200/JCO.2010.28.7045
Kaylan KB, Gentile SD, Milling LE et al (2016) Mapping lung tumor cell drug responses as a function of matrix context and genotype using cell microarrays. Integr Biol 8(12):1221–1231. https://doi.org/10.1039/c6ib00179c
Low WS, Wan Abas WA (2015) Benchtop technologies for circulating tumor cells separation based on biophysical properties. Biomed Res Int 2015:239362. https://doi.org/10.1155/2015/239362
Chen J, Li J, Sun Y (2012) Microfluidic approaches for cancer cell detection, characterization, and separation. Lab Chip 12(10):1753–1767. https://doi.org/10.1039/c2lc21273k
Shields CW, Reyes CD, Lopez GP (2015) Microfluidic cell sorting: a review of the advances in the separation of cells from debulking to rare cell isolation. Lab Chip 15(5):1230–1249. https://doi.org/10.1039/c4lc01246a
Hao SJ, Wan Y, Xia YQ et al (2018) Size-based separation methods of circulating tumor cells. Adv Drug Deliv Rev 125:3–20. https://doi.org/10.1016/j.addr.2018.01.002
Li T, Liu Y, Zhang W et al (2020) A rapid liquid biopsy of lung cancer by separation and detection of exfoliated tumor cells from bronchoalveolar lavage fluid with a dual-layer “PERFECT” filter system. Theranostics 10(14):6517–6529. https://doi.org/10.7150/thno.44274
Tomlinson MJ, Tomlinson S, Yang XB et al (2013) Cell separation: terminology and practical considerations. J Tissue Eng 4:2041731412472690. https://doi.org/10.1177/2041731412472690
van den Driesche S, Rao V, Puchberger-Enengl D et al (2012) Continuous cell from cell separation by traveling wave dielectrophoresis. Sens Actuat B Chem 170:207–214. https://doi.org/10.1016/j.snb.2011.01.012
Lien KY, Chuang YH, Hung LY et al (2010) Rapid isolation and detection of cancer cells by utilizing integrated microfluidic systems. Lab Chip 10(21):2875–2886. https://doi.org/10.1039/c005178k
Wu M, Ozcelik A, Rufo J et al (2019) Acoustofluidic separation of cells and particles. Microsyst Nanoeng 5:32. https://doi.org/10.1038/s41378-019-0064-3
Khojah R, Stoutamore R, Di Carlo D (2017) Size-tunable microvortex capture of rare cells. Lab Chip 17(15):2542–2549. https://doi.org/10.1039/c7lc00355b
McFaul SM, Lin BK, Ma H (2012) Cell separation based on size and deformability using microfluidic funnel ratchets. Lab Chip 12(13):2369–2376. https://doi.org/10.1039/c2lc21045b
Sarioglu AF, Aceto N, Kojic N et al (2015) A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat Methods 12(7):685–691. https://doi.org/10.1038/nmeth.3404
Zhou W, Liu Y, Ran M et al (2019) Rapid liquid biopsy for Mohs surgery: rare target cell separation from surgical margin lavage fluid with a high recovery rate and selectivity. Lab Chip 19(6):974–983. https://doi.org/10.1039/C8LC01335G
Adams DL, Zhu P, Makarova OV et al (2014) The systematic study of circulating tumor cell isolation using lithographic microfilters. RSC Adv 9:4334–4342. https://doi.org/10.1039/C3RA46839A
Lee A, Park J, Lim M et al (2014) All-in-one centrifugal microfluidic device for size-selective circulating tumor cell isolation with high purity. Anal Chem 86(22):11349–11356. https://doi.org/10.1021/ac5035049
Chiu PL, Chang CH, Lin YL et al (2019) Rapid and safe isolation of human peripheral blood B and T lymphocytes through spiral microfluidic channels. Sci Rep 9:8145. https://doi.org/10.1038/s41598-019-44677-3
Kim EH, Lee JK, Kim BC et al (2013) Enrichment of cancer cells from whole blood using a microfabricated porous filter. Anal Biochem 440(1):114–116. https://doi.org/10.1016/j.ab.2013.05.016
Zheng S, Lin HK, Lu B et al (2011) 3D microfilter device for viable circulating tumor cell (CTC) enrichment from blood. Biomed Microdev 13(1):203–213. https://doi.org/10.1007/s10544-010-9485-3
Harouaka RA, Zhou MD, Yeh YT et al (2014) Flexible micro spring array device for high-throughput enrichment of viable circulating tumor cells. Clin Chem 60(2):323–333. https://doi.org/10.1373/clinchem.2013.206805
Zheng S, Lin H, Liu JQ et al (2007) Membrane microfilter device for selective capture, electrolysis and genomic analysis of human circulating tumor cells. J Chromatogr A 1162(2):154–161. https://doi.org/10.1016/j.chroma.2007.05.064
Kang YT, Doh I, Byun J et al (2017) Label-free rapid viable enrichment of circulating tumor cell by photosensitive polymer-based microfilter device. Theranostics 7(13):3179–3191. https://doi.org/10.7150/thno.19686
Yusa A, Toneri M, Masuda T et al (2014) Development of a new rapid isolation device for circulating tumor cells (CTCs) using 3D palladium filter and its application for genetic analysis. PLoS ONE 9(2):e88821. https://doi.org/10.1371/journal.pone.0088821
Liu Y, Li T, Xu M et al (2018) A high-throughput liquid biopsy for rapid rare cell separation from large-volume samples. Lab Chip 19(1):68–78. https://doi.org/10.1039/c8lc01048j
Fleischer RL, Alter HW, Furman SC et al (1972) Particle track etching: diverse technological uses range from virus identification to uranium exploration. Science 178(4058):255–263. https://doi.org/10.1126/science.178.4058.255
George JH, Waller ST, Ye H (2017) 4.13 Use and manipulation of microporous membranes in mammalian cell cultures. Compr Membr Sci Eng. 4:272–292. https://doi.org/10.1016/B978-0-12-409547-2.12267-X
Vona G, Sabile A, Louha M et al (2000) Isolation by size of epithelial tumor cells. Am J Pathol 156(1):57–63. https://doi.org/10.1016/S0002-9440(10)64706-2
Lin PP, Gires O, Wang DD et al (2017) Comprehensive in situ co-detection of aneuploid circulating endothelial and tumor cells. Sci Rep 7(1):9789. https://doi.org/10.1038/s41598-017-10763-7
Lin P, Fischer T, Weiss T et al (2000) Calnuc, an EF-hand Ca2+ binding protein, specifically interacts with the C-terminal alpha 5-helix of Gαi3. Proc Natl Acad Sci USA 97(2):674–679. https://doi.org/10.1073/pnas.97.2.674
Antonangelo L, Sales RK, Cora AP et al (2015) Pleural fluid tumour markers in malignant pleural effusion with inconclusive cytologic results. Curr Oncol 22(5):336–341. https://doi.org/10.3747/co.22.2563
Tang Y, Shi J, Li S et al (2014) Microfluidic device with integrated microfilter of conical-shaped holes for high efficiency and high purity capture of circulating tumor cells. Sci Rep 4:6052. https://doi.org/10.1038/srep06052
Tsou PH, Chiang PH, Lin ZT et al (2020) Rapid purification of lung cancer cells in pleural effusion through spiral microfluidic channels for diagnosis improvement. Lab Chip 20(21):4007–4015. https://doi.org/10.1039/d0lc00663g
Si LL, Lv L, Zhou WH et al (2015) Establishment and identification of human primary lung cancer cell culture in vitro. Int J Clin Exp Pathol 8(6):6540–6546 (PMID: 26261533; PMCID: PMC4525867)
Gabriela AL, Daniela ME (2016) Treatment for small cell lung cancer, where are we now?-a review. Transl Lung Cancer Res 5(1):26–38. https://doi.org/10.3978/j.issn.2218-6751.2016.01.13
Hou X, Hu Y, Grinthal A et al (2015) Liquid-based gating mechanism with tunable multiphase selectivity and antifouling behaviour. Nature 519(7541):70–73. https://doi.org/10.1038/nature14253
Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2018YFC2001100), the National Natural Science Foundation of China (Nos. 61774167 and 61801477), the Instrument Development Program of the Chinese Academy of Sciences, Beijing Municipal Natural Science Foundation (Nos. 4192062 and 4182072), Beijing Municipal Administration of Hospitals Incubating Program (No. PX2017050), and Youth Innovation Promotion Association of Chinese Academy of Sciences.
Author information
Authors and Affiliations
Contributions
JC was involved in conceptualization, methodology, validation, investigation, and writing—original draft. LNZ was involved in resources, data curation, and writing—original draft. YRZ was involved in resources and validation. YFY was involved in validation and data curation. WJZ was involved in validation and resources. LQZ was involved in investigation, writing—review and editing. YAL was involved in validation and data curation. YL was involved in visualization and software. WCZ was involved in validation, investigation, and resources. HYG was involved in resources and conceptualization. MXL was involved in writing—review and editing and funding acquisition. YZ was involved in conceptualization, supervision, project administration, writing—review and editing, funding acquisition. CJH was involved in writing—review and editing, project administration, and funding acquisition.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Pleural effusion samples from non-small cell lung cancer patients and peripheral blood samples from healthy donors were obtained in Beijing Chest Hospital with written informed consent. The clinical sample collection and experiment were carried out in accordance with guidelines and protocols (BJXKYYFADL-V2) approved by the Ethics and Scientific Committees of the Beijing Chest Hospital.
Additional information
Jie Cheng and Lina Zhang contributed equally to this work.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (MP4 2645 kb)
Rights and permissions
About this article

Cite this article
Cheng, J., Zhang, L., Zhang, Y. et al. 3D spiral channels combined with flexible micro-sieve for high-throughput rare tumor cell enrichment and assay from clinical pleural effusion samples. Bio-des. Manuf. 5, 358–370 (2022). https://doi.org/10.1007/s42242-021-00167-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42242-021-00167-y