
Abstract
Raspberry (Rubus idaeus L.) is a crop with increasing commercial value due to its nutritional properties and demand in national and international markets. Throughout the growing process, there have been reports of the presence of galls on roots and stems, which reduce plant size and yields. The current study used phylogenetic reconstruction to identify bacteria associated with these symptoms in raspberry plantations in the Mexican states of Jalisco and Michoacán. A total of 69 representative strains selected according to their colony morphotype were identified based on partial sequencing of the 16S ribosomal gene. Forty-nine of them were tested for pathogenicity, including of which three strains induced gall symptoms in raspberry and tomato plants. A set of 28 strains identified within the Agrobacterium and Rhizobium genera were chosen for amplification of the atpD, glnA, gyrB, and rpoB genes. Strain CPO 2.419 was selected for whole genome sequencing, with a total length of 5,679,921 base pairs assembled into 46 contigs. This strain was identified as A. tumefaciens (GenBank JAVIYJ000000000.1) using phylogenetic reconstruction. According to the OrthoANIu and dDDH percentage values, the CPO 2.419 strain belongs to the A. tumefaciens complex, along with the closely related strains of A. tumefaciens LMG 232 (99.4, 89.1), A. tumefaciens CNPSo 675 (98.5, 84.8), and A. tumefaciens ATCC 4720T (98.1, 78), respectively. This study contributes to the understanding of the genomic Agrobacterium diversity in raspberry-producing areas in Mexico and highlights the relevance of genomics for accurate bacterial identification, with important implications for agriculture.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Raspberry (Rubus idaeus L.) is a valuable plant due to its distinctive fruit and nutritional value. Its consumption has grown noticeably in recent years, leading to an increase in global production. This species is not only consumed fresh but also has applications in the food, pharmaceutical, and cosmetic industries (Beekwilder et al. 2005; Krzepiłko et al. 2021). However, raspberry production is hindered by the presence of crown gall, a bacterial disease commonly associated with the genus Agrobacterium. These bacteria enter the plant through wounds and induce the formation of galls on the roots and stems. Over time, these galls become woody, impairing the plant’s ability to absorb nutrients and water, weakening it, and reducing its yield. Symptoms include stunted growth, wilting, leaf deformities, and premature leaf drop (Martin et al. 2017; Lacroix and Citovsky 2022).
The use of contaminated soil and propagation material is a prevalent means of transmission of the bacteria and represents a significant risk for producers who acquire susceptible varieties or those from non-certified nurseries (Kerr 2015). Agrobacterium may move from galls to the surrounding roots and soil, where it may persist and spread to new plants during propagation or planting (Paret et al. 2011). The ability of the bacteria to induce galls varies depending on the host plant and the bacterial strain involved (Puławska et al. 2015).
Accurate identification of the strains responsible for crown gall is challenging due to the presence of pathogenic and non-pathogenic bacteria from different phylogenetic groups in the galls. Furthermore, determining which strain is the causative agent of a specific gall is challenging since, once the plant tissue has been transformed, the continued presence of the bacteria is not required for disease development (Lacroix and Citovsky 2022). The presence of the tumour-inducing plasmid (pTi), which contains virulence genes, is essential for the pathogenicity of the strains (Gordon and Christie 2014). However, pTi can be lost or gained over time (Kerr 2015).
Since 2015, several new species of Agrobacterium have been identified, isolated from tumours and plant tissues using genetic analysis and whole genome sequencing. More species are expected to be described in the future, including recently proposed species like A. tomkonis, A. vaccinii, A. burrii, and A. shirazense (Singh et al. 2021; Puławska et al. 2022; Mafakheri et al. 2022). The genetic diversity and relationship between the strains responsible for crown gall disease outbreaks provide crucial information for understanding the pathogen’s epidemiology, ecology, and evolution.
Therefore, in the current study, bacterial strains were isolated from symptomatic crown gall tissues obtained from raspberry samples from the Mexican states of Jalisco and Michoacán. The goal of this study was to identify the causal agent through a phylogenetic approach along with bacterial isolate characterization and pathogenicity tests on raspberry and tomato seedlings, under the assumption that the pathogen responsible for the galls was present in the tissues collected.
Materials and methods
Bacterial isolation
Symptomatic plants were collected from commercial raspberry plantations in the Mexican states of Jalisco and Michoacán between 2019 and 2021. Twenty-five plants were washed with water to remove soil particles adhered to the root. The plant material was disinfested in a 1% sodium hypochlorite solution to remove microorganisms and other contaminants on the outside of the sample, rinsed with sterile water, and allowed to dry in a biosafety chamber on sterile absorbent paper to remove excess humidity.
On the stems, longitudinal tissue cuts ranging from 5 to 8 mm were made in sections close to the crown where the gall develops, exposing part of the vascular system. For the galls, the outer bark was removed to expose the interior. The inner tissue was cut into fragments the same size as the stems and lightly pressed against a sterile surface. Likewise, several root segments were excised and immersed into Eppendorf tubes containing 1 mL of sterile distilled water for 12 h to allow the microorganisms to emerge from the vascular bundles.
Disinfested tissue fragments were placed on Petri dishes with D1-M selective medium (Perry and Kado 1982) and King’s B medium (Schaad et al. 2001). Similarly, 30 µL of the bacterial suspension contained in the Eppendorf tubes were placed in the same culture medium. The Petri dishes were incubated at a temperature of 28 °C for 48 h. Bacterial colonies that emerged from the tissues were selected based on their morphological traits and purified through multiple transfers in King’s B medium. Gram staining, 3% KOH solubility test (Schaad et al. 2001), and oxidative-fermentative metabolism using thioglycolate broth were used to classify the isolates. Purified isolates were stored in 15% v/v glycerol at −80 °C.
DNA extraction
For DNA extraction, 69 representative strains were selected based on colony morphology, including size, shape, surface appearance, and colour of colonies after 48 h of incubation. Isolates were reactivated in Petri dishes with King’s B medium and incubated at 28 °C for 48 h. The DNA was extracted from each isolate using the 2% hexadecyltrimethylammonium bromide (CTAB) method (Doyle and Doyle 1990).
Molecular identification
The 16S ribosomal gene was amplified using universal primers 8F (5′- AGAGTTTGATCCTGGCTCAG- 3′) (Edwards et al. 1989) and 1492R (5′- GGTTACCTTGTTACGACTT- 3′) (Stackebrandt and Liesack 1993). The PCR reaction mixture contained 60 ng genomic DNA, 0.8 mM dNTPs, 0.3 U Taq DNA polymerase (Promega, WI, USA), 5× GoTaq reaction buffer (Promega, WI, USA), and 20 pmol of each primer in a final volume of 15 µL. Amplification was carried out in a C1000 Touch thermocycler (BIO-RAD, CA, USA). The amplification conditions were as follows: an initial denaturation temperature of 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 1 min, annealing at 58 °C for 1 min, extension at 72 °C for 2 min, and a final cycle at 72 °C for 10 min.
For a more detailed phylogenetic reconstruction, a subset of 28 strains initially identified within the Agrobacterium and Rhizobium genera were selected for the amplification of four housekeeping genes (atpD, glnA, gyrB, and rpoB) that have been demonstrated to be informative in previous studies (Aujoulat et al. 2011; Martens et al. 2008) (Supplementary Table 1). The partial gene amplification was achieved in a C1000 Touch thermocycler (BIO-RAD, CA, USA). The amplification conditions were as follows: an initial denaturation temperature of 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 1 min; alignment at 55 °C for 1 min for rpoB, 58 °C for atpD and gyrB, and 60 °C for glnA; extension at 72 °C for 2 min; and a final cycle of 72 °C for 10 min.
Horizontal electrophoresis in a 1.5% agarose gel stained with GelRed TM (Biotium, CA, USA) in a Sub-Cell GT chamber (BIO-RAD, CA, USA) operated at 90 V for an hour was used to confirm all amplified products. The amplicons were cleaned using the ExoSAP-IT enzymatic reaction protocol (Affymetrix, CA, USA). Sanger sequencing of the amplified fragments was performed by Psomagen (Psomagen Inc., Rockville, MD, USA).
Pathogenicity tests
Pathogenicity tests were performed for a subset of 49 representative strains from the previously selected bacteria according to morphological characteristics (Table 1). Tests were conducted on two types of crops: pathogen-free raspberry Adelita variety seedlings obtained from tissue culture and 3-week-old tomato (Solanum lycopersicum L.) seedlings. One hundred and fifty plants from each crop were subjected to 50 treatments, comprising 49 bacterial isolates and a distilled water control. The toothpick method was used for inoculation, producing wounds in the plants’ stems and allowing bacteria to enter. After 48 h, the stick was removed and the wounds were wrapped in Parafilm. The seedlings were grown in a greenhouse with high relative humidity (75%) and an average temperature of 28 °C. The plants were monitored for two months to detect incidence (the formation of galls as a symptom of pathogenicity), with the infection considered positive only if galls were observed.
Phylogenetic analysis
Raw data from the forward and reverse DNA fragments was assembled with the Bioedit 7.1.9.0 software (Hall 1999). The consensus sequences from each gene were aligned with the MUSCLE algorithm (Edgar 2004) using MEGA7 software (Tamura et al. 2012). Each set of aligned sequences of atpD, glnA, gyrB, and rpoB was concatenated using the Mesquite v3.6 software (https://www.mesquiteproject.org). The assembled sequences were deposited in GenBank from the National Centre for Biotechnology Information database (Table 2).
The reference sequences of each gene for the Agrobacterium and Rhizobium species mentioned in the List of Prokaryotic names with Standing in Nomenclature (LPSN) database (Parte et al. 2020) and those of relevance used in previous research by other authors (Velázquez et al. 2020; Kuzmanović et al. 2015b) were obtained from GenBank and included in the final alignment.
The phylogenetic reconstruction (for both 16S rRNA and MLSA) was performed using Bayesian statistics and the Mr Bayes program (Ronquist and Huelsenbeck 2003) by implementing the MCMC algorithm with four exchangeable Markov chains. The nucleotide substitution model used was the General Time-Reversible (GTR + I), which assumes different substitution rates for each pair of nucleotides as well as different frequencies of appearance of nucleotides with sites. The analysis was terminated when the standard deviations of the split frequencies reached a value of 0.01. Trees were sampled every 1000 generations. The ‘burn in-phase’ option was used to discard 25% of the total trees generated; the remaining trees were used to calculate the posterior probability of support at each node. The final trees were visualised using the FigTree v1.4.4 program (http://tree.bioed.ac.uk/software/figtree/).
Whole genome sequencing and assembly
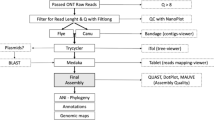
Based on the pathogenicity test results, DNA from the selected Agrobacterium sample (CPO 2.419) was used for whole genome sequencing. The Picogreen Victor X2 fluorometry method (Life technologies, Thermo Fisher Scientific, MA, USA) was used to calculate the DNA concentration in the sample. The Psomagen Next Generation Sequencing service (Psomagen Inc., Rockville, MD, USA) and Hi-Seq 2000 technologies (Illumina, CA, USA) were used to sequence paired-end reads, yielding fragments of 151 base pairs (bp). The Illumina bcl2fastq program was used to convert binary base calling to FASTQ files. Adapters were removed from the final reads.
FastQC version 0.11.9 (www.bioinformatics.babraham.ac.uk/projects/) was considered to evaluate the quality of raw sequences. Trimmomatic v0.39 software (Bolger et al. 2014) was used to eliminate low-quality reads (with parameters leading: 3, trailing: 3, sliding window: 4:15, minlen: 75). The high-quality lecture pairs were processed using the BV-BRC assembly service (https://www.bv-brc.org/), via a predefined process (pipeline) (unicycler -t 12 -o. --min_fasta_length 300 --keep 2 --no_pilon) in Unicycler v0.4.8 (Wick et al. 2017). The annotations were created using the NCBI Prokaryotic Annotation Pipeline (PGAP).
Genome analysis
Amino acid and nucleotide sequences from 100 global protein families (PGFams) were obtained using the BV-BRC platform (Davis et al. 2016). An alignment was constructed, and a tree was generated based on differences within these sequences. Protein sequences were aligned with MUSCLE (Edgar 2004), whereas nucleotide-coding gene sequences were aligned with BioPython’s Codon_align function (Cock et al. 2009). A concatenated alignment of all proteins and nucleotides was generated in a PHYLIP formatted file, followed by a partition file for RaxML 8.0.0 program (Stamatakis 2014) with the ‘AUTO’ model to find the best substitution model and conditions -PTHREADS-SSE3 -m GTRCAT -p 12,345 -T 12 -f a -x 12,345 -N 100 to find the best tree. Strain genomes used for comparison were also obtained from GenBank (Table 3). Support values were generated after using 100 rounds of RaxML’s “quick” start-up option (Stamatakis et al. 2008).
Average nucleotide identity (ANI) values were calculated by comparing the genome obtained from strain CPO 2.419 to A. tumefaciens LMG 232, A. tumefaciens CNPSo 675, A. tumefaciens ATCC 4720T, and A. arsenijevicii KFBT 330 using OrthoANIu (Yoon et al. 2017). Furthermore, the digital DNA-DNA hybridization (dDDH) values were computed and compared using the Genome-to-Genome Distance Calculator 2.1 (Meier-Kolthoff et al. 2013) on the Type Strain Genomic Server (TYGS) platform.
Furthermore, CPO 2.419 contig 7 with sequences corresponding to the vir genes and others were compared against CFBP2712 pTi using the tblastx tool (NCBI).
Results
Sample processing
The processed plant material from commercial plantations in Jalisco and Michoacán states showed characteristic crown galls, which were distinguished by the thickening of anomalous irregularly shaped protuberances with a rough appearance. The galls had a tumorous appearance and were mostly light brown in colour, ranging from reddish to greenish depending on the stage of disease development (Fig. 1). In contrast to the surrounding healthy tissue, these had a soft consistency to the touch. The roots and stems near the galls were observed to be normal in appearance, with no visible signs of disease. In some cases, longitudinal sections of the area revealed necrosis of the adjacent tissue.

Galls on raspberry plants from production fields in the Mexican states of Jalisco and Michoacán. Root segment with a compact appearance, showing galls along the tissue (A), young plant with the presence of greenish-brown tumours on the crown (B), gall in an advanced state, with brown, greenish and reddish coloration (C)
Bacterial identification
The isolation and purification of bacteria from symptomatic raspberry plant tissue revealed the presence of several types of colonies. These were selected based on their morphological characteristics on D1-M and King’s B medium, with a focus on possible Agrobacterium strains but not excluding other types of growth. The partial 16S ribosomal gene sequence of the 69 strains revealed the bacteria belong to different genera (Table 1). Similarly, genetic heterogeneity amongst the 28 Agrobacterium/Rhizobium strains was shown (Table 2), with most sequences recognised at the species level.
Pathogenicity tests
In the case of raspberry, symptoms appeared 5 weeks after inoculation for three strains (CPO 2.418, 2.419, and 2.404) for pathogenicity tests; the three of them were identified as Agrobacterium. The development of symptoms included the appearance of galls at the wound sites where the toothpick puncture was performed. They were small and light brown-greenish in colour at first, turning dark brown near the end of the evaluation (60 days post-inoculation) (Fig. 2a). In the case of tomato, symptoms showed up 2 weeks after inoculation for the same strains as in raspberry. The galls, which were light brown in colour and soft in texture, formed at the wound points where the toothpick inoculation was done. Toward the end of the evaluation, symptoms of wilting and general yellowing of the plant began to appear, indicating obstruction of the vascular system by gall growth (Fig. 2b).

Pathogenicity test exhibiting the gall symptoms on raspberry (A) and on tomato seedlings (B)
Phylogenetic analysis
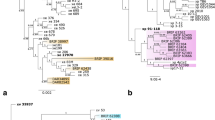
The phylogenetic tree of the 28 strains identified within the Agrobacterium and Rhizobium genera constructed using the 16S rRNA sequences (1335 bp) revealed the formation of four clades: within the tumefaciens complex, closely related to A. tumefaciens and A. arsenijevicii (CPO 2.303, 2.312, 2.314, 2.315, 2.322, 2.327, 2.329, 2.336, 2.350, 2.371, 2.418, and 2.419, the last two which resulted pathogenic strains) and related to A. leguminum and A. shirazense (CPO 2.302, 2.311, 2.334, 2.337, 2.352, 2.355, 2.372, 2.374, 2.381, and 2.205); within the Rhizobium genus (CPO 2.404 and 2.343), with CPO 2.404 standing out as a pathogenic isolate quite related to R. rhizogenes, and CPO 2.343 not clearly defined but close to R. grahamanii; and finally, a group related to A. pusense and A. salinotolerans (CPO 2.104, 2.113, 2.114, and 2.138) (Fig. 3).

Phylogenetic tree based on 16S rDNA sequences (1,335 bp) constructed using the Bayesian inference method. The nucleotide substitution model GTR + I was used. The isolated strains were divided into three major clades, with 22 of them belonging to the tumefaciens complex (marked in blue)
The multilocus phylogenetic tree of the four concatenated housekeeping genes (3806 bp) showed more precise grouping (Fig. 4). The strains clustered in the tumefaciens complex were more clearly defined as A. tumefaciens genomic species 1, leaving a clear separation from A. arsenijevicii. The pathogenic strains CPO 2.418 and 2.419 stand out as being closely related to A. tumefaciens LMG 232. The identity of the A. leguminum and A. shirazense strains could be defined better, with CPO 2.302, 2.311, 2.334, 2.337, 2.352, 2.355, 2.372, 2.374, and 2.381 being more related to A. leguminum and CPO 2.205 being closely related to A. shirazense, remaining relatively separated from the clade. Likewise, CPO 2.104, 2.113, 2.114, and 2.138 were confirmed to be related to A. pusense. Finally, CPO 2.404 was still grouped within the R. rhizogenes clade, confirming its identity, while CPO 2.343 remained close to R. grahamanii.

Multilocus Bayesian phylogenetic tree constructed from the concatenated partial sequences of the atpD, glnA, gyrB, and rpoB genes (3806 bp) using the GTR + I + G substitution model for all genes. Within the tumefaciens complex, CPO strains clustered with A. tumefaciens (genomic species 1), and A. arsenijevici (marked in blue) with greater definition
Whole genome sequencing
The whole genome sequencing files generated by the Illumina paired-end sequencing of the CPO 2.419 sample were 6180 Gb in size when decompressed. A total of 5,158,916,208 reads were produced. The average quality score (Q-score) of the reads was 36.896, and the average genome cover value was 443, which means that each base in the genome was sequenced an average of 5,754,873.65 times. The GC and AT contents were 59.15 and 40.85%, respectively. The high precision of each sequenced nucleotide is indicated by the Phred quality values (Supplementary Table 2). The sequencing reads were used to generate the whole-genome consensus sequence, which has a total length of 5,679,921 bp assembled into 46 contigs with an approximate N50 (average contig length) of 305 kb (Fig. 5). The average GC content was found to be 58.45%. Contig 7 corresponds to the Ti plasmid. The final sequence was deposited in the GenBank database, with the accession number JAVIYJ000000000.1.

Graphical circular map of the Agrobacterium tumefaciens strain CPO 2.419 genome. From outside to the centre: Circle 1 shows genes on the forward strand in green; Circle 2 shows genes on the reverse strand in purple; Circle 3 shows G + C% content; and the innermost ring shows GC skew (A). Circular graphical shows the genome annotations distribution: the contigs are present in black, CDS on the forward strand is green, and CDS on the reverse strand is purple, Non-CDS features are in turquoise, AMR genes are in red, VF genes are in orange, blue for the transporter genes, black for the drug target genes, a black line on pink for background GC content, and black on cream for GC skew (B)
The 100-housekeeping protein phylogenetic analysis for the strain CPO 2.419 revealed that it belongs to the tumefaciens complex, closest to A. tumefaciens LMG 232 (Fig. 6). The high similarity between strains LMG 232, CNPSo 675 (formerly the type strain of A. fabacearum), and ATCC 4720T is appreciated. The taxonomic position of the CPO 2.419 strain at the species level was determined by comparing OrthoANIu and dDDH values (Table 4). The high OrthoANIu percentage values obtained when compared to A. tumefaciens LMG 232, A. tumefaciens CNPSo 675, and A. tumefaciens ATCC 4720T (99.43, 98.56 and 98.1%, respectively) suggest a high level of genetic similarity. Meanwhile, a dDDH value of 89.1% was obtained for LMG 232, indicating the strongest relationship with CPO 2.419. A. arsenijevicii KFB 330, on the other hand, obtained OrthoANIu and dDDH values of 90.60 and 57.5%, respectively, against CPO 2.419, which were lower than the values of 90 and 70% for species delineation, reaffirming its taxonomic separation.

Phylogenetic tree constructed using sequences that code for 100 housekeeping proteins genes and information from 25 complete genomes belonging to the genus Agrobacterium. CPO 4.219 was placed in the A. tumefaciens clade (marked in blue)
Comparison of CPO 2.419 contig 7 (GenBank JAVIYJ010000007.1) with CFBP2712 pTi (NCBI) exhibited high similarity (Fig. 7) with the sequences corresponding to the vir genes (virA, virB1–11, virC1–2, virD1–4, virE1–3, virG, virK, and virA/G); opine synthases such as agrocinopine synthase and tnp; TraA–F family protein; transposases such as Tn3 family transposase; ABC transporter ATP-binding protein; ABC transporter permease; HipA domain-containing protein; transcriptional regulator TraR; repA–C; and trbB–L.

Plasmids comparison between Agrobacterium tumefaciens CPO 2.419 contig 7 (205,933 nt) and A. tumefaciens pTi CFBP2 (193,011 nt) depicts high sequence similarity using tBLASTxS
Discussion
Isolation and purification of bacteria from symptomatic tissue showed the diversity of the bacterial microbiota, with Agrobacterium genus isolates standing out. The identification of the 69 representative strains (Table 1) showed the genus diversity found within the tissue of the symptomatic raspberry plants. Commonly found bacterial genera in this study included Burkholderia, Delftia, Enterobacter, Flavobacterium, Klebsiella, Pantoea, Pseudomonas, Stenotrophomonas, Agrobacterium, and Rhizobium. Sánchez-Jiménez et al. (2022) reported that part of this bacterial diversity could also be found in raspberry gall tissue from Tlaxcala, Mexico, with Delftia and Pseudomonas being the most frequent genera found in their study.
Pathogenicity tests revealed that three strains (CPO 2.418, 2.419, and 2.404) demonstrated the ability to induce symptoms in both raspberry and tomato. Similarly, Agrobacterium strains with no ability to induce symptoms were frequently isolated in this study.
This 16S rRNA analysis provides an overview of the genetic diversity and phylogenetic relationships among the strains under consideration (Fig. 3). The results reveal their classification into specific clades within the Agrobacterium/Rhizobium genus. The first group of bacteria comprises 22 strains belonging to the tumefaciens species complex, divided into two clades: the first within the A. tumefaciens group and the second within the A. leguminum and A. shirazense groups. A third clade formed with the Rhizobium genus, with two strains, and a fourth with A. pusense and A. salinotolerans, with four strains. The Agrobacterium tumefaciens species complex largely predominates, as reported in previous studies for isolates obtained from raspberry (Puławska et al. 2015; Kuzmanović et al. 2015a; Mafakheri et al. 2019).
This information is critical for understanding the strains’ population structure and evolutionary relationships. The presence of different phylogenetic groupings suggests changes in the original population structure and the formation of cryptic species (species in the process of separation), which may explain the presence of highly similar strains in different lineages within the genus. From a single gall, pathogenic and non-pathogenic bacteria from different phylogenetic groups could be isolated. This makes determining which strain caused the gall difficult. Furthermore, bacteria are not required for disease development after the plant has been infected (Puławska et al. 2015).
The partial sequencing of the 16S ribosomal gene is useful for studying the population diversity of the strains in the symptomatic sample. However, for the tumefaciens complex, which includes several closely related species, this molecular marker lacks sufficient resolution to distinguish between species, which was clearly the case between A. tumefaciens and A. arsenijevicii. The high conservation of this gene in some bacteria may make species distinction difficult. This is evidenced by the clustering of strains within the aforementioned clades, where identification may be inaccurate despite genetic variability.
To address these limitations, the multilocus analysis using the housekeeping genes atpD, glnA, gyrB, and rpoB allowed for a more accurate identification. The strains were more precisely classified, particularly those in the tumefaciens complex (Fig. 4), with all strains in the first clade clustering better with A. tumefaciens. The 100-housekeeping protein analysis, which provides even better resolution for identification, reaffirmed that strain CPO 2.419 belongs to the tumefaciens complex and is most closely related to A. tumefaciens LMG 232 (Fig. 6).
This study shows the Agrobacterium strain variety found in disease-struck plantations in the region, some of which can be found even within the same sample (Llop et al. 2009). These findings add significantly to our understanding of microbial diversity and the evolution of phytopathogenic strains associated with crown gall in raspberry-producing areas of Mexico. This is relevant as common bacterial diseases spread between nurseries via plant material distribution and remain in contaminated soil. Producers in Mexico’s berry-growing sector may be at risk from the trade of contaminated plant material.
The OrthoANIu values provide information about genetic similarity (Table 4). The high percentages obtained for strain CPO 2.419 compared to A. tumefaciens LMG 232 (99.43%) suggest a high level of genetic similarity, while lower values with A. arsenijevicii KFB 330 indicate a clear taxonomic separation. Such high ANI values are frequently associated with strains of the same species or closely related species. Meanwhile, dDDH values reflect the degree of DNA-DNA hybridization and may be related to genetic relatedness and taxonomic classification. The highest value obtained was 89.1% for A. tumefaciens LMG 232, indicating the strongest relationship with CPO 2.419; therefore, considering both OrthoANIu and dDDH values, strain CPO 2.419 was identified as A. tumefaciens.
Prior to the taxonomic reclassification of A. tumefaciens and A. radiobacter, several strains were recorded with an incorrect name and are now classified as A. tumefaciens NCPPB 3001, A. tumefaciens CFBP 5877, and A. tumefaciens IIF1SW-B1. According to Velázquez et al. (2020), when compared to A. tumefaciens ATCC 4720T with the genomes of different strains of A. fabacearum have ANIb and dDDH values higher than what is recommended for species differentiation, implying that both may belong to the same species. The recent taxonomic change for A. tumefaciens CNPSo 675 (which was the former type strain for A. fabacearum) reflect the need for constant taxonomic re-evaluation of the existent and new Agrobacterium species and strains using whole genome analysis.
The findings of this study support the revaluation of the taxonomic position of the studied strains and provide valuable information on the identity and phylogenetic relationships of the CPO 2.419 strain in the context of related Agrobacterium species. These findings add to the knowledge of the genomic diversity of this bacterial genus in raspberry plantations in Mexico. Therefore, it is necessary to develop management strategies to reduce the impact that tumorigenic Agrobacterium can cause on raspberry plantations.
References
Aujoulat F, Jumas-Bilak E, Masnou A et al (2011) Multilocus sequence-based analysis delineates a clonal population of Agrobacterium (Rhizobium) radiobacter (Agrobacterium tumefaciens) of human origin. J Bacteriol 193(10):2608–2618. https://doi.org/10.1128/jb.00107-11
Beekwilder J, Jonker H, Meesters P et al (2005) Antioxidants in raspberry: on-line analysis links antioxidant activity to a diversity of individual metabolites. J Agric Food Chem 53(9):3313–3320. https://doi.org/10.1021/jf047880b
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina Sequence Data. Bioinformatics btu170.
Cock PJA, Antao T, Chang JT et al (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinform 25(11):1422–1423. https://doi.org/10.1093/bioinformatics/btp163
Davis JJ, Gerdes S, Olsen GJ et al (2016) PATtyFams: protein families for the microbial genomes in the PATRIC Database. Front Microbiol 7:118. https://doi.org/10.3389/fmicb.2016.00118
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Edwards U, Rogall T, Blöcker H et al (1989) Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res 17(19):7843–7853. https://doi.org/10.1093/nar/17.19.7843
Gordon JE, Christie PJ (2014) The Agrobacterium Ti plasmids. Microbiol Spectr 2(6). https://doi.org/10.1128/microbiolspec.plas-0010-2013
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Kerr A (2015) Biological control of crown gall. Australas Plant Pathol 45(1):15–18. https://doi.org/10.1007/s13313-015-0389-9
Krzepiłko A, Prażak R, Święciło A (2021) Chemical composition, antioxidant and antimicrobial activity of raspberry, blackberry and raspberry-blackberry hybrid leaf buds. Molecules 26(2):327. https://doi.org/10.3390/molecules26020327
Kuzmanović N, Prokić A, Ivanović M et al (2015a) Genetic diversity of tumorigenic bacteria associated with crown gall disease of raspberry in Serbia. Eur J Plant Pathol 142(4):701–713. https://doi.org/10.1007/s10658-015-0645-4
Kuzmanović N, Puławska J, Prokić A et al (2015b) Agrobacterium arsenijevicii sp. nov., isolated from crown gall tumors on raspberry and cherry plum. Syst Appl Microbiol 38(6):373–378. https://doi.org/10.1016/j.syapm.2015.06.001
Lacroix B, Citovsky V (2022) Agrobacterium. Life Sci. https://doi.org/10.1016/b978-0-12-822563-9.00066-4
Llop P, Murillo J, Lastra B, López MM (2009) Recovery of nonpathogenic mutant bacteria from tumors caused by several Agrobacterium tumefaciens strains: a frequent event? Appl Environ Microbiol 75(20):6504–6514. https://doi.org/10.1128/aem.01867-08
Mafakheri H, Taghavi SM, Puławska J et al (2019) Two novel genomospecies in the Agrobacterium rhizogenes species complex associated with rose crown gall. Phytopathology 109(11):1859–1868. https://doi.org/10.1094/phyto-05-19-0178-r
Mafakheri H, Taghavi SM, Zarei S et al (2022) Phenotypic and molecular-phylogenetic analyses reveal distinct features of crown gall-associated Xanthomonas strains. Microbiol Spectr 10(1). https://doi.org/10.1128/spectrum.00577-21
Martens M, Dawyndt P, Coopman R et al (2008) Advantages of multilocus sequence analysis for taxonomic studies: a case study using 10 housekeeping genes in the genus Ensifer (including former Sinorhizobium). Int J Syst Evol Microbiol 58(1):200–214. https://doi.org/10.1099/ijs.0.65392-0
Martin RR, Ellis MA, Williamson B, Williams RN (2017) Compendium of Raspberry and Blackberry diseases and pests, 2nd edn. APS, St. Paul, MN, USA. https://doi.org/10.1094/9780890545720
Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform 14(1):60. https://doi.org/10.1186/1471-2105-14-60
Paret ML, Momol T, Ritchie L, Dankers H (2011) 2011 Florida plant disease management guide: Apple (Malus domestica): PDMG-V3-05/PG004, Rev. 7/2011. EDIS 2011(7 https://doi.org/10.32473/edis-pg004-2011
Parte AC, Sardà Carbasse J, Meier-Kolthoff JP et al (2020) List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int J Syst Evol Microbiol 70(11):5607–5612. https://doi.org/10.1099/ijsem.0.004332
Perry KL, Kado CI (1982) Characteristics of Ti plasmids from broad-host-range and ecologically specific biotype 2 and 3 strains of Agrobacterium tumefaciens. J Bacteriol 151(1):343–350. https://doi.org/10.1128/jb.151.1.343-350.1982
Puławska J, Warabieda W, Ismail E (2015) Identification and characterization of bacteria isolated from crown galls on stone fruits in Poland. J Plant Pathol 65(6):1034–1043. https://doi.org/10.1111/ppa.12482
Puławska J, Kuzmanović N, Trzciński P (2022) Agrobacterium vaccinii sp. nov. isolated from galls on blueberry plants (Vaccinium corymbosum). Syst Appl Microbiol 45(3):126319. https://doi.org/10.1016/j.syapm.2022.126319
Ronquist F, Huelsenbeck JP (2003) MRBAYES 3: bayesian phylogenetic reference under mixed models. Bioinform 19:1572–1574
Sánchez-Jiménez E, Aranda-Ocampo S, Ochoa-Martínez DL, Mejía-Sánchez D (2022) Native bacteria in raspberry crown gall reduce the severity of Agrobacterium tumefaciens. Agrociencia 56(8):1–12. https://doi.org/10.47163/agrociencia.v56i8.2871
Schaad NW, Jones JB, Chum W (2001) Laboratory Guide for Identification of Plant pathogenic Bacteria. APS, St. Paul, MN, USA
Singh NK, Lavire C, Nesme J et al (2021) Comparative genomics of novel Agrobacterium G3 strains isolated from the international space station and description of Agrobacterium tomkonis sp. nov. Front Microbiol 12. https://doi.org/10.3389/fmicb.2021.765943
Stackebrandt E, Liesack W (1993) Nucleic acids and classification. In: Goodfellow M, O’Donnell AG (eds) Handbook of New Bacterial Systematics. Academic, London, UK, pp 152–189
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinform 30(9):1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 57(5):758–771. https://doi.org/10.1080/10635150802429642
Tamura K, Battistuzzi FU, Billing-Ross P et al (2012) Estimating divergence times in large molecular phylogenies. Proc Natl Acad Sci 109(47):19333–19338. https://doi.org/10.1073/pnas.1213199109
Velázquez E, Flores-Félix JD, Sánchez-Juanes F et al (2020) Strain ATCC 4720T is the authentic type strain of Agrobacterium tumefaciens, which is not a later heterotypic synonym of Agrobacterium radiobacter. Int J Syst Evol Microbiol 70(9):5172–5176. https://doi.org/10.1099/ijsem.0.004443
Wick RR, Judd LM, Gorrie CL, Holt KE (2017) Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput Biol 13(6):e1005595. https://doi.org/10.1371/journal.pcbi.1005595
Yoon SH, Ha S, Lim J et al (2017) A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110(10):1281–1286. https://doi.org/10.1007/s10482-017-0844-4
Acknowledgements
The authors wish to express their gratitude to the National Council of Humanities, Science and Technology of Mexico (CONAHCyT) for financial assistance to the first author.
Funding
This work was partially supported by the Biotechnology and Seed Pathology Laboratory of the Postgraduate College at Montecillo Campus (external project PS 20-4002, PS 21-4002, and PS 22-4001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This article does not contains any studies requiring ethical approval.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Godínez-Alemán, J.E., Silva-Rojas, H.V., Aguilar-Granados, A. et al. Genomic insights into Agrobacteria inducing galls on raspberry. J Plant Pathol (2024). https://doi.org/10.1007/s42161-024-01676-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42161-024-01676-2