
Abstract
Verticillium wilt caused by the soil-borne fungus Verticillium dahliae is a devastating disease of cotton, leading to serious loss of lint yield in China. It is important to characterize the prevailing populations of V. dahliae for understanding disease development. In this study, the representative 74 V. dahliae isolates from 28 different cotton regions in three major cotton-production areas in China were identified for their pathotypes, races, vegetative compatibility groups (VCGs) and genetic diversity by specific PCR, amplified fragment length polymorphism (AFLP) and inter simple sequence repeat (ISSR) analysis. Of 74 isolates, 60 isolates (81%) were defoliating (D) pathotype strains, especially in Yellow River/Yantze River Valley region. Seventy isolates belonged to race 2, whereas no race 1 specific fragment was amplified. These isolates were grouped into five VCGs: VCG1A (48), VCG2A/VCG4B (3), VCG2B824 (6), VCG6 (5). The D pathotype and VCG1A isolates have become the dominant strains, moderately and highly virulent isolates were widely distributed in three major cotton-growing areas in China. High level of polymorphic markers was obtained by AFLP and ISSR analyses. Through comparison of the obtained dendrograms by unweighted paired group method with arithmetic averages cluster analysis (UPGMA), the V. dahliae isolates were divided into two major groups and five subgroups. The obvious correlations with the geographic origin of the isolates between AFLP and ISSR were revealed, with significant diversity among V. dahliae isolates. In addition, AFLP dendrogram showed relationships with pathotypes. The results offered a better understanding of the epidemiology of Verticillium wilt in China.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Verticillium wilt, a devastating disease of cotton, is caused by the soil-borne, destructive plant fungus Verticillium dahliae Kleb. This pathogenic fungus is widely distributed in agricultural soils with a broad host range of more than 400 plant species, including many economically important crops (Bell 1992). The disease was first reported on upland cotton in Virginia, USA and then widely spread to most of the cotton-growing countries worldwide. It was firstly introduced to China via imported American cotton in 1935 (Ma 2007). Initially, the disease just occurred in limited areas in China. However, since 1990s, due to the widespread of V. dahliae, more than 70% of cotton-planting areas have been subjected to Verticillium wilt, resulting in a tremendous economic loss yearly (Ma 2007). Therefore, characterization of genetic variations among populations of the prevailing V. dahliae is an essential and important step to understand disease development and further to formulate the appropriate strategies for disease management.
Based on symptomatology, V. dahliae is generally classified into two pathotypes, namely defoliating pathotype (D) and non-defoliating pathotype (ND) (Schnathorst and Mathre 1966). The former causes defoliation of the cotton and is highly virulent while the latter causes mild wilt symptoms without defoliation (Schnathorst and Mathre 1966; Joaquim and Rowe 1990). Using random amplified polymorphic DNA (RAPD) and specific polymerase chain reaction (PCR) analyses, Perez-Artes et al. (2000) clearly differentiated D strains from ND ones. It is important to determine the V. dahliae pathotypes that infect cotton in order to manage the disease by planting V. dahliae-tolerant/resistant cotton cultivars to overcome this highly virulent D pathotype.
V. dahliae isolates have also been classified by vegetative compatibility groups (VCGs) (Leslie 1993). After surveys of a large collection of isolates, six VCGs from different hosts and geographic origins were identified (Daayf et al. 1995; Jimenez-Diaz et al. 2006). Due to the strong linkage disequilibrium in clonal populations, VCGs in V. dahliae are highly correlated with molecular markers and clonal lineages (Jimenez-Diaz et al. 2012; Jimenez-Gasco et al. 2014; Jimenez-Diaz et al. 2017). VCGs are informative, and isolates from different VCGs are thought to be genetically distinct populations which may vary in a number of ecological, physiological, virulence, and host-range traits (Angela et al. 2013).
Molecular markers are able to provide a direct measure of genetic diversity. Therefore, molecular markers, such as those based on amplified fragment length polymorphism (AFLP), inter-simple sequence repeat (ISSR), random amplification of polymorphic DNA (RAPD), simple sequence repeats (SSRs or microsatellites), the intergenic spacer region of the ribosomal DNA (IGS rDNA) and restriction fragment length polymorphism (RFLP) analyses, have been applied in the analysis of genetic diversity of V. dahliae (Atallah et al. 2010; Collado-Romero et al. 2006, 2009; ElSharawy et al. 2015; Erdogan et al. 2013; Gharbi et al. 2014, 2015; Pramateftaki et al. 2000). Among the molecular markers, AFLP (Vos et al. 1995) and ISSR (Zietkiewicz et al. 1994) are the highly polymorphic ones, which provide highly discriminating information with good reproducibility. In the present study, we applied both AFLP and ISSR markers to analyze the genetic diversity among V. dahliae populations in samples collected from three major cotton planting areas in China. We also tested the pathogenicity of all the isolates and used the specific PCR analysis to confirm the pathotypes, race and VCGs of V. dahliae in China (Perez-Artes et al. 2000; Collado-Romero et al. 2009; de Jonge et al. 2012; Short et al. 2014). The results obtained in this study will provide further information about the pathogenic diversity of V. dahliae in China, which can be helpful for forecasting disease development and formulating management measures for effective control and prevention of Verticillium wilt of cotton in China.
Materials and methods
Collection of isolates and culture
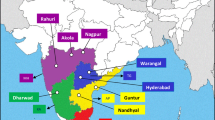
The 74 V. dahliae isolates used in this study were collected from infected cotton showing Verticillium wilt symptoms from 28 different cotton-planting regions in three traditional major cotton production areas across 11 provinces of China (Fig. 1) from 2014 to 2016. The three major cotton production regions areas are: (a) Yellow River Valley region, which includes Hebei (Baoding, Langfang, Xingtai), Henan (Kaifeng, Xinxiang), Shandong (Heze), Shanxi (Yuncheng) provinces; (b) Yantze River Valley region, which includes Anhui (Anqing, Chizhou, Wuhu), Hubei (Qianjiang, Jingzhou, Tianmen, Xiantao, Zhijiang), Hunan (Yirang, Yueyang), Jiangsu (Dafeng, Nantong, Xuzhou), and Jiangxi (Jiujiang) provinces; and (c) inland of Northwest region, which includes Gansu (Dunhuang) province and Xinjiang Autonomous region (Akesu, Bole, Kashi, Kuitun, Manasi, Shawan). The V. dahliae isolates from these regions are described in Table 1 and Fig. 1.

Geographical distribution of the collected V. dahliae isolates
To isolate V. dahliae, cotton stems showing wilt symptoms were cut into small pieces (0.5-2 cm) with a sterilized razor. The stem surface was sterilized with 5% sodium hypochorite (12%) followed by rinsing twice in sterile distilled water. The washed pieces were dried on sterile filter paper, then placed onto potato dextrose agar (PDA) plates. After incubation at 25 °C for one week, the emerging fungi were transferred onto new PDA plates. Single spore was isolated and stored in a 20% glycerol solution at −80 °C.
Extraction of fungal DNA and specific PCR assay
Single spore isolates were grown on PDA plates at 25 °C for one week. Mycelia were collected and ground into powder in mortar with liquid nitrogen. Total fungal DNA was extracted from each isolate of V. dahliae essentially as described by Lee and Taylor (1990). The DNA integrity was assessed using agarose gel electrophoresis and NanoDrop ND1000 spectrophotometer (NanoDrop Technologies Inc., USA). All V. dahliae isolates in the study were characterized by PCR assays using eight primer pairs. Two pairs of primers, VDF/VDR and VNDF/VNDR, described by Perez-Artes et al. (2000) were used to determine the pathotype of the collected isolates. Primer pair VDF/VDR yields a specific PCR product (548 bp) related to the D pathotype, whereas primer pair VNDF/VNDR yields a specific PCR product (1410 bp) associated with the ND pathotype. Four pairs of primers were used to measure the VCGs of all V. dahliae isolates (Collado-Romero et al. 2009). Primer pair DB19/espdef01 (Mercado-Blanco et al. 2003) yields a specific 334 bp product which was amplified from VCG1A, VCG1B or VCG2B334 isolates. Primer pair INTND2F/INTND3R (Collado-Romero et al. 2009) yields a 688 bp marker which was amplified from VCG2A, VCG4B or VCG2B824 isolates. Primer pair INTND2F/INTND2R (Mercado-Blanco et al. 2001) yields a 824 bp marker which was amplified from VCG2A, VCG2B824, VCG4B isolates. Primer pair INTND2F/MCR2B (Collado-Romero et al. 2009) yields a 964 bp marker, which was amplified from VCG2B824 isolates. Primer pair VdAve1F/ VdAve1R was used to identify V. dahliae race 1 (de Jonge et al. 2012), primer pair VdR2F/VdR2R was used to identify V. dahliae race 2 (Short et al. 2014). All isolates were tested separately using these eight primer pairs.
AFLP and ISSR analysis
AFLP assays were performed according to the fluorescent procedures of the method described by Vos et al. (1995) with modifications. Adapters, pre-amplification primers, nine EcoRI: MseI AFLP selective primer combinations were shown in Supplementary Table S1. The MseI primer was labeled with FAM dye. The selective PCR amplification consisted of one cycle of denaturation at 94 °C for 30 s, annealing at 65 °C for 30 s and extension at 72 °C for 80 s, followed by 11 cycles under the same conditions except that the annealing temperature was decreased by 0.7 °C per cycle. Then, the last 23 cycles were carried out at 94 °C for 30 s, 55 °C for 30 s and 72 °C for 80 s. Nine primers (Supplementary Table S1) were used for ISSR analysis. PCR amplification was performed by initial denaturation at 94 °C for 5 min, followed by 45 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C for 45 s and extension at 72 °C for 2 min, and a final extension of 10 min at 72 °C. The amplicons of AFLP and ISSR were separated by polyacrylamide gel electrophoresis in an automatic DNA sequencer (ABI337), respectively. The data were collected by GeneScan analysis software. Amplification products were scored as presence (1) or absence (0) based on the AFLP/ISSR pattern obtained for each isolate and the data were compiled as a binary matrix. Dendrograms were generated by unweighted pair-group mean arithmetic method analysis (UPGMA) and clustered with the Ntsys software (Rohlf 1997). The Mantel significance test was used to compare each pair of similarity matrices produced with the Ntsys 2.2 software.
Pathogenicity assay
Six different resistant varieties were used in virulence assays in a growth chamber under long-day condition (16 h photoperiod) with 26/20 °C day/night temperatures to determine the pathogenicity of all the isolates. Cotton cultivars included two resistant cultivars: Zhongzhimian No. 2 (Ren et al. 2018) and Jimian 958 (Geng et al. 2006); two tolerant cultivars: Luyanmian 28 (Ren et al. 2018) and Zhongmiansuo 35 (Zhu et al. 2012); two susceptible cultivars: 86–1 (Zhang et al. 2013) and Xinluzao No. 7 (Ren et al. 2018). Cotton growth and inoculation with V. dahliae isolates by the root dipping method were as in Zhang et al. (2013). Seedlings dipped in sterile distilled water were used as non-inoculated controls. For each V. dahliae isolate, 10 plants were inoculated per experiment and the whole experiment was conducted three times. According to the percentage of affected plant tissues, such as chlorosis, necrosis, wilt and defoliation, a Verticillium wilt disease index (DI) scaling from 0 to 4 was introduced to evaluate the disease severity (Zhang et al. 2013). The DI of Verticillium wilt was determined using the following formula (Zhang et al. (2017):
Then, the pathogenicity of the isolates was evaluated by the mean DI value on six cotton varieties. The mean DI value lower than 20.0 designated a weakly virulence isolate, the mean DI value between 20.01 to 35.0 designated a moderately virulent isolate, the mean DI value higher than 35.0 designated highly virulent isolates (Zhang et al. 2012). Statistical analyses were conducted by ANOVA using the SAS 9.4. The confidence level of all analyses was set at 95%, and P < 0.05 was considered as statistically significant.
Results
Pathotypes and race characterization
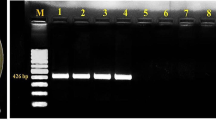
Seventy-four representative isolates were analyzed to detect pathotypes and races by specific PCR assay based on the presence of 548 bp (D pathotype), 1410 bp (ND pathotype), 900 bp (race 1), and 256 bp (race 2) amplicons (Fig. 2a, b, c). Most of the isolates produced the 548 bp specific fragment and they were thus, designated as D pathotype. Fourteen isolates produced the 1410 bp specific fragment and thus, were designated as ND pathotype (Table 1), whereas 60 isolates belonged to D pathotype, which accounted for 81% of all the isolates.


PCR products obtained with the different primer pairs used in this study. a PCR products obtained with non-defoliating specific primers VNDF/VNDR (1410 bp). b defoliating specific primers VDF/VDR (548 bp). c race 2 specific primers VdR2F/VdR2R (256 bp). d primer pairs DB19/espdef01 (334 bp). e primer pairs INTND2F/INTND3R (688 bp). f primer pairs INTND2F/INTND2R (824 bp). g primer pairs INTND2F/MCR2B (964 bp). Lane M: 200 bp ladder. Lanes 1 to 24: VD11, VD18, VD47, VD48, VD50, VD51, VD53, VD55, VD65, VD73, VD1, VD4, VD6, VD9, VD10, VD19, VD22, VD23, VD25, VD31, VD38, VD40, VD62, VD71
Seventy isolates produced the 256 bp specific fragment and were designated as race 2. Four isolates (VD23, VD47, VD53, VD72) could not be amplified with race 2-specific primer pair (Table 1). However, no isolate could be amplified with race 1-specific VdAve1F/VdAve1R primers. A previous study showed that defoliating pathotype is more virulent than ND pathotype (Joaquim and Rowe 1990). VD23 and VD72 were D isolates, while VD47 and VD53 were ND isolates. There is no obvious relationship between race 1 and race 2 with D and ND pathotypes.
VCGs characterization
Amplification of both the 334 bp and 548 bp specific fragments was obtained from 48 isolates, which were designated as VCG1A isolates. VD48, VD54 and VD58 amplified both the 688 bp and 824 bp specific fragments, indicating them as VCG2A or VCG4B isolates. VD11, VD18, VD50, VD51, VD53 and VD73 amplified three specific markers including 688 bp, 824 bp and 964 bp, which designated them as VCG2B824 isolates. VD47, VD49, VD55, VD63 and VD65 only produced 824 bp specific fragments, which designated them as VCG6 isolates. Moreover, 12 isolates only amplified the 548 bp specific fragment, but could not amplify the 334 bp specific fragment. The results are listed in Table 1 and Fig. 2a, d–g. It can be concluded that the VCG1A isolates are dominant across China cotton production areas.
Fingerprinting
In order to produce clear and reproducible polymorphic banding patterns from all V. dahliae isolates tested, nine primer pairs were used for AFLP and nine primers for ISSR analysis. The sizes of amplified markers were in the range of 70–500 bp for AFLP and 50–1000 bp for ISSR. These primers generated a large number of polymorphic amplification markers. In the case of AFLP, 881 unambiguous and easily discernible polymorphic markers were obtained from the 74 isolates, including 91 markers with EcoRI-AAC/MseI-CAA, 88 with EcoRI-AAC/MseI-CTA, 85 with EcoRI-AAG/MseI-CTC, 75 with EcoRI-AAG/MseI-CTT, 129 with EcoRI-ACA/MseI-CTC, 112 with EcoRI-ACG/MseI-CTC, 87 with EcoRI-AGC/MseI-CAG, 94 with EcoRI-AGG/Mse I-CTC, and 120 with EcoRI-AGG/MseI-CTG. In the case of ISSR, 1676 markers were obtained from the 74 isolates, including 235 markers with ISSR807, 193 with ISSR808, 153 with ISSR809, 157 with ISSR811, 172 with ISSR835, 170 with ISSR836, 178 with ISSR840, 234 with ISSR881, and 184 with ISSR886.
Genetic diversity and comparison between AFLP and ISSR
To evaluate the effectiveness of the two different molecular markers (AFLP and ISSR) in studying genetic relationships between V. dahliae isolates, genetic similarity coefficient was calculated. The genetic similarity value for AFLP ranged from 0.26 (between VD32 and VD23, 55) to 0.61 (between VD37 and VD38). For ISSR, it ranged from 0.13 (between VD23 and VD55) to 0.56 (between VD39 and VD40).
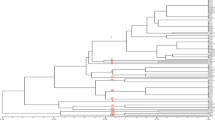
The dendrogram generated from UPGMA clustering of two marker data divided all V. dahliae isolates into two major clusters (Fig. 3). Each of their dendrograms was unique with some evident similarities. The AFLP-based dendrogram divided the isolates into two major groups at a similarity index of 0.26 (Fig. 3a), placing VD23, VD32, and VD55 in one cluster and the remaining 71 isolates in the other cluster. The latter cluster was further divided into five distinct subgroups. Isolates collected from Yellow River Valley region and Yantze River Valley region were mainly grouped together in subgroup A. The ISSR-based dendrogram was discriminated at a higher level of diversity, two major clusters were divided at a similarity index of 0.13 (Fig. 3b), placing VD23 in one cluster and the remaining 73 isolates in the other cluster. The larger group was further separated into five subgroups which were grouped mainly according to the geographical origin. There is an obvious correlation with the geographic origin of isolates between the dendrograms of AFLP and those of ISSR.


Dendrograms of the 74 V. dahliaeisolates generated from AFLP and ISSR. a Dendrograms generated from AFLP data of 74 Verticillium dahliae isolates from cotton in China. b Dendrograms generated from ISSR data. UPGMA was based on DICE similarity distance
Furthermore, the results of AFLP dendrogram showed a relationship to pathotypes. In AFLP clusters, the ND isolates were separated from D isolates. Isolates of subgroup B and E were ND pathotype while the isolates of subgroups A, C and D belonged to D pathotype. However, in ISSR clustering patterns, ND pathotype showed a mixed distribution between them. In ISSR, ND isolates VD11 and VD18 collected from Anhui and Hebei were placed in subgroup B. Subgroup C included seven ND isolates from inland of Northwest region (VD47–51, VD53 and VD54), whereas four ND isolates from inland of Northwest region (VD58, VD63, VD65 and VD73) were placed in subgroup D. ND isolate VD55 constituted subgroup E by itself.
In addition, goodness of fit test was performed for DICE similarity distances by Mantel test in both AFLP and ISSR analyses. The strongest correlation coefficients (r) were found to correspond to the dendrograms produced by AFLP and ISSR at a threshold of P < 0.001. ISSR’s correlation coefficient value was the highest with r being 0.94, followed by AFLP with r being 0.85.
Virulence of isolates
All the isolates showed variation in virulence on the six cotton varieties. Based on the results of mean DI values on six cotton varieties, these isolates were classified into three main virulence categories: weakly virulent, moderately virulent and highly virulent isolates. Five isolates were weakly virulent isolates, that means DI value lower than 20.0. Thirty-nine isolates were moderately virulent isolates, with DI value between 20.01 and 35.0. Thirty-nine isolates were highly virulent isolates, that means DI value higher than 35.0 (Table 1, Supplementary Table S2). Among them, highly virulent isolates were distributed in nine of 11 provinces, including Anhui (3), Hebei (4), Henan (2), Jiangsu (7), Jiangxi (1), Hubei (4), Hunan (1), Shanxi (2), Xinjiang (6). As for defoliating isolates, only VD23, VD46 and VD60 were weakly virulent isolates, whereas the other isolates were moderately or highly virulent isolates. Among ND isolates, VD47 and VD73 were weakly virulent isolates, VD18 and VD50 were highly virulent isolates, the other 10 isolates were moderately virulent. This indicated that moderately and highly virulent isolates were widely distributed in three major cotton production areas of China.
Discussion
As the most important cotton pathogen, V. dahliae infecting cotton can result in substantial yield loss in China. Therefore, it is important to know the disease development in China. In this study, we selected 74 representative isolates from three major cotton production areas distributed across 11 provinces in China from 2014 to 2016, which represented more than 70% of cotton-planting areas in the country. The prevailing V. dahliae isolates were molecularly tested for their pathotype, races, vegetative compatibility, genetic diversity. These results, together with those from our experiment on virulence of isolates of V. dahliae on a group of six genotypes, some of them carrying genes for resistance against the disease, will allow a better understanding of Verticillium wilt of cotton in China.
The pathotypes of V. dahliae were classified as either D or ND based on disease symptomotology. In the last century, the ND pathotype of V. dahliae was widespread (Perez-Artes et al. 2000), while the D pathotype was found only from a few places (Bejarano-Alcazar et al. 1996; Xia et al. 1998). On the contrary, and according to our molecular results, the D pathotype dominates in China. In this study, 81% of all the isolates belonged to D pathotype. Especially in Yellow River Valley region and Yantze River Valley region, 47 isolates were D pathotypes, and only two isolates were ND pathotypes. In inland of Northwest region, D pathotype isolates accounted for 52%, while ND pathotype isolates accounted for 48%. It was reported that only small numbers of D pathotypes (about 8.6%) were scattered at inland of Northwest region (Zhang et al. 2006). The D pathotype seems to have expanded rapidly in inland of Northwest region, what can be due to the wide spread of infected cotton seeds.
Several pathogenic races of V. dahliae have been described on lettuce, pepper, strawberry, sunflower and tomato (Maruthachalam et al. 2010; Short et al. 2014; Martín-Sanz et al. 2018). Race 2 lacks the virulence gene (Ave 1) and is not recognized in plants by resistant Ve gene, resulting in disease (Jonge et al. 2012; Short et al. 2014). In previous studies, nearly 37% of the V. dahliae isolates from cotton were characterized as race 1/ND isolates, and 61% of the V. dahliae isolates from cotton were characterized as race 2/D isolates (Hu et al. 2015). But in this study, no race 1 bands were amplified. The V. dahliae isolates from cotton were only characterized as race 2 isolates. Moreover, D type had significant relationship with race 2 and ND type correlated with race 1 (Hu et al. 2015). Nearly 86% ND isolates were characterized as race 2, and 97% D isolates were characterized as race 2. In disagreement with Hu et al. (2015), there is no significant relationship between race and pathotypes in cotton.
There are six main VCGs in V. dahliae (Jimenez-Diaz et al. 2006). In China, isolates from cotton were usually grouped into VCG1 and VCG2. Xia et al. (1998) found that 98 isolates from cotton belonged to VCG2, and 11 isolates belonged to VCG1. Xu et al. (2012) showed that 332 (97%) isolates from Yantze River Valley region (including Province Hubei, Hunan, Jiangxi, Anhui and Jiangsu) belonged to VCG1A and the remaining nine (3%) isolates belonged to VCG2. Consistently with previous results, VCG1A isolates were dominant in Yellow River Valley region and Yantze River Valley region. However, a diversity of VCGs was molecularly detected in inland of Northwest region getting our attention. In inland of Northwest region, four isolates were identified as VCG1A, four isolates belonged to VCG2B824, three isolates belonged to VCG2A or VCG4B, and five isolates belonged to VCG6. These results suggest that the genetic exchange between different strains of V. dahliae (Jimenez-Diaz et al. 2006) might be more probable in inland of Northwest region than in Yellow River region and Yantze River Valleys region.
Molecular determination of VCGs provided useful information, but the overall genetic diversity among V. dahliae strains still needed further molecular analyses. In previous studies, several molecular-based techniques have been successfully used to detect genetic diversity among isolates of V. dahliae (Erdogan et al. 2013; Fahleson et al. 2003; Gharbi et al. 2014, 2015; ElSharawy et al. 2015; Collado-Romero et al. 2006, 2009). In the present study, the applications of both AFLP and ISSR markers revealed a high level of polymorphism. The isolates were clustered using UPGMA method to determine their relationships in the dendrogram and a clear correlation between clusters was found. Within each cluster, individuals collected from the same geographical origin tended to be grouped together as shown in both AFLP and ISSR dendrograms. These results agreed well with those obtained in other studies conducted on V. dahliae using AFLP and ISSR methods (Erdogan et al. 2013; Gharbi et al. 2015; ElSharawy et al. 2015). Based on the dendrograms of AFLP and ISSR, isolates from Anhui, Hebei, Henan and Hubei were clustered to one subgroup while isolates from Hunan, Jiangsu, Jiangxi, Shanxi and Shandong were clustered to another subgroup. Such consistent results obtained from both AFLP and ISSR analyses showed the high reliability results of genetic diversity of isolates. Based on the dendrograms results, the minimum variations of AFLP and ISSR were 0.26 and 0.13, and the maximum variations were 0.61 and 0.56, respectively. The results showed that large genetic diversity existed among the isolates studied by both AFLP and ISSR assays. Erdogan et al. (2013) analyzed 33 V. dahliae isolates from cotton in western Turkey by AFLP, and the genetic distance of these isolates was between 0.24 to 0.79. This rate was also similar to the study of Collado-Romero et al. (2006) on V. dahliae diversity. The AFLP analyses also revealed some correlations with pathotypes, which can separate ND isolates from D isolates. Our previous study showed that primer pairs E64/M53 and E49/M65 could yield 433 bp and 110 bp fragments from ND isolates, respectively, which could differentiate ND pathotype from D pathotype (Zou et al. 2003). Collado-Romero et al. (2006) reported that AFLP analysis could cluster the isolates according to their assignment to VCG. Moreover, no association was found between the virulence phenotype, races characterization (data not shown) and AFLP/ISSR clustering of the isolates.
In summary, we tested the pathotypes, races, VCGs and genetic diversity of representative V. dahliae isolates from three major cotton-growing areas in China. The results offered a better understanding of the epidemiology of Verticillium wilt in China and will help formulating the appropriate strategies for cotton wilt management in production practice.
References
Angela M, Iglesias-Garcia MI et al (2013) Pathogenicity, virulence, and vegetative compatibility grouping of Verticillium isolates from spinach seed. Plant Dis 97(11):1457–1469
Atallah ZK, Maruthachalam K, du Toit L et al (2010) Population analyses of the vascular plant pathogen Verticillium dahliae detect recombination and transcontinental gene flow. Fugal Genet Biol 47:416–422
Bejarano-Alcazar J, Blanco-Lopeza MA, Melero-Vara JM et al (1996) Etiology, importance, and distribution of Verticillium wilt of cotton in southern Spain. Plant Dis 80:1233–1238
Bell AA (1992) Verticillium wilt. In: Hillocks RJ (ed) Cotton diseases. C.A.B. International, Oxford, pp 87–126
Collado-Romero M, Mercado-Blanco J, Olivares-Garcia C et al (2006) Molecular variability within and among Verticillium dahliae vegetative compatibility groups determined by fluorescent amplified fragment length polymorphism and polymerase chain reaction markers. Phytopathology 96:485–495
Collado-Romero M, Berbegal M, Jimenez-Diaz RM et al (2009) A PCR-based ‘molecular tool box’ for in planta differential detection of Verticillium dahliae vegetative compatibility groups infecting artichoke. Plant Pathol 58:515–526
Daayf F, Nicole M, Geiger JP (1995) Differentiation of Verticillium dahliae populations on the basis of vegetative compatibility and pathogenicity on cotton. Eur J Plant Pathol 101:69–79
ElSharawy AA, Yang G, Hu X et al (2015) Genetic relationships between virulence, vegetative compatibility and ISSR marker of Verticillium dahliae isolated from cotton. Arch Phytopathol Plant Protect 48(8):646–663
Erdogan O, Nemli S, Oncu T et al (2013) Genetic variation among pathotypes of Verticillium dahliae Kleb. from cotton in western Turkey revealed by AFLP. Can J Plant Pathol 35:354–352
Fahleson J, Lagercrantz U, Hu Q et al (2003) Estimation of genetic variation among Verticillium isolates using AFLP analysis. Eur J Plant Pathol 109:361–371
Geng JY, Zhang XY, Cui RM et al (2006) The development of high-yielding and insect resistant cotton cultivar Jimian 958. J Hebei Agric Sci 10(4):9–13
Gharbi Y, Triki MA, Jolodar A et al (2014) Genetic diversity of Verticillium dahliae from olive trees in Tunisia based on RAMS and IGS-RFLP analyses. Can J Plant Pathol 36:491–500
Gharbi Y, Triki MA, Trabelsi R et al (2015) Genetic structure of Verticillium dahliae isolates infecting Oliver tress in Tunisia using AFLP, pathogenicity and PCR markers. Plant Pathol 64:871–879
Hu XP, Gurung S, Short SPG et al (2015) Nondefoliating and defoliating strains from cotton correlated with races 1 and 2 of Verticillium dahliae. Plant Dis 99(12):1713–1720
Jimenez-Diaz RM, Mercado-Blanco J, Olivares-Garcia C et al (2006) Genetic and virulence diversity in Verticillium dahliae populations infecting artichoke in eastern-Central Spain. Phytopathology 96:288–298
Jimenez-Diaz RM, Cirulli M, Bubici G (2012) Verticillium wilt, a major threat to olive production: current status and future prospects for its management. Plant Dis 96:304–329
Jimenez-Diaz RM, Olivares-Garcia C, Trapero-Casas JL et al (2017) Variation of pathotypes and races and their correlations with clonal lineages in Verticillium dahliae. Plant Pathol 66:651–666
Jimenez-Gasco MM, Malcolm GM, Berbegal M et al (2014) Complex molecular relationship between vegetative compatibility groups (VCGs) in Verticillium dahliae: VCGs do not always align with clonal lineages. Phytopathology 104:650–659
Joaquim TR, Rowe RC (1990) Reassessment of vegetative compatibility relationships among strains of Verticillium dahliae using nitrate-nonutilizing mutants. Phytopathology 80:1160–1166
de Jonge R, van Esse HP, Maruthachalam K, et al, 2012. Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. PNAS, 109 (13): 5110–5115
Lee SB, Taylor JW. 1990. Isolation of DNA from fungal mycelia and single spores. Pages 282–287 in: PCR protocols: A guide to methods and applications. eds. Innis, M.A., D.H. Gelfand, J.J. Sninsky, and T.J. White. Academic Press, Inc, New York
Leslie JK (1993) Fungal vegetative compatibility. Annu Rev Phytopathol 31:127–150
Ma C (2007) Verticillium wilt. In: Ma C (ed) Fusarium wilt and Verticillium wilt of cotton. China Agriculture Press, Beijing, pp 61–70
Martín-Sanz A, Rueda S, García-Carneros Ana B et al (2018) Genetics, host range, and molecular and pathogenic characterization of Verticillium dahliae from sunflower reveal two differentiated groups in Europe. Front Plant Sci 9:288. https://doi.org/10.3389/fpls.2018.00288
Maruthachalam K, Atallah Z, Vallad G et al (2010) Molecular variation among isolates of Verticillium dahliae and polymerase chain reaction-based differentiation of races. Phytopathology 100:1222–1230
Mercado-Blanco J, Rodriguez-Jurado D, Perez-Artes E et al (2001) Detection of the nondefoliating pathotype of Verticillium dahliae in infected olive plants by nested PCR. Plant Pathol 50:609–619
Mercado-Blanco J, Rodriguez-Jurado D, Parrilla-Araujo S et al (2003) Simultaneous detection of the defoliating and nondefoliating Verticillium dahliae pathotypes in infected olive plants by duplex, nested polymerase chain reaction. Plant Dis 87:1487–1494
Perez-Artes E, Garcia-Pedrajas MD, Bejarano-Alcazar et al (2000) Differentiation of cotton-defoliating and nondefoliating pathotypes of Verticillium dahliae by RAPD and specific PCR analyses. Eur J Plant Pathol 106:507–517
Pramateftaki PV, Antoniou PP, Typas MA (2000) The complete DNA sequence of the nuclear ribosomal RNA gene complex of Verticillium dahliae: intraspecific heterogeneity within the intergenic spacer region. Fungal Genet Biol 29:19–27
Ren YH, Zhang WW, Si N et al (2018) Research on integrated management strategy for controlling cotton Verticillium wilt. China Cotton 45(2):28–30
Rohlf FJ (1997) NTSYS-pc: numerical taxonomy and multivariate analysis system. Exeter Publishing, LTD, New York
Schnathorst WC, Mathre DE (1966) Host range and differentiation of a severe form of Verticillium albo-atrum in cotton. Phytopathology 56:1155–1161
Short DP, Gurung S, Maruthachalam K et al (2014) Verticillium dahliae race 2-specific PCR reveals a high frequency of race 2 strains in commercial spinach seed lots and delineates race structure. Phytopathology 104:779–785
Vos P, Hogers R, Bleeker M et al (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Xia ZJ, Achar PN, Gu BK (1998) Vegetative compatibility groupings of Verticillium dahliae from cotton in mainland China. Eur J Plant Pathol 104:871–876
Xu F, Yang L, Zhang J et al (2012) Prevalence of the defoliating pathotype of Verticillium dahliae on cotton in Central China and virulence on selected cotton cultivars. J Phytopathol 160:369–376
Zhang L, Duan WJ, Li GY et al (2006) Population monitoring study of Verticillium dahliae in cotton in Xinjiang. J of Northwest A & F University (Natural Science) 34(11):189–193
Zhang WW, Zhao L, Wang SZ et al (2012) Stability analysis on Verticillium wilt resistance of ten cotton cultivars in disease nursery. China Cotton 39(5):20–22
Zhang WW, Jiang TF, Cui X et al (2013) Colonization in cotton plants by a green fluorescent protein labeled strain of Verticillium dahliae. Eur J Plant Pathol 135:867–876
Zhang WW, Zhang HC, Liu K et al (2017) Large-scale identification of Gossypium hirsutum genes associated with Verticillium dahliae by comparative transcriptomic and reverse genetics analysis. PLoS One 12(8):e0181609
Zhu HQ, Feng ZL, Yin ZX et al (2012) Pathogenicity differentiation and ISSR fingerprint analysis of cotton Verticillium dahliae in China. Acta Phytopathol Sin 42:225–235
Zietkiewicz E, Rafalski A, Labuda D (1994) Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics 20(2):176–183
Zou YF, Jian GL, Ma C et al (2003) AFLP analysis of pathotype of Verticillium dahliae of cotton. Acta Phytopathol Sin 33:135–141
Acknowledgements
This study was supported by the Special Fund for Agro-scientific Research in the Public Interest (201503109), National Key R&D Program of China (No. 2017YFD0201900).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 21 kb)
Rights and permissions
About this article

Cite this article
Zhang, W., Ren, Y., Zhang, H. et al. Genetic variations of prevailing Verticillium dahliae isolates from cotton in China. J Plant Pathol 101, 565–578 (2019). https://doi.org/10.1007/s42161-018-00236-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42161-018-00236-9